Rho-kinase-dependent F-actin rearrangement is involved in the inhibition of PI3-kinase/Akt during ischemia–reperfusion-induced endothelial cell apoptosis (original) (raw)
Introduction
Experimental ischemia and reperfusion (I/R) induces endothelial cell apoptosis in several organs [1, 2]. We and others have previously shown that activation of RhoA and downstream effector Rho-kinase during experimental I/R plays a major role in I/R injury and associated apoptosis [3–7]. Indeed, Rho-kinase inhibition has beneficial effects on I/R injury in several organs [4, 6–12]. Endothelial cells may be essential for mediating the beneficial effects of Rho-kinase inhibition on I/R injury, because, for instance inhibition of endothelial nitric oxide synthase blocked the beneficial effects of Rho-kinase inhibition on rat myocardial infarct size [12].
Rho-kinase activation during the execution of apoptosis results in increased myosin activity, bundling of F-actin by activated myosins, actin–myosin contractile force generation, cell contraction and ultimately membrane blebbing [13–15]. Recent in vivo and in vitro studies suggest that Rho-kinase and the effects of Rho-kinase on the actin–myosin cytoskeleton may also be involved in the initiation of apoptosis [4, 12, 16–20]. Rho-kinase is a negative regulator of the pro-survival phosphatidylinositol 3-kinase (PI3-kinase)/Akt pathway and it upregulates mitochondrial death pathway protein Bax [4, 12, 16, 19]. The effects of Rho-kinase on the cytoskeleton may therefore be important in linking Rho-kinase activation and PI3-kinase/Akt inactivation.
We hypothesized that (1) inhibition of Rho-kinase can prevent I/R-induced endothelial cell apoptosis by maintaining PI3-kinase and Akt activity and (2) prevention of F-actin rearrangement facilitates the protective effects of Rho-kinase inhibition (Fig. 1). To test this hypothesis, we assessed whether specific inhibition of Rho-kinase with Y-27632 [21] attenuates apoptosis via phosphorylation of Akt in an in vitro model of simulated I/R. The involvement of the F-actin cytoskeleton was explored by the use of F-actin depolymerizers cytochalasinD and latrunculinA and F-actin stabilizer jasplakinolide [22].
Fig. 1
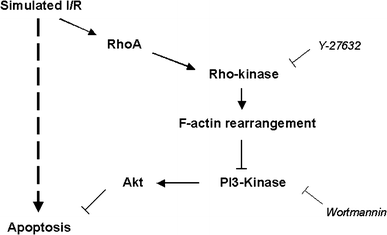
Proposed mechanism for modulation of I/R-induced apoptosis involving RhoA/Rho-kinase and PI3-kinase/Akt. Simulated I/R activates RhoA/Rho-kinase [3–7], which leads to F-actin rearrangement. F-actin rearrangement facilitates reduction of PI3-kinase/Akt activity and induction of apoptosis. This can be prevented by inhibition of Rho-kinase with Y-27632. The protective effects of Y-27632 can be blocked with PI3-kinase inhibitor wortmannin. Several other pathways for I/R-induced apoptosis are represented by the dashed arrow [2, 19]
Materials and methods
Cell culture
Human Umbilical Vein Endothelial Cells (HUVECs) were isolated, cultured and characterized as previously described [23]. Briefly, HUVECs were cultured on gelatin-coated wells/glass cover slips until confluent at 37°C under 5% CO2/95% air atmosphere. Culture medium was renewed every other day (medium 199 supplemented with 10% heat-inactivated human serum, 10% heat-inactivated new born calf serum, 150 μg/ml crude endothelial cell growth factor, 2 mM l-glutamine, 5 U/ml heparin, 100 IU/ml penicillin, and 100 μg/ml streptomycin).
Simulation of I/R
We used an established in vitro endothelial cell model for simulation of ischemia and reperfusion [24, 25]. Figure 2 shows a schematic representation of the experimental protocol. Cells were washed twice with washing buffer (1.2 mM MgSO4 · 7H2O, 116 mM NaCl, 5.3 mM KCl, 1.13 mM NaH2PO4 · H2O, 1.8 mM CaCl2 · 2H2O, 20 mM HEPES) and treated for 1 h with washing buffer with or without 10 μM Y-27632 (Tocris Cookson Ltd., Bristol, United Kingdom (UK)), 250 nM cytochalasinD (Sigma-Aldrich, Saint Louis, Missouri, United States of America (USA)), 150 nM latrunculinA (Calbiochem, Darmstadt, Germany), 20 nM jasplakinolide (Calbiochem) and 50 nM wortmannin (Sigma-Aldrich). This was followed by 1 h of simulated ischemia by covering cells with 1 ml mineral oil (nitrogen bubbled) and subsequently simulated reperfusion by replacement of culture medium for different time points: no reperfusion for adenosine tri-phosphate (ATP) measurement and F-actin staining, 1 h for ATP measurement, F-actin staining and western blotting to analyze phospho Akt (pAkt) levels and 24 h for quantification of apoptosis. Drugs were also present during simulated reperfusion. As controls, we used cells treated for 1 h with washing buffer followed by treatment with medium (control) or medium supplemented with 10% mineral oil (control oil) to assess any injurious effects of products within the oil.
Fig. 2
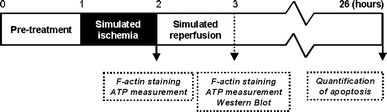
Schematic representation of the experimental protocol. Cells were pre-treated for 1 h with Y-27632, cytochalasinD, latrunculinA, jasplakinolide or wortmannin. One hour of simulated ischemia was followed by 1–24 h of simulated reperfusion. No reperfusion for ATP measurement and F-actin staining, 1 h for ATP measurement, F-actin staining and analysis of Akt activity by western blotting and 24 h for quantification of apoptosis. The drugs were also present during the reperfusion phase
ATP measurement
We measured ATP after ischemia and I/R (1 h of reperfusion) in order to confirm ischemic substrate deprivation. Cells were grown in a gelatin-coated 12-well chamber and treated according to the simulated I/R protocol. ATP was measured using the ENLITEN ATP Assay System Bioluminiscence Detection Kit for ATP (Promega Corporation, Madison, Wisconsin, USA). Proteins in lysate were measured with BCA Protein Assay Kit (Pierce, Rockford, Illinois, USA) in order to calculate nmol ATP/μg protein.
Quantification of apoptosis
The cells were grown in a 24-well chamber on gelatin-coated glass cover slips and treated according to the simulated I/R protocol (24 h of reperfusion). Cells were fixed in 4% formaldehyde and permeabilized with 0.2% triton X-100 (Sigma-Aldrich). The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield® Mounting medium (Vector Laboratories, Inc., Burlingame, California, USA). Apoptosis on each glass cover slip was quantified using a DeadEndTM Fluorometric TdT-mediated dUTP Nick End Labeling (TUNEL) System (Promega Corporation) with fluorescence microscopy using a MarianasTM digital imaging microscope and Slidebook 4.2 software (Intelligent Imaging Innovations, Inc., Denver, Colorado, USA). The number of apoptotic nuclei (TUNEL-positive) and the total number of nuclei (DAPI-positive) were counted in three non-overlapping microscope fields/glass cover slip using a 10× air lens (Carl Zeiss B.V., Sliedrecht, The Netherlands) and averaged. The number of apoptotic nuclei was expressed as percentage of the total number of nuclei.
F-actin cytoskeleton staining
The cells were grown in a 24-well chamber on gelatin-coated glass cover slips and treated according to the simulated I/R protocol (1 h of reperfusion). Cells were fixed in 4% formaldehyde and permeabilized with 0.2% triton X-100. F-actin was stained with rhodamin-phalloidin (1:100) (Molecular Probes, Inc., Eugene, Oregon, USA) and the nucleus with DAPI in Vectashield® Mounting medium. Cells were visualized with fluorescence microscopy and Slidebook software.
Analysis of Akt phosphorylation
Cells were grown in a gelatin coated 6-well chamber, treated according to the simulated I/R protocol (1 h of reperfusion) and lysated with lysis buffer (20 mM Tris/HCl pH 8.0, 150 mM NaCl, 90 mM KCl, 2 mM EDTA/NaOH pH 8.0, igepal (1:200), triton X-100 (1:200), 1 mM Na3VO4, 10 mM NaF, protease inhibitors (1:100), phosphatase inhibitors (1:100)). Protein concentration was measured (BCA method) and equal amounts of protein from each sample were separated by SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, California, USA). Membranes were incubated with polyclonal antibodies against pAkt (serine-473, rabbit, 1:1,000) and total Akt (tAkt, rabbit, 1:1,000) (Cell Signaling Technology, Inc., Danvers, Massachusetts, USA). tAkt was used as loading control. Goat anti-rabbit immunoglobulins horseradish peroxidase (HRP) from DakoCytomation (Glostrup, Denmark) was used for the detection of the primary antibodies at 1:1,000. Detection of the HRP reaction was performed with ECL plus Western Blotting Detection System (Amersham Biosciences, Little Chalfont Buckinghamshire, UK). Imaging and analysis were performed with LAS-3000 (Fuji Photo Film Co., Ltd., Tokyo, Japan) and AIDA Image Analyzer (Raytest GmbH, Straubenhardt, Germany). Data were expressed as pAkt/tAkt as percentage of control.
Statistical analysis
A one-way analysis of variance (ANOVA) was conducted to explore the effects of I/R, Y-27632, cytochalasinD or wortmannin treatment, and their interaction on apoptosis. The _t_-test was used to further analyse the differences between the groups when ANOVA indicated statistical significance. We adjusted for multiple testing with help of the Bonferroni Holm method. The paired _t_-test was used for paired ATP data and logarithmic transformed absolute pAkt/tAkt data. Absolute pAkt/tAkt data were logarithmic transformed to obtain a Gaussian distribution. We adjusted for multiple testing with help of the Bonferroni Holm method. The Pearson coefficient of determination (R 2) was used to express the relation between the percentage of apoptotic nuclei and the logarithmic transformed absolute pAkt/tAkt data. Differences were considered significant at the P < 0.05 level. Data are presented as means ± standard error of the mean (SEM).
Results
ATP levels and apoptosis following simulated ischemia and I/R
In order to confirm ischemic substrate deprivation in simulated I/R, we measured intracellular ATP levels. Ischemia was simulated by covering the monolayer of cells with nitrogen bubbled mineral oil. Reperfusion was simulated by replacement of culture medium (Fig. 2). Intracellular ATP levels decreased following simulated ischemia compared to the levels in cells treated with medium supplemented with 10% mineral oil (control oil) (0.15 ± 0.04 nmol ATP/μg protein versus 0.22 ± 0.04 nmol ATP/μg protein, P < 0.05, n = 7). Intracellular ATP decreased even further during the reperfusion phase (0.10 ± 0.04 nmol ATP/μg protein, P < 0.05 versus control oil, n = 7). ATP levels of control oil cells were comparable to those of control cells (0.20 ± 0.04 nmol ATP/μg protein, n = 7). Simulated I/R induced a significant percentage of apoptotic nuclei (ANOVA for the effect of I/R on apoptosis, P < 0.0001), while hardly any apoptosis was observed in control and control oil cells (Fig. 3).
Fig. 3
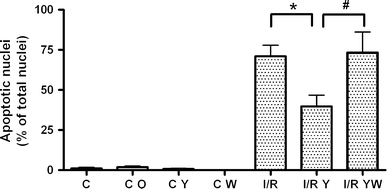
Rho-kinase inhibition reduces the degree of apoptosis during I/R via PI3-kinase. Non-treated control (C, n = 11), control oil (C O, n = 2), Y-27632-treated control (C Y, n = 11) and wortmannin-treated control (C W, n = 5) showed hardly any apoptosis. Simulated I/R induced a significant percentage of apoptotic nuclei (minimum P < 0.005 versus the controls, n = 12). Y-27632 treatment reduced the percentage of apoptotic nuclei during simulated I/R (I/R Y, *P < 0.01, n = 12). This protective effect was completely blocked by wortmannin (I/R YW, # P < 0.05, n = 5). The number of apoptotic nuclei is expressed as percentage of total number of nuclei. Data are presented as means ± SEM
The effect of Rho-kinase inhibition on apoptosis
To investigate the role of Rho-kinase during I/R-induced apoptosis, HUVECs were treated with inhibitor Y-27632 during simulated I/R (Fig. 3). Y-27632 had no effect on basal apoptosis, but reduced the degree of apoptosis during I/R (ANOVA for the effects of Y-27632 and the interaction between I/R and Y-27632 on apoptosis, both P < 0.01). To determine whether the protective effects of Rho-kinase inhibition were mediated by PI3-kinase, cells were co-treated with PI3-kinase inhibitor wortmannin. Wortmannin had no effect on basal apoptosis, but blocked the protective effect of Rho-kinase inhibition during I/R (ANOVA for the effect of wortmannin on apoptosis, P < 0.05).
Akt activity in Y-27632-mediated protection against apoptosis
To investigate the effect of Y-27632 treatment during I/R on Akt activity, Akt phosphorylation was analyzed by western blotting (Fig. 4). Simulated I/R reduced Akt activity compared to non-treated or Y-27632-treated control cells. Y-27632 treatment maintained Akt activity in I/R cells. To determine whether this effect was mediated by PI3-kinase, cells were co-treated with wortmannin. Wortmannin blocked the Y-27632-induced maintenance of Akt activity. Akt activity in Y-27632- (Fig. 4) or wortmannin-treated control cells (preliminary data) was comparable to the activity in non-treated control cells.
Fig. 4
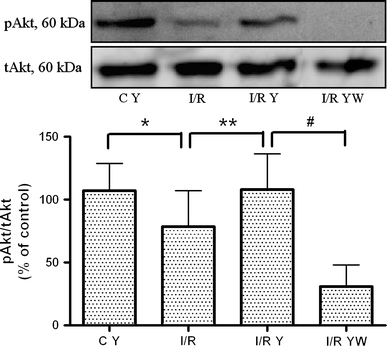
Rho-kinase inhibition maintains Akt activity during I/R. A representative western blot of pAkt and tAkt is shown in the upper panel and a graph of pAkt/tAkt as percentage of control in the lower panel. Simulated I/R (I/R, n = 15) induced a decrease in pAkt compared to the control and the Y-27632-treated control (C Y, *P < 0.05, n = 15). Y-27632 treatment during simulated I/R maintained pAkt levels (I/R Y, **P < 0.05, n = 14). Wortmannin blocked the Y-27632-induced maintenance of Akt activity (I/R YW, # P < 0.05, n = 12). Data are presented as means ± SEM. Absolute pAkt/tAkt data in arbitrary units (a.u.): control: 0.75 a.u.; C Y: 0.64 a.u.; I/R: 0.32 a.u.; I/R Y: 0.36 a.u.; I/R YW: 0.04 a.u
The F-actin cytoskeleton in Y-27632-mediated protection against apoptosis
To investigate whether Y-27632-mediated protection against apoptosis paralleled the absence of Rho-kinase-mediated F-actin rearrangement, the F-actin cytoskeleton was visualized with rhodamin-phalloidin in non-treated or Y-27632-treated control, simulated ischemia and simulated I/R cells (n = 9, representative pictures are shown in Fig. 5). Y-27632-treated control cells showed a slight decrease in peripheral F-actin bundles compared non-treated control cells, indicating that in vitro inhibition of Rho-kinase was able to reduce F-actin bundle formation. After simulated ischemia in non-treated cells, some F-actin bundles appeared in the cytoplasm. Y-27632-treated ischemic cells showed only F-actin clumps. After simulated reperfusion, even more F-actin bundles appeared in the periphery and the cytoplasm of the non-treated cells. In contrast, Y-27632-treated I/R cells, showed no F-actin bundles.
Fig. 5
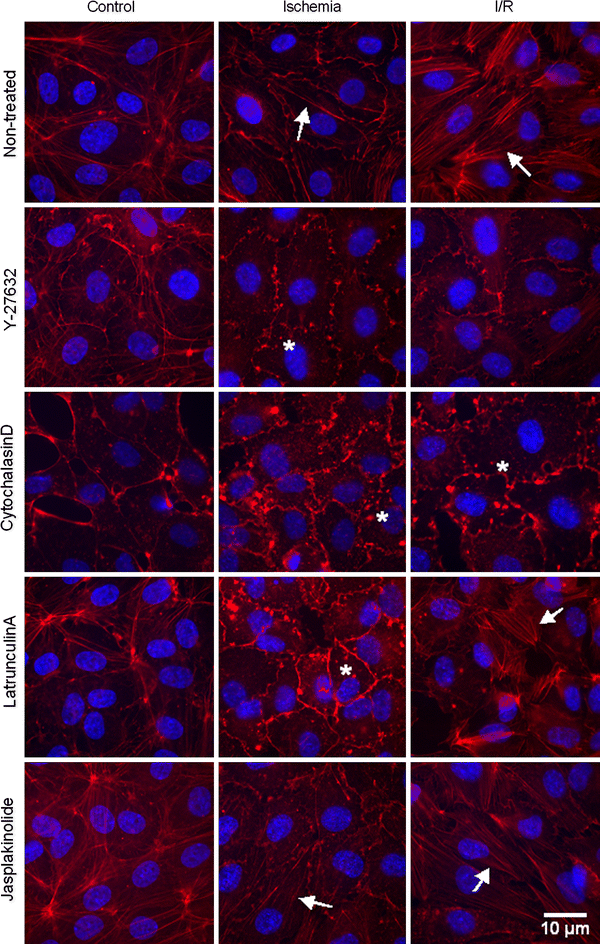
Y-27632 and cytochalasinD prevent F-actin bundle formation during I/R. Shown is fluorescent imaging of rhodamin-phalloidin stained F-actin structures and DAPI stained nuclei in non-treated, Y-27632-, cytochalasinD-, latrunculinA- and jasplakinolide-treated control, simulated ischemia and simulated I/R cells. The arrow and * indicate F-actin bundles and F-actin clumps, respectively. Control cells: In non-treated cells, most F-actin bundles were seen in the periphery of the cell. Y-27632-treated cells showed a slight decrease in peripheral F-actin bundles. CytochalasinD-treated cells showed only F-actin clumps. Cells treated with latrunculinA or jasplakinolide showed a slight decrease and increase in F-actin bundles, respectively. Ischemic cells: Non-treated cells showed F-actin bundles throughout the whole cell. Cells treated with Y-27632, cytochalasinD or latrunculinA showed only F-actin clumps. Jasplakinolide-treated cells showed F-actin bundles. I/R cells: Non-treated cells showed F-actin bundles throughout the whole cell. In Y-27632-treated cells, no F-actin bundles were seen. CytochalasinD-treated cells showed also no F-actin bundles, only F-actin clumps were visible. LatrunculinA-treated cells showed some F-actin bundles. Jasplakinolide-treated cells showed as many F-actin bundles as non-treated I/R cells
To further investigate the role of the F-actin cytoskeleton in I/R-induced apoptosis, we used F-actin depolymerizers cytochalasinD and latrunculinA and F-actin stabilizer jasplakinolide (Fig. 5). CytochalasinD-treated control cells showed no F-actin bundles. LatrunculinA-treated control cells showed a slight decrease and jasplakinolide-treated control cells showed a slight increase in F-actin bundles compared to non-treated control cells. CytochalasinD- and latrunculinA-treated ischemic cells showed no F-actin bundles, only F-actin clumps were visible. Jasplakinolide-treated ischemic cells showed F-actin bundles throughout the whole cell. In cytochalasinD-treated I/R cells, no F-actin bundles were visible, only F-actin clumps could be seen. In contrast, latrunculinA-treated I/R cells still showed some F-actin bundles and jasplakinolide-treated I/R cells showed as many F-actin bundles as non-treated I/R cells.
Since cytochalasinD treatment prevented F-actin rearrangement similarly to Y-27632 treatment, we investigated whether cytochalasinD treatment could also prevent apoptosis during simulated I/R (Fig. 6). CytochalasinD had no effect on basal apoptosis, but reduced the degree of apoptosis during I/R (ANOVA for the effects of cytochalasinD and the interaction between I/R and cytochalasinD on apoptosis, both P < 0.05). Wortmannin blocked the protective effect of cytochalasinD in I/R (ANOVA for the effect of wortmannin on apoptosis, P < 0.0001). In contrast to the protective effect of Y-27632 and cytochalasinD on apoptosis, latrunculinA and jasplakinolide did not affect I/R-induced apoptosis, also not in co-treatment with wortmannin (data not shown). LatrunculinA and jasplakinolide had no effect on basal apoptosis (data not shown).
Fig. 6
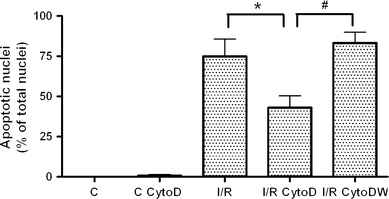
CytochalasinD reduces the degree of apoptosis during I/R via PI3-kinase. Non-treated control (C, n = 6) and cytochalasinD-treated control (C CytoD, n = 6) showed hardly any apoptosis. Simulated I/R induced a significant percentage of apoptotic nuclei (P < 0.0001 versus the controls, n = 6). Treatment with cytochalasinD reduced the percentage of apoptotic nuclei in simulated I/R (I/R CytoD, *P < 0.05, n = 6). This protective effect was completely blocked by wortmannin (I/R CytoDW, # P < 0.005, n = 6). The number of apoptotic nuclei is expressed as percentage of total number of nuclei. Data are presented as means ± SEM
Furthermore, cytochalasinD maintained Akt activity during simulated I/R (Fig. 7). Co-treatment with wortmannin completely blocked the cytochalasinD-induced maintenance of Akt activity. Akt activity in cytochalasinD-treated control cells was comparable to the activity in non-treated control cells.
Fig. 7
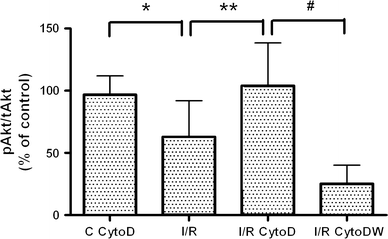
CytochalasinD treatment maintains Akt activity during I/R. Simulated I/R (I/R, n = 11) induced a decrease in pAkt compared to the control and the cytochalasinD-treated control (C CytoD, *P < 0.01, n = 11). CytochalasinD treatment during simulated I/R maintained pAkt levels (I/R CytoD, **P < 0.05, n = 11). Wortmannin blocked the cytochalasinD-induced maintenance of Akt activity (I/R CytoDW, # P < 0.01, n = 7). Data are presented as means ± SEM. Absolute pAkt/tAkt data in arbitrary units (a.u.): control: 0.61 a.u.; C CytoD: 0.84 a.u.; I/R: 0.36 a.u.; I/R CytoD: 0.54 a.u.; I/R CytoDW: 0.05 a.u
Interestingly, the percentage of apoptotic nuclei inversely correlated to Akt activity (Fig. 8), indicating that a decrease in Akt activity correlated to an increase in apoptosis.
Fig. 8
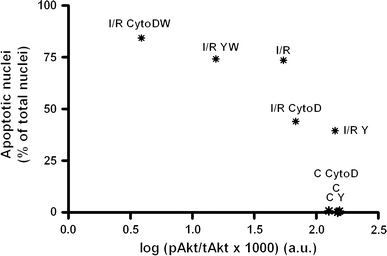
The percentage of apoptotic nuclei inversely correlates to Akt activity (R 2 = 0.68, P < 0.05). Mean percentage of apoptotic nuclei and mean logarithmic transformed absolute pAkt/tAkt levels in non-treated (C), Y-27632 (C Y)- and cytochalasinD (C CytoD)-treated control cells and in non-treated (I/R), Y-27632 (I/R Y)-, cytochalasinD (I/R CytoD)- and wortmannin (I/R YW and I/R CytoDW)- treated I/R cells are shown as *
Discussion
This study shows that (1) inhibition of Rho-kinase attenuates I/R-induced endothelial cell apoptosis by maintaining PI3-kinase and Akt activity and (2) the effects of Rho-kinase inhibition on PI3-kinase/Akt and apoptosis are facilitated by prevention of F-actin rearrangement.
Our data point to a novel mechanistic link between activation of Rho-kinase during I/R and negative regulation of PI3-kinase/Akt, namely the effect of Rho-kinase on F-actin rearrangement (Fig. 1). Prevention of F-actin rearrangement by Rho-kinase inhibition or by treatment with actin depolymerizer cytochalasinD, independent of Rho-kinase, both attenuated I/R-induced endothelial cell apoptosis by maintaining PI3-kinase and Akt activity. Indeed, it has been reported previously that cytochalasins inhibited in vivo and in vitro apoptosis induced by a variety of stimuli, including I/R [5, 13, 26]. This study is therefore an extension of and a link between reports on Rho-kinase as negative regulator of the PI3-kinase/Akt pathway and reports on the involvement of Rho-kinase-dependent cytoskeletal rearrangement in the initiation of apoptosis [4, 12, 16–20].
Actin depolymerizer latrunculinA did not affect I/R-induced endothelial cell apoptosis. This may be explained by the nature and degree of F-actin rearrangement during I/R, since F-actin bundles were completely absent in Y-27632- and cytochalasinD-treated I/R cells, while latrunculinA-treated cells still showed some bundles. Indeed, cytochalasinD and latrunculinA depolymerize F-actin by different mechanisms and induce different patterns in vitro [22, 27]. CytochalasinD caps the barbed end of actin filaments, severs actin filaments, sequesters actin monomers or dimers, promotes nucleation and stimulates the ATPase activity of G-actin. In vitro in hamster fibroblasts, it caused a complete collapse of the stress fibers at concentrations comparable to ours [22, 27]. LatrunculinA on the other hand binds to monomeric G-actin to form a non-polymerizable complex and caused concentration-dependent shortening and thickening of stress fibers in the fibroblasts at concentrations comparable to ours [22, 27]. Jasplakinolide, which promotes actin polymerization and stabilizes actin filaments by binding to F-actin [22], induced as many F-actin bundles in I/R as non-treated I/R cells and consequently did not affect I/R-induced endothelial apoptosis. However, in vivo it has been demonstrated that jasplakinolide prevented I/R-induced apoptosis in the rat kidney, while latrunculinB increased I/R-induced apoptosis in rat kidney [28, 29]. Furthermore, we observed hardly any apoptosis in our Y-27632-, cytochalasinD-, latrunculinA- and jasplakinolide-treated control cells, while some reports describe the induction of apoptosis in control cells by latrunculinA or jasplakinolide at much higher concentrations than ours [30, 31], by cytochalasinD in airway epithelial cells, but not in canine kidney epithelial cells [32] or by Y-27632 in airway epithelial cells [32]. Therefore, these discrepancies may be explained by the use of different concentrations, cell types or the use of an in vivo/in vitro model.
Akt activation after 1 h of reperfusion inversely correlated to the percentage of apoptosis after 24 h of reperfusion. Therefore, early reduced Akt activity may have a predictive value for the development of late I/R-induced apoptosis. The preventive effect of Y-27632 and cytochalasinD treatment probably takes place early in the process of apoptosis, because the Y-27632-induced increase in Akt activity is rapid and transient with a maximum after 30 min [12, 16]. Indeed, we could neither detect increased Akt activity in our Y-27632-treated control cells after 3 h nor in our Y-27632-treated I/R cells after 24 h of reperfusion (pilot study, data not shown), possibly because this was to late to detect the peak in Akt activity. I/R-induced apoptosis was maximal as late as after 24–48 h of reperfusion [25]. Therefore, apoptosis was quantified after 24 h of reperfusion. Indeed, we did not see any TUNEL-positive nuclei after 1 h of reperfusion (pilot study, data not shown). Our percentage of apoptotic nuclei was comparable to the percentage reported by the developers of the simulated I/R model [25]. However, less endothelial cell apoptosis was observed in a model of hypoxia/reoxygenation [33], possibly because this induces less severe metabolic deprivation. In addition, PI3-kinase inhibition did not decrease basal Akt activity nor increased basal apoptosis, while it blocked the effects of Rho-kinase inhibition on I/R-induced apoptosis and Akt activity. This suggests that the basal situation is independent of PI3-kinase, while the effects Rho-kinase inhibition during I/R are dependent on PI3-kinase.
Although it is unclear by which mechanism F-actin rearrangement facilitates the effects of Rho-kinase on PI3-kinase/Akt and apoptosis, several mechanisms can be proposed. First, RhoA and Rho-kinase may regulate pAkt levels by regulating phosphatase and tensin homologue (PTEN) activity. PTEN is in inactive state in the cytosol and translocates to the membrane upon RhoA or Rho-kinase-induced activation, where it can inhibit the phosphorylation of Akt via PI3-kinase [34]. Disruption of the F-actin bundles by Rho-kinase inhibition or cytochalasinD treatment or the direct effects of Rho-kinase inhibition on PTEN may prevent the translocation and activation of PTEN, thereby preventing its inhibitory effect on Akt phosphorylation. Furthermore, there may be some PI3-kinase independent mechanisms involved in the Y-27632-induced protection against apoptosis. It is reported that F-actin polymerization and actin–myosin contractility are necessary for membrane blebbing and chromatin condensation during execution of apoptosis induced by tumor necrosis factor-α or overexpression of Rho-kinase [13, 26]. Moreover, the actin–myosin cytoskeleton may be involved in facilitating the cellular localization of specific components of the apoptotic machinery and be required for caspase-3 activation [17, 18].
Our study carries some limitations. First, we cannot conclude on the mechanisms by which F-actin rearrangement facilitates the effects of Rho-kinase on PI3-kinase/Akt and apoptosis. Second, Rho-kinase inhibition and F-actin depolymerization could establish only a partial protection against apoptosis, in agreement with the literature [4, 5, 8, 12], possibly because other mechanisms are also involved and the I/R-induced injury is too extensive. Third, we may have underestimated the number of apoptotic cells, because endothelial cells that detached from the basement membrane during simulated I/R and did not reattach later were not counted in the quantification of apoptosis. Indeed, Prahalad et al. [35] describe this reversible cell detachment during ATP depletion of Madin-Darby canine kidney cells.
Conclusion
Prevention of F-actin rearrangement or F-actin bundle formation by Rho-kinase inhibition or by treatment with actin depolymerizator cytochalasinD, independent of Rho-kinase, attenuated I/R-induced endothelial cell apoptosis by maintaining PI3-kinase and Akt activity. The present finding suggests that the changes in the F-actin cytoskeleton play a pivotal role in the negative regulation of PI3-kinase and Akt by Rho-kinase. Further studies are required to conclude on the mechanisms by which F-actin rearrangement facilitates the effects of Rho-kinase on PI3-kinase/Akt and apoptosis.
References
- Horbelt M, Lee SY, Mang HE et al (2007) Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol 293:F688–F695
Article PubMed CAS Google Scholar - Scarabelli TM, Stephanou A, Pasini E et al (2002) Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ Res 90:745–748
Article PubMed CAS Google Scholar - Bailly K, Ridley AJ, Hall SM, Haworth SG (2004) RhoA activation by hypoxia in pulmonary arterial smooth muscle cells is age and site specific. Circ Res 94:1383–1391
Article PubMed CAS Google Scholar - Hamid SA, Bower HS, Baxter GF (2007) Rho kinase activation plays a major role as a mediator of irreversible injury in reperfused myocardium. Am J Physiol Heart Circ Physiol 292:H2598–H2606
Article PubMed CAS Google Scholar - Sanada S, Asanuma H, Tsukamoto O et al (2004) Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 110:51–57
Article PubMed CAS Google Scholar - Versteilen AM, Korstjens IJ, Musters RJ, Groeneveld AB, Sipkema P (2006) Rho kinase regulates renal blood flow by modulating eNOS activity in ischemia–reperfusion of the rat kidney. Am J Physiol Renal Physiol 291:F606–F611
Article PubMed CAS Google Scholar - Manintveld OC, Verdouw PD, Duncker DJ (2007) The RISK of ROCK. Am J Physiol Heart Circ Physiol 292:H2563–H2565
Article PubMed CAS Google Scholar - Bao W, Hu E, Tao L et al (2004) Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 61:548–558
Article PubMed CAS Google Scholar - Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E (2005) Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci 238:31–39
Article PubMed CAS Google Scholar - Ikeda F, Terajima H, Shimahara Y, Kondo T, Yamaoka Y (2003) Reduction of hepatic ischemia/reperfusion-induced injury by a specific ROCK/Rho kinase inhibitor Y-27632. J Surg Res 109:155–160
Article PubMed CAS Google Scholar - Teraishi K, Kurata H, Nakajima A, Takaoka M, Matsumura Y (2004) Preventive effect of Y-27632, a selective Rho-kinase inhibitor, on ischemia/reperfusion-induced acute renal failure in rats. Eur J Pharmacol 505:205–211
Article PubMed CAS Google Scholar - Wolfrum S, Dendorfer A, Rikitake Y et al (2004) Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol 24:1842–1847
Article PubMed CAS Google Scholar - Song Y, Hoang BQ, Chang DD (2002) ROCK-II-induced membrane blebbing and chromatin condensation require actin cytoskeleton. Exp Cell Res 278:45–52
Article PubMed CAS Google Scholar - Coleman ML, Olson MF (2002) Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death Differ 9:493–504
Article PubMed CAS Google Scholar - Van Nieuw Amerongen GP, Van Hinsbergh VW (2001) Cytoskeletal effects of rho-like small guanine nucleotide-binding proteins in the vascular system. Arterioscler Thromb Vasc Biol 21:300–311
PubMed Google Scholar - Ming XF, Viswambharan H, Barandier C et al (2002) Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 22:8467–8477
Article PubMed CAS Google Scholar - Petrache I, Crow MT, Neuss M, Garcia JG (2003) Central involvement of Rho family GTPases in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res Commun 306:244–249
Article PubMed CAS Google Scholar - Lai JM, Hsieh CL, Chang ZF (2003) Caspase activation during phorbol ester-induced apoptosis requires ROCK-dependent myosin-mediated contraction. J Cell Sci 116:3491–3501
Article PubMed CAS Google Scholar - Del Re DP, Miyamoto S, Brown JH (2007) RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem 282:8069–8078
Article PubMed Google Scholar - Kureishi Y, Luo Z, Shiojima I et al (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004–1010
Article PubMed CAS Google Scholar - Ishizaki T, Uehata M, Tamechika I et al (2000) Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol 57:976–983
PubMed CAS Google Scholar - Spector I, Braet F, Shochet NR, Bubb MR (1999) New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc Res Tech 47:18–37
Article PubMed CAS Google Scholar - Draijer R, Atsma DE, Van der Laarse A, Van Hinsbergh VW (1995) cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res 76:199–208
PubMed CAS Google Scholar - Henry P, Popescu A, Puceat M, Hinescu ME, Escande D (1996) Acute simulated ischaemia produces both inhibition and activation of K+ currents in isolated ventricular myocytes. Cardiovasc Res 32:930–939
Article PubMed CAS Google Scholar - Meldrum KK, Meldrum DR, Hile KL, Burnett AL, Harken AH (2001) A novel model of ischemia in renal tubular cells which closely parallels in vivo injury. J Surg Res 99:288–293
Article PubMed CAS Google Scholar - Suarez-Huerta N, Mosselmans R, Dumont JE, Robaye B (2000) Actin depolymerization and polymerization are required during apoptosis in endothelial cells. J Cell Physiol 184:239–245
Article PubMed CAS Google Scholar - Spector I, Shochet NR, Blasberger D, Kashman Y (1989) Latrunculins—novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton 13:127–144
Article PubMed CAS Google Scholar - Genesca M, Sola A, Hotter G (2006) Actin cytoskeleton derangement induces apoptosis in renal ischemia/reperfusion. Apoptosis 11:563–571
Article PubMed CAS Google Scholar - Vinas JL, Hotter G, Pi F, Palacios L, Sola A (2007) Role of peroxynitrite on cytoskeleton alterations and apoptosis in renal ischemia–reperfusion. Am J Physiol Renal Physiol 292:F1673–F1680
Article PubMed CAS Google Scholar - Rao JY, Jin YS, Zheng Q, Cheng J, Tai J, Hemstreet GP (1999) Alterations of the actin polymerization status as an apoptotic morphological effector in HL-60 cells. J Cell Biochem 75:686–697
Article PubMed CAS Google Scholar - Martin SS, Leder P (2001) Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol 21:6529–6536
Article PubMed CAS Google Scholar - Moore M, Marroquin BA, Gugliotta W, Tse R, White SR (2004) Rho kinase inhibition initiates apoptosis in human airway epithelial cells. Am J Respir Cell Mol Biol 30:379–387
Article PubMed CAS Google Scholar - Mold C, Morris CA (2001) Complement activation by apoptotic endothelial cells following hypoxia/reoxygenation. Immunology 102:359–364
Article PubMed CAS Google Scholar - Li Z, Dong X, Wang Z et al (2005) Regulation of PTEN by Rho small GTPases. Nat Cell Biol 7:399–404
Article PubMed CAS Google Scholar - Prahalad P, Calvo I, Waechter H, Matthews JB, Zuk A, Matlin KS (2004) Regulation of MDCK cell-substratum adhesion by RhoA and myosin light chain kinase after ATP depletion. Am J Physiol Cell Physiol 286:C693–C707
Article PubMed CAS Google Scholar