Phase I and pharmacokinetic study of dacomitinib (PF-00299804), an oral irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese patients with advanced solid tumors (original) (raw)
Summary
Background Dacomitinib (PF-00299804) is an oral, irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases. Methods This phase I, open-label, dose-escalation study (clinicaltrials.gov: NCT00783328) primarily evaluated the safety and tolerability of dacomitinib by dose-limiting toxicity (DLT), and determined the clinically recommended phase II dose (RP2D) in Japanese patients with advanced solid tumors. Dacomitinib was administered orally at three dose levels (15, 30, or 45 mg once daily [QD]). Patients initially received a single dose, and after 9 days of follow-up, continuously QD in 21-day cycles. Endpoints included pharmacokinetics (PK) and antitumor activity. Results Thirteen patients were assigned to the three dose levels (15 mg cohort: n = 3; 30 mg cohort: n = 3; 45 mg cohort: n = 7) according to a traditional ‘3 + 3’ design. None of the treated patients experienced a DLT. Toxicities were manageable and similar in type to those observed in other studies. PK concentration parameters increased with dose over the range evaluated, with no evidence of accumulation over time. Of 13 evaluable patients, one with NSCLC (adenocarcinoma) had a partial response and nine patients had stable disease. Conclusions Dacomitinib 45 mg QD was defined as the RP2D and demonstrated preliminary activity in Japanese patients with advanced solid tumors.
Similar content being viewed by others
Introduction
The epidermal growth factor receptor (EGFR) is a membrane receptor tyrosine kinase. EGFR belongs to the human epidermal growth factor receptor (HER) family, comprising the following four types of highly similar membrane receptors: EGFR (HER1), HER2, HER3, and HER4 [1]. The role of the HER family members in many cancer types is well documented. One or more members of this receptor family are expressed in over 90% of solid tumors, and approximately 60% of those tumors possess abnormalities in this family that potentially contribute to their neoplastic phenotype [2, 3]. EGFR is activated by binding of selective ligands, with subsequent receptor dimerization. This results in autophosphorylation which is known to lead to the regulation of various biological processes, including cell proliferation, angiogenesis, invasion/metastasis, and suppression of apoptosis [4]. The simultaneous activation of different HER family members through dynamic hetero- and homodimerization could compromise the therapeutic efficacy by inhibition of a single receptor. Therefore, an inhibitor that blocks the tyrosine kinase activity of the entire HER family, and hence the signaling of both hetero- and homodimers, could have a significant therapeutic effect, even in tumors that have not previously responded to conventional single-receptor inhibitors.
Dacomitinib (PF-00299804; Pfizer Inc., New York, NY, USA) is an orally administered, highly selective irreversible small molecule inhibitor of the HER family of tyrosine kinases (HER1, HER2, and HER4) [5, 6]. In preclinical human tumor xenograft models that express and/or overexpress HER family members, dacomitinib showed antitumor effects. Antitumor effects were shown against tumors with _EGFR_-activating mutations, found in 30–50% of Asian patients with lung cancer [7], and also against tumors with T790M, a mutation detected in the tumors of approximately 50% of patients with lung adenocarcinoma who develop acquired resistance to gefitinib or erlotinib [8–10].
In a phase I, dose-escalation study [11], the safety of dacomitinib (0.5–60 mg) was studied in Western patients with advanced solid tumors. Dose-limiting toxicities (DLTs) included stomatitis (n = 2; 1 each at 30 mg once daily [QD] and 60 mg QD), rash (n = 2; at 45 mg QD), palmar–plantar erythrodysesthesia syndrome, dehydration, and diarrhea in the same patient (n = 1 at 60 mg QD). The maximum tolerated dose (MTD) was defined as the highest dose level at which the proportion of patients with DLTs did not exceed the protocol-defined rate of 33%. Thus, dacomitinib 45 mg QD was determined as the MTD, and also chosen as the clinically recommended phase II dose (RP2D). Subsequent phase II studies have been conducted with a starting dose of 45 mg QD in advanced non-small cell lung cancer (NSCLC) and in squamous cell cancer of the head and neck [12–14].
The aim of this phase I, open-label, dose-escalation study (NCT00783328) was primarily to evaluate the safety and tolerability of dacomitinib in Japanese patients with advanced solid tumors at doses up to the RP2D of 45 mg QD, which had previously been determined in a Western study [11, 15]. Secondary objectives were to evaluate the pharmacokinetic (PK) profile of dacomitinib following single and multiple dosing in Japanese patients, and to assess antitumor activity.
Materials and methods
Patient population
Patients were aged 20–75 years (male or female), with histologic or cytopathologic diagnosis of solid tumors. Other key inclusion criteria included: Eastern Cooperative Oncology Group (ECOG) performance status of 0/1; resolution of acute toxicities caused by prior therapy or surgery to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3.0 grade 1; previous drug treatment for cancer completed and no further treatment for at least 3 weeks prior to initiation of study treatment; and adequate organ function, including left ventricular ejection fraction (LVEF) ≥50%.
Key exclusion criteria included: clinically significant abnormalities of the cornea; brain/central nervous system metastases (symptomatic and/or requiring treatment); any clinically significant gastrointestinal abnormalities; uncontrolled or significant cardiovascular disease; grade 3/4 diarrhea or skin rash toxicity and/or history of pneumonitis related to treatment with a tyrosine kinase inhibitor; and requirement for drugs that are highly dependent on cytochrome P450 (CYP) 2D6 for metabolism.
Study design and treatment
This was a phase I, open-label, dose-escalation study. Dacomitinib was administered at three dose levels (15, 30, and 45 mg) orally. After single-dose administration, and 9 days of follow-up, dacomitinib was given continuously QD in 21-day cycles. Treatment was repeated in 21-day cycles until progression, unacceptable toxicity, or withdrawal of patient consent.
A traditional 3 + 3 dose-escalation design was used to escalate dacomitinib 15 mg QD to 45 mg QD, and an additional three patients were enrolled into the 45-mg cohort for the safety analysis. Dose interruptions or reductions were permitted in patients experiencing grade 3/4 toxicity, with treatment resumed at the same or next lowest dose level upon adequate recovery (grade ≤1). Tolerability was evaluated according to the type and frequency of DLTs observed from single-dose administration to day 21 of the first cycle of continuous dosing. DLTs were defined as: grade ≥3 non-hematologic toxicity uncontrollable with standard supportive care; delayed recovery from toxicity related to dacomitinib treatment which postponed scheduled retreatment for >14 days; patients unable to take at least 50% of planned doses due to toxicity related to dacomitinib; grade 4 neutropenia or febrile neutropenia; and grade 4 thrombocytopenia (<25,000 cells/mm3) or bleeding requiring a platelet transfusion.
If a patient’s dose was delayed due to a DLT, treatment could be resumed at the next lowest dose level. Dose reductions (two dose reductions for patients treated at 30 mg QD, one dose reduction for patients treated at 15 mg QD) were permitted. However, if a dose was delayed due to toxicity for ≥21 days, treatment was not to be resumed and the patient was withdrawn from the study.
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization (ICH) Good Clinical Practice (GCP) Guidelines. In addition, all local regulatory requirements were followed, in particular, those affording greater protection for the safety of trial participants. The final protocol, any amendments, and informed consent documentation were reviewed and approved by the Institutional Review Board at the Shizuoka Cancer Center. The study was conducted at one center in Japan. All patients provided written, signed informed consent prior to entry into the trial.
Assessments and analysis
LVEF, slit lamp ophthalmologic examination, vital signs, ECOG performance status, laboratory tests (hematology, blood chemistry, coagulation, and urinalysis), chest X-ray, and electrocardiogram were performed at baseline and at regular intervals throughout the study. Safety was assessed according to CTCAE version 3.0.
Analyses for detection of HER1 and KRAS mutations in tumor tissue were performed as optional at baseline. Tumor assessments were performed at baseline, cycle 2, cycle 4, and every 6 weeks thereafter. Evaluation of antitumor activity was based on objective tumor assessments using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 [16].
Evaluation of best overall response (BOR) was determined as the most favorable overall response confirmed as partial response (PR) or complete response (CR) during the treatment period, or as stable disease (SD) if a response of SD, PR or CR was achieved without subsequent confirmation at a response evaluation at least 6 weeks after initiation of multiple-dose administration. An evaluation of PR or CR required that changes in tumor measurements were confirmed by repeated assessments performed no less than 4 weeks after the criteria for the response had first been met.
Pharmacokinetic assessments
Serial blood samples for PK assessment were collected after a single dose on any day between 9 and 1 days prior to the start of continuous dosing (referred to as D-9 throughout this manuscript), and on day 14 of cycle 1 (C1D14; steady state). Pre-dose blood samples were collected on day 1 of cycles 2–4 (plasma trough concentrations [Ctrough]). Plasma samples were analyzed for dacomitinib concentrations at Alta Analytical Laboratory (El Dorado Hills, CA, USA) using a validated analytical assay (validated, sensitive, and a specific high-performance liquid chromatography tandem mass spectrometric method [LC/MS/MS]) in compliance with Pfizer standard operating procedures.
Pharmacokinetic parameters were derived from dacomitinib plasma concentration after single and multiple dosing using non-compartmental analysis. For single-dose administration (D-9), the following PK parameters were calculated: maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), terminal half-life (t1/2), area under the plasma concentration–time curve from 0 to 24 h after a single dose (AUC24), the area under the plasma concentration–time curve from 0 to infinity (AUCinf), and clearance (CL). For multiple-dose administration (C1D14), the following PK parameters were calculated: Cmax, Tmax, CL, area under the plasma concentration–time curve from 0 to 24 h at steady state (AUCτ), trough concentration (Ctrough), mean plasma concentration (Cave), accumulation ratio (Rac, the ratio of AUCτ to AUC24), and the linearity ratio (Rss, the ratio of AUCτ to AUCinf).
For both single- and multiple-dose administration, descriptive statistics were calculated (arithmetic mean, standard deviation, coefficient of variation, median, and geometric mean). Trough concentration data from cycle 2 day 1, cycle 3 day 1, and cycle 4 day 1 were analyzed together with the trough concentration data from cycle 1 day 14 to assess whether the PK steady-state had been achieved.
Dynamic model of tumor size
Change in size of tumor target lesions over time was recorded as the sum of the longest dimensions; all target lesions were measured using spiral computed tomography (CT) or magnetic resonance imaging (MRI) according to RECIST version 1.0 [16]. The longitudinal tumor size data were analyzed using nonlinear mixed effect models (NONMEM® 7.12, Globomax). The time course of tumor growth was described using two parameters based on a previous report [[17](/article/10.1007/s10637-011-9789-z#ref-CR17 "FDA Advisory Committee Briefing Document: Pharmaceutical Science and Clinical Pharmacology Advisory Committee, March 18−19, 2008 ( http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4351b1-00-index.htm
)")\]: shrinkage rate (SR) following an exponential tumor growth decline, and a linear progression rate growth (TPR):textTtextStextileft(texttright)=textBAStextEtexti.texte−−textSRtexti.textt+textTPtextRtexti.textt{\text{T}}{{\text{S}}_{\text{i}}}\left( {\text{t}} \right){ } = {\text{ BAS}}{{\text{E}}_{\text{i}}}.{{\text{e}}^{{--{\text{SR}}}}}{{\text{i}}^{{.{\text{t}}}}} + {\text{TP}}{{\text{R}}_{\text{i}}}.{\text{t}}textTtextStextileft(texttright)=textBAStextEtexti.texte−−textSRtexti.textt+textTPtextRtexti.textt
where TS i (t) is the tumor size at time t for the i th individual, Base i is the observed individual tumor size at baseline, SR i is the tumor shrinkage rate constant, and TPR i is the linear tumor progression rate.
Inter-individual variability (IIV) was accounted for in the population mean parameters using an exponential error model:
thetai=thetacdoteetai{\theta_i} = \theta \cdot {e^{{{\eta_i}}}}thetai=thetacdoteetai
where θ i is the individual parameter estimate, θ is the mean population value of the parameter (SR or TPR), and η is a random variable to describe the IIV. The IIV has a normal probability distribution, with a mean of 0 and variance ω2. The estimates of IIV were presented as standard deviations calculated as √ω2.
The residual variability was modeled using a homoscedastic, exponential error model of the form:
Yij=Fijcdotevarepsilonij{Y_{{ij}}} = {F_{{ij}}} \cdot {e^{{{\varepsilon_{{ij}}}}}}Yij=Fijcdotevarepsilonij
where Y ij is the observed data for the i th individual at time t j , F ij is the prediction based on the pharmacodynamic model, and ε ij is the exponential error that is a normally distributed random variable with a mean of 0 and variance σ2.
The purpose of this tumor model analysis was to explore the potential effect of several covariates on tumor growth parameters (dose, gender, baseline ECOG, and EGFR mutation status). Full model development involved testing for covariates (both continuous and categorical) with the goal of explaining inter-individual variability and improving predictive performance.
Parameterization of covariate models was guided by examination of the plots of the ηs on SR and TPR versus covariates (gender, ECOG PS, dose cohort, and EGFR mutation status) from the final base model.
Dichotomous covariates were modeled as follows:
theta=theta0cdotleft(1+,,thetatextxcdottextxright)\theta = {\theta _{0}} \cdot \left( {1 + \,\,{\theta _{{\text{x}}}} \cdot {\text{x}}} \right)theta=theta0cdotleft(1+,,thetatextxcdottextxright)
where θ0 denotes the population value of the parameter for the null value of the covariate x (i.e. x = 0). The parameter θx denotes the fractional change in θ0 when x = 1.
A full-model approach was performed after the selection of the covariates based on the assessment of the ηs plots or assessment of the individual parameter estimates versus the covariates of interest. The final model was constructed after removing all covariates that were not statistically significant (e.g. α = 0.01) and also those covariates that did not have a clinically significant effect.
Results
Patient characteristics and disposition
Thirteen patients were enrolled across three dacomitinib dose levels: 15 mg QD, n = 3; 30 mg QD, n = 3; 45 mg QD, n = 7. The most common tumor type in the 13 treated patients was NSCLC (n = 9, 69%). All patients were Japanese; baseline characteristics are summarized in Table 1.
Table 1 Patient characteristics at baseline
The median number of cycles received at 15, 30, and 45 mg QD was 2 (range, 1–37), 6 (range, 4–9), and 6 (range, 1–13), respectively.
All 13 patients were finally withdrawn from the study; the most common reason for discontinuation was disease progression (n = 10). Other reasons were global deterioration (n = 2) and adverse event (AE) (rash; n = 1). One patient in the 45 mg cohort required dose reduction due to AE (grade 2 rash).
Safety and tolerability
One patient in the 45 mg cohort was excluded from the DLT analysis set due to poor treatment compliance (28.6% of planned doses received) in cycle 1 of multiple-dose administration due to disease progression. DLT was evaluated in the other 12 patients. There were no DLTs observed at any dose during the DLT evaluation period.
Adverse events were generally of grade 1/2 severity and manageable with standard supportive care. There were no grade 4 AEs reported. One grade 5 AE occurred in a patient in the 15 mg cohort; this was not considered related to the study drug. This patient died of respiratory failure due to disease progression 25 days after the last dose of study drug. Most common all-causality AEs across all dose levels were rash (n = 13), diarrhea (n = 12), paronychia (n = 9), dry skin (n = 8), stomatitis (n = 8), and fatigue (n = 6).
Table 2 lists treatment-related AEs (TRAEs) by grade occurring in ≥2 patients in all cycles. Overall, there were eight treatment-related grade 3 AEs: one in the 15 mg cohort: transient ischemic attack; and seven in the 45 mg dosing cohort: rash (n = 2), device-related infection, decreased appetite, alanine aminotransferase increased, aspartate aminotransferase increased, and blood bilirubin increased (each n = 1). AEs observed in ≥8 of 13 patients (≥61.5%) were rash (n = 13), diarrhea (n = 12), paronychia (n = 9), dry skin (n = 8), and stomatitis (n = 8). AEs observed in ≥1 patient during the single-dose administration phase of dacomitinib were rash and diarrhea.
Table 2 Number of patients with treatment-related adverse events by maximum CTCAE gradea (occurring in ≥2 patients in all cycles)
No patient had an absolute change of ≥500 ms or a ≥60 ms change from baseline in QTc, QTcB, or QTcF interval. Two cases of change in QT interval were recorded as AEs; one patient (15 mg cohort) experienced a prolonged QT interval (grade 2) 8 days after being discontinued from study due to disease progression, and another patient (15 mg cohort) had a prolonged QTcF interval (26 ms from baseline; grade 1) at day 1 cycle 1, but was not considered to have clinically significant changes.
There were two serious AEs (SAEs) considered by investigators to be possibly related to the study drug, both in the 45 mg dose cohort; one case of hemobilia occurred in a patient with liver metastases shown to have progression on imaging at day 6 cycle 1—the drug was temporarily withdrawn and the patient recovered. The other SAE was an instance of device-related infection (an episode of bacteremia following catheter placement at a skin site involved with a treatment-induced skin reaction). One patient permanently discontinued the study due to a TRAE (grade 2, treatment-related rash). Rash was first reported on day 14 of cycle 1, and the patient discontinued on day 1 of cycle 6 before progressive disease. There were no deaths during the study.
Temporary discontinuations due to TRAEs were required by five patients (all in the 45 mg dosing cohort); grade 3 rash occurred in two patients, and the remaining AEs were of grade 2 severity. One patient with a rash required a dose reduction from 45 mg to 30 mg.
Pharmacokinetics
Mean plasma concentration–time profiles and PK parameters for dacomitinib following single (D-9) and multiple (C1D14) dosing are shown in Fig. 1 and Table 3.
Fig. 1
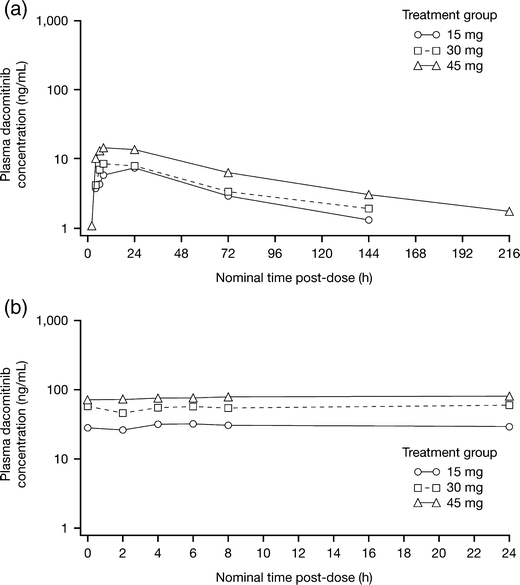
Mean (SD) plasma concentration–time profiles of dacomitinib following single (panel a; D-9) and multiple (panel b; C1D14) dosing
Table 3 Preliminary mean (%CV) dacomitinib plasma pharmacokinetic parameters following single (D-9) and multiple dosing (C1D14)
Following single oral dose administration (D-9), median Tmax ranged from 6 to 24 h over a dose range of 15–45 mg. The mean values for the PK parameters representing systemic exposure in rate (Cmax) and extent (AUCinf) increased along with dose in a proportional manner. Dose-proportionality was confirmed by consistency in the mean values for dose-normalized Cmax and AUCinf across the dose range studied. Mean apparent clearance ranged from 23.7 to 32 L/h across the 15–45 mg dose levels.
Mean Cmax and AUCτ increased after multiple dosing (C1D14). The geometric mean accumulation ratio ranged from 6.32 to 7.37 over the dose range. These calculated values are within an anticipated range based on the estimated half-life of dacomitinib and the daily dosing schedule. The linearity ratio was close to 1 across the studied dose levels. These reported parameters suggest that dacomitinib has linear kinetics after single- and multiple-dose administration in the dose range studied.
Antitumor activity
Of 13 evaluable patients, one experienced a PR as a BOR per RECIST. The PR (treatment duration 767 days) was observed in a patient with NSCLC (adenocarcinoma, male, 70 years) in the 15 mg cohort (Table 4, Fig. 2a). No patient had a CR. SD (≥6 weeks) was observed in nine patients: three in the 30 mg QD cohort and six in the 45 mg QD cohort (NSCLC, n = 6; colorectal carcinoma, n = 1; breast cancer, n = 1; squamous cell neoplasm, n = 1). One patient with SD (28.7% reduction) was a 66-year-old never-smoking female who, following a PR on gefitinib, was found via analysis of pleural effusion to have adenocarcinoma of lung characterized by both L858R and secondary_T790M_ mutations; she started dacomitinib 30 mg 79 days after discontinuation of gefitinib and continued for 134 days (8 cycles) before progression (Fig. 2b). Progressive disease (PD) was observed in three patients (15 mg QD cohort, n = 2; 45 mg QD cohort, n = 1). Four of eight patients with NSCLC had tumor size reductions (Table 4).
Table 4 Summary of profiles for patients with NSCLC (n = 8) and reductions in tumor size
Fig. 2
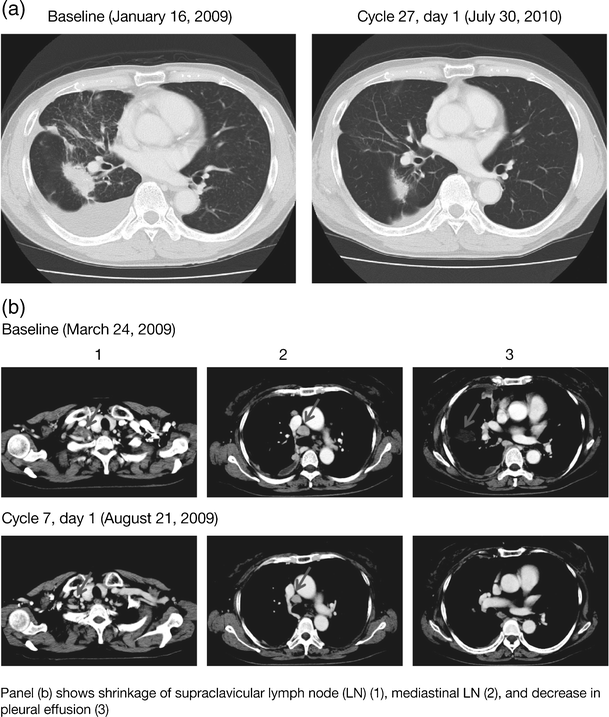
Pre- and post-treatment computed tomography: a 70-year-old patient with NSCLC (Table 4, patient No. 1) (a) receiving dacomitinib at 15 mg QD; and a 66-year-old patient with NSCLC (Table 4, patient No. 4) (b) receiving dacomitinib at 30 mg QD
Tumor growth model
The model of tumor growth with the parameters for shrinkage and progression rates fitted the data for tumor size fairly well (Fig. 3). The observations versus population and individual predictions were uniformly and closely distributed around the line of identity. Table 5 lists the parameter and precision estimates. The covariate analysis identified that the shrinkage rate was significantly affected by EGFR mutation status. Figure 4 shows the individual model parameter estimates plots versus gender, ECOG status, dose level, and EGFR mutation status for the base model (without any covariates) and final model (EGFR on shrinkage rate). Shrinkage rate was 85% less in EGFR wild type versus EGFR mutant subjects. For the 15 mg cohort, although the variability around individual SR and TPR parameters was larger than for the other two dose groups, the available data did not support the addition of dose cohort as covariate, probably due to the small sample size evaluated in this study. No correlation with duration of treatment, progression-free survival, or overall survival was undertaken at this time.
Fig. 3
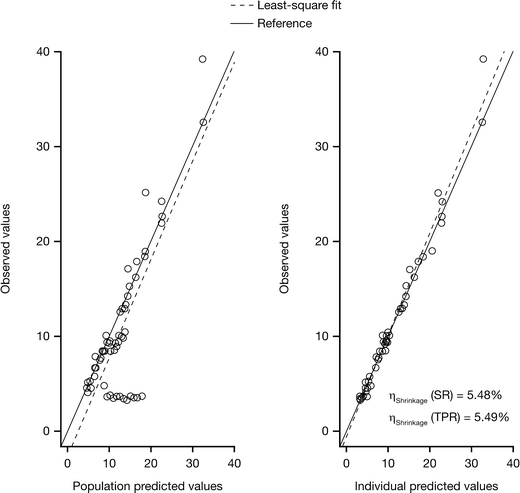
Observed tumor size versus population and individual predictions
Table 5 Parameter and precision estimates in the tumor growth model
Fig. 4
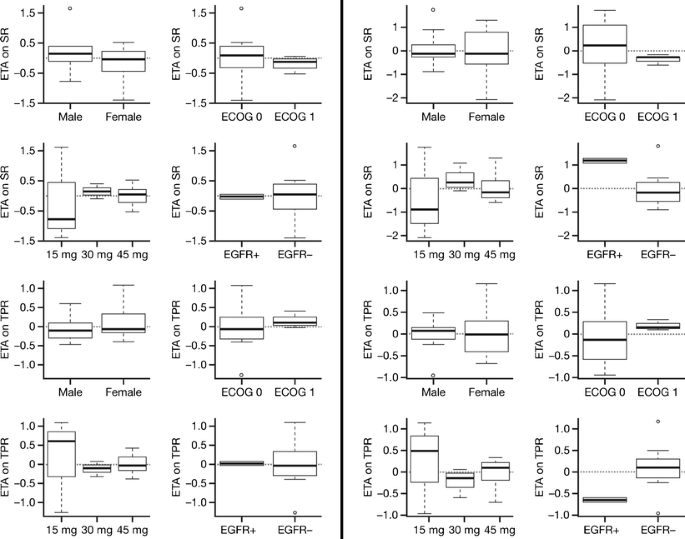
Individual variability in parameter estimates (ETA) for SR and TPR versus gender, ECOG status, dose group, and EGFR mutation status for the final model with EGFR on SR as covariate (left) and base model with no covariates added (right)
Discussion
This is the first report of the safety, PK profile, and antitumor activity of dacomitinib in Japanese patients with advanced solid tumors.
Treatment with dacomitinib in phase I and II studies of Western and Korean patients with advanced solid tumors has previously been associated with an acceptable safety profile and evidence of preliminary activity [11–13, 18]. The RP2D was established as 45 mg QD (given continuously in 21-day cycles) in these patient populations.
Dacomitinib appeared to be generally well tolerated in Japanese patients; the safety profile was consistent with that observed in Western and Korean studies [11–13, 18]. There were no DLTs observed at any dose, and AEs were generally of grade 1/2 severity and manageable. No grade 4 AEs were reported. Most common AEs (primarily skin and gastrointestinal toxicities of rash, diarrhea, paronychia, stomatitis, and dry skin) were consistent with the toxicities of other EGFR tyrosine kinase inhibitors [19–21]. For comparison, the most commonly reported AEs in the phase II dacomitinib versus erlotinib study of advanced NSCLC [13] were diarrhea, dermatitis acneiform, stomatitis, mucosal inflammation, and paronychia; similarly, the Korean phase I/II study in refractory NSCLC [18] reported diarrhea, dermatitis acneiform, paronychia, and stomatitis as the most common AEs. In each of these studies, AEs were primarily of grade 1 or 2 severity and manageable with standard supportive care.
Dacomitinib systemic exposure increased with increasing dose in this study, and the evaluated parameters suggested linear PK after both single- and multiple-dose administration over the dose range studied. PK exposure parameters for dacomitinib appeared to be comparable between the Japanese patients evaluated in this study and patients included in two separate study populations (A7471003, Korean patients with advanced NSCLC; and A7471001, Western patients with advanced solid malignancies) [11, 18]. In the Korean study, dacomitinib showed linear kinetics at the doses studied and the PK parameters were similar to those previously observed in Western populations after single- and multiple-dose administration. For example, substantial overlap was seen in the AUCtau between Asian and Western patients after multiple dosing at 30 mg QD and 45 mg QD [22].
While these data are limited by the small sample size, antitumor activity was suggested in these Japanese patients. One of eight patients with NSCLC achieved a PR and six patients experienced SD (≥6 weeks) as best response. One patient with NSCLC (adenocarcinoma, ex-smoker, male, 70 years [Table 4]) who had an exon 19 deletion and who was EGFR tyrosine kinase inhibitor treatment-naïve experienced a PR (57.7% reduction) as best response following treatment with dacomitinib 15 mg QD, and had an extended treatment duration of 25 months (767 days) (Fig. 2a). One further patient with NSCLC, with an exon 21 mutation (L858R) and T790M secondary mutation, had sustained SD (179 days, 28.7% reduction) (Fig. 2b). Approximately 50% of the acquired resistance to EGFR tyrosine kinase inhibitors such as gefitinib and erlotinib has been considered to be due to the presence of the T790M EGFR mutation [23] and, as treatment with pan-HER inhibitors (such as dacomitinib, afatinib, and neratinib) has not previously been reported to be beneficial in patients with known T790M resistance, the degree of benefit observed in this patient is noteworthy.
Antitumor activity in early clinical studies is generally evaluated using objective response rate or progression-free survival. However, in typical small, non-comparative phase I or II studies, these estimates are generally imprecise and do not effectively inform ‘go, no-go’ decisions and the subsequent design of phase III clinical trials. In addition, change in tumor size can be seen as a biomarker of drug effect that is further predictive of a clinical endpoint (i.e. survival). There is clearly a need for additional quantitative approaches to improve success rates in oncology drug development, consistent with recent Food and Drug Administration initiatives [[24](/article/10.1007/s10637-011-9789-z#ref-CR24 "Innovation/stagnation: challenge and opportunity on the critical path to new medical products. March 2004 ( http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm
)"), [25](/article/10.1007/s10637-011-9789-z#ref-CR25 "End-of-phase 2a meetings. FDA guidance for Industry. September 2009 (
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079690.pdf
) (accessed April 16, 2010)")\]. The exploratory dynamic tumor growth model analysis performed in this study \[[26](/article/10.1007/s10637-011-9789-z#ref-CR26 "Wang Y, Sung C, Dartois C et al (2009) Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Therapeutics 86:167–174")\] provides support for the importance of molecular markers in patient selection for treatment with targeted agents. In the present study, this model confirmed a better response to dacomitinb in _EGFR_ mutant compared with _EGFR_ wild-type tumors, although no other markers were identified to be related to response. The small sample size in this study precluded identification of other markers, but further tumor growth analyses, using pooled data from several dacomitinib trials, could enable us to better explore the impact of dacomitinib exposure and other covariates such as _KRAS_ mutant status, race, etc., on response to treatment. Moreover, by modeling tumor growth patterns we may be better able to understand differences between irreversible and reversible tyrosine kinase inhibitors.In conclusion, this first report of dacomitinib in Japanese patients with advanced solid tumors suggests that the treatment is well tolerated and has potential for therapeutic benefit in Japanese patients. Safety and PK data were consistent with previous clinical experience in Western and Korean patients with advanced malignancies. Antitumor activity was also suggested, particularly in NSCLC where efficacy has previously been shown in other phase I and II clinical trials in refractory NSCLC and in a randomized phase II study versus erlotinib in second-/third-line treatment [11, 13, 18, 27]. On the basis of these data, phase III clinical trials of dacomitinib are ongoing in two settings of advanced NSCLC: after failure of one or two prior chemotherapy regimens and prior EGFR tyrosine kinase inhibitor therapy (NCT01000025), and after failure of one (and no more than two) prior chemotherapies versus erlotinib (NCT01360554). Both studies include collection of tissue for analysis of key molecular markers that may be predictive of benefit, including EGFR and KRAS, and include an analysis of data for East Asian versus other patients as a stratification factor.
References
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Article PubMed CAS Google Scholar - Salomon DS, Brandt R, Ciardiello F, Normanno N (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19:183–232
Article PubMed CAS Google Scholar - Woodburn JR (1999) The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 82:241–250
Article PubMed CAS Google Scholar - Mendelsohn J, Baselga J (2003) Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 21:2787–2799
Article PubMed CAS Google Scholar - Engelman JA, Zejnullahu K, Gale CM et al (2007) PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 67:11924–11932
Article PubMed CAS Google Scholar - Gonzales AJ, Hook KE, Althaus IW et al (2008) Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther 7:1880–1889
Article PubMed CAS Google Scholar - Shigematsu H, Takahashi T, Nomura M et al (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97:339–346
Article PubMed CAS Google Scholar - Kobayashi S, Boggon TJ, Dayaram T et al (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792
Article PubMed CAS Google Scholar - Pao W, Miller VA, Politi KA et al (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:e73
Article PubMed Google Scholar - Balak MN, Gong Y, Riely GJ et al (2006) Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 12:6494–6501
Article PubMed CAS Google Scholar - Janne PA, Boss DS, Camidge DR et al (2011) Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin Cancer Res 17:1131–1139
Article PubMed CAS Google Scholar - Janne P, Reckamp K, Koczywas M, et al. (2009) Efficacy and safety of PF-00299804 (PF299) in patients (pt) with advanced NSCLC after failure of at least one prior chemotherapy regimen and prior treatment with erlotinib (E): A two-arm, phase II trial. J Clin Oncol 27(suppl.) (abstr. 8063)
Google Scholar - Ramalingam SS, Boyer MJ, Park K, et al. (2010) Randomized phase 2 study of PF299804, an irreversible human epidermal growth factor receptor (EGFR) inhibitor, versus (v) erlotinib (E) in patients (pts) with advanced non-small cell lung cancer (NSCLC) after chemotherapy (CT) failure: quantitative and qualitative benefits. Ann Oncol 21(suppl. 8) (abstr. 365PD)
- Siu LL, Hotte SJ, Laurie SA, et al. (2011) Phase II trial of the irreversible oral pan-human EGF receptor (HER) inhibitor PF-00299804 (PF) as first-line treatment in recurrent and/or metastatic (RM) squamous cell carcinoma of the head and neck (SCCHN). J Clin Oncol 29(suppl.) (abstr. 5561)
Google Scholar - Schellens JH, Britten CD, Camidge DR, et al. (2007) First-in-human study of the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of PF-00299804, a small molecule irreversible panHER inhibitor in patients with advanced cancer. J Clin Oncol 25(suppl.) (abstr. 3599)
Google Scholar - Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Article PubMed CAS Google Scholar - FDA Advisory Committee Briefing Document: Pharmaceutical Science and Clinical Pharmacology Advisory Committee, March 18−19, 2008 (http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4351b1-00-index.htm)
- Park K, Heo DS, Cho BC, et al. (2010) PF299804 in Asian patients with non-small cell lung cancer refractory to chemotherapy and erlotinib or gefitinib—a phase 1/2 study. J Clin Oncol 28(suppl.) (abstr. 7599)
Google Scholar - Hartmann JT, Haap M, Kopp HG, Lipp HP (2009) Tyrosine kinase inhibitors—a review on pharmacology, metabolism and side effects. Curr Drug Metab 10:470–481
Article PubMed CAS Google Scholar - Li T, Perez-Soler R (2009) Skin toxicities associated with epidermal growth factor receptor inhibitors. Target Oncol 4:107–119
Article PubMed Google Scholar - Ouwerkerk J, Boers-Doets C (2010) Best practices in the management of toxicities related to anti-EGFR agents for metastatic colorectal cancer. Eur J Oncol Nurs 14:337–349
Article PubMed Google Scholar - Heo DS, Kim JH, Cho BC, et al. (2009) First report of PF-00299804 in Asian patients: a Korean phase I/II study in KRAS wild-type, stage IIIb/IV non-small cell lung cancer, refractory to chemotherapy and erlotinib or gefitinib. Oral presentation at the 13th World Conference on Lung Cancer, San Francisco, CA, USA, 2009
- Engelman JA, Zejnullahu K, Mitsudomi T et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Article PubMed CAS Google Scholar - Innovation/stagnation: challenge and opportunity on the critical path to new medical products. March 2004 (http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm)
- End-of-phase 2a meetings. FDA guidance for Industry. September 2009 (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079690.pdf) (accessed April 16, 2010)
- Wang Y, Sung C, Dartois C et al (2009) Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Therapeutics 86:167–174
Article CAS Google Scholar - Campbell A, Reckamp KL, Camidge DR, et al. (2010) PF-00299804 (PF299) patient (pt)-reported outcomes (PROs) and efficacy in adenocarcinoma (adeno) and nonadeno non-small cell lung cancer (NSCLC): A phase (P) II trial in advanced NSCLC after failure of chemotherapy (CT) and erlotinib (E). J Clin Oncol 28(suppl.) (abstr. 7596)
Google Scholar
Acknowledgments
We would like to thank all of the participating patients and their families, as well as the network of investigators, research nurses, study coordinators, and operations staff. Medical writing support was provided by Rachel Mason at ACUMED® (Tytherington, UK) with funding from Pfizer Inc. This study was sponsored by Pfizer Inc.
Conflict of interest
T. Takahashi, N. Boku, H. Murakami, T. Naito, A. Tsuya, Y. Nakamura, A. Ono, N. Machida, K. Yamazaki, J. Watanabe, N. Yamamoto: nothing to disclose.
A. Ruiz-Garcia, K. Imai, E. Ohki: employees of Pfizer and hold stock in Pfizer.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Shizuoka Cancer Center, Shizuoka, Japan
Toshiaki Takahashi, Narikazu Boku, Haruyasu Murakami, Tateaki Naito, Asuka Tsuya, Yukiko Nakamura, Akira Ono, Nozomu Machida, Kentaro Yamazaki, Junichiro Watanabe & Nobuyuki Yamamoto - Pfizer Oncology, La Jolla, CA, USA
Ana Ruiz-Garcia - Pfizer Japan Inc., Tokyo, Japan
Keiji Imai & Emiko Ohki - Division of Thoracic Oncology, Shizuoka Cancer Center, 1007 Shimonagakubo, Nagaizumi-cho, Sunto-gun, Shizuoka, 411-8777, Japan
Toshiaki Takahashi
Authors
- Toshiaki Takahashi
You can also search for this author inPubMed Google Scholar - Narikazu Boku
You can also search for this author inPubMed Google Scholar - Haruyasu Murakami
You can also search for this author inPubMed Google Scholar - Tateaki Naito
You can also search for this author inPubMed Google Scholar - Asuka Tsuya
You can also search for this author inPubMed Google Scholar - Yukiko Nakamura
You can also search for this author inPubMed Google Scholar - Akira Ono
You can also search for this author inPubMed Google Scholar - Nozomu Machida
You can also search for this author inPubMed Google Scholar - Kentaro Yamazaki
You can also search for this author inPubMed Google Scholar - Junichiro Watanabe
You can also search for this author inPubMed Google Scholar - Ana Ruiz-Garcia
You can also search for this author inPubMed Google Scholar - Keiji Imai
You can also search for this author inPubMed Google Scholar - Emiko Ohki
You can also search for this author inPubMed Google Scholar - Nobuyuki Yamamoto
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toToshiaki Takahashi.
Additional information
Acknowledgment of research support: This study was sponsored by Pfizer Inc.
Prior presentation: Takahashi T, Boku N, Murakami H, et al. First report of the safety, pharmacokinetics, and preliminary activity of PF-00299804 in Japanese patients with advanced solid tumors. Poster presentation at the 35th Congress of the European Society for Medical Oncology (ESMO), Milan, Italy, October 8–12, 2010
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Takahashi, T., Boku, N., Murakami, H. et al. Phase I and pharmacokinetic study of dacomitinib (PF-00299804), an oral irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese patients with advanced solid tumors.Invest New Drugs 30, 2352–2363 (2012). https://doi.org/10.1007/s10637-011-9789-z
- Received: 30 November 2011
- Accepted: 27 December 2011
- Published: 17 January 2012
- Issue Date: December 2012
- DOI: https://doi.org/10.1007/s10637-011-9789-z