Extrinsic Fluorescent Dyes as Tools for Protein Characterization (original) (raw)
- 25k Accesses
- 963 Citations
- 6 Altmetric
- Explore all metrics
Abstract
Noncovalent, extrinsic fluorescent dyes are applied in various fields of protein analysis, e.g. to characterize folding intermediates, measure surface hydrophobicity, and detect aggregation or fibrillation. The main underlying mechanisms, which explain the fluorescence properties of many extrinsic dyes, are solvent relaxation processes and (twisted) intramolecular charge transfer reactions, which are affected by the environment and by interactions of the dyes with proteins. In recent time, the use of extrinsic fluorescent dyes such as ANS, Bis-ANS, Nile Red, Thioflavin T and others has increased, because of their versatility, sensitivity and suitability for high-throughput screening. The intention of this review is to give an overview of available extrinsic dyes, explain their spectral properties, and show illustrative examples of their various applications in protein characterization.
Similar content being viewed by others
INTRODUCTION
In recent years, recombinant proteins and antibodies have established themselves on the pharmaceutical market for the treatment and prevention of numerous diseases (1). A prerequisite for their clinical application is the development of stable formulations, because proteins are complex molecules, susceptible to various degradation reactions (2,3). In order to assure protein integrity during bioprocessing, formulation, storage and handling, the use of analytical techniques to detect and quantify degraded protein is inevitable. Especially the characterization of conformational variants, e.g. induced by environmental stress or chemical changes of the protein such as oxidation or deamidation, and various types of aggregates, which can differ in shape, morphology and size (4–6) and which may be immunogenic (7–9), requires a selection of complementary methods (10,11). Ideally, these methods should selectively detect minute amounts of conformationally altered or aggregated protein molecules that can function as nuclei for severe aggregation (12) or fibrillation (13–15).
Fluorescence spectroscopy can be employed as a highly sensitive method for protein analysis. Intrinsic protein fluorescence deriving from the naturally fluorescent amino acid tryptophan, and to a lesser extent from tyrosine, can provide information on conformational changes of proteins as reviewed elsewhere (16,17). Furthermore, various extrinsic fluorescent dyes offer additional possibilities for protein characterization. Extrinsic dyes can be covalently attached to proteins, e.g. via the ɛ-amino group of lysine, the α-amino group of the N-terminus, or the thiol group of cystein. More interesting for the analysis of pharmaceutical formulations are extrinsic dyes that interact noncovalently with proteins and protein degradation products, e.g. via hydrophobic or electrostatic interactions. The focus of this review is on the application of noncovalent, extrinsic fluorescent dyes for protein characterization. After a brief history of commonly used dyes, the spectral properties and characteristics of the dyes and practical considerations for their application are presented, followed by various applications of the use of fluorescent dyes for protein characterization illustrated with examples.
BRIEF HISTORY AND OVERVIEW OF AVAILABLE DYES AND THEIR USE
Already in the 1950s it was discovered that the introduction of various substituted aniline groups to naphthalene derivatives resulted in molecules like 1-anilinonaphthalene-8-sulfonate (ANS), which are hardly fluorescent in aqueous environment, but become highly fluorescent in apolar, organic solvents or upon adsorption to solid phases (18). Interactions of ANS with hydrophobic binding sites within apomyoglobin and apohemoglobin, accompanied by an increase in fluorescence and a blue shift of the peak maximum, were described by Stryer (19). Until now, ANS and its dimeric analogon 4,4′-bis-1-anilinonaphthalene-8-sulfonate (Bis-ANS), which was first described by Rosen and Weber in 1969 (20), have been amongst the most frequently used dyes for protein characterization. Another important dye for protein characterization is Nile Red, which had been first applied for the detection of intracellular lipid droplets (21) and shortly after for monitoring conformational changes of different proteins (22). The dye 9-(dicyanovinyl)-julolidine (DCVJ) has been used to probe tubulin structure (23) and the formation of hydrophobic microdomains, e.g. in protein aggregation (24). In the field of fibrillation and amyloid fibril characterization, Congo Red (25,26) and Thioflavin T (27) are commonly used dyes.
Until now, extrinsic fluorescent dyes have been used for numerous applications in protein characterization, e.g. to monitor refolding and unfolding processes (28), to detect molten globule intermediates (29), to assess surface hydrophobicity (30), to probe active sites of enzymes (31), to characterize aggregation and fibrillation (32,33), to monitor conformational changes induced by chemical degradation (34), to probe protein–surfactant interactions (35) and to analyze protein crystals (36). In the last years, an increasing number of research groups have been making use of extrinsic fluorescent dyes for protein characterization, which is reflected by the large number of recent publications in this field. This development was prompted by the emerging focus on protein aggregation during the manufacturing, storage and handling of biopharmaceuticals and the related need for sensitive detection methods.
Steady-state fluorescence spectroscopy is the most commonly used technique in conjunction with fluorescent dyes. Besides the collection of spectral information and fluorescence intensity, other fluorescence techniques, which can provide useful additional information, have been used in connection with extrinsic dyes (Table I). For an explanation of the different fluorescence techniques, the reader is referred to the literature, as this is out of the scope of this review (16,37,38).
Table I Fluorescence Methods and Their Application with Extrinsic Fluorescent Dyes for Protein Characterization
PROPERTIES OF COMMONLY USED EXTRINSIC FLUORESCENT DYES
Mechanisms of Dye Fluorescence
For a correct interpretation of fluorescence signals, knowledge of the underlying mechanisms at the molecular level and the spectral properties of the dyes are of great importance. For detailed explanations about fluorescence in general the reader is referred to the literature (16,37,38),while selected basic principles that govern the fluorescence properties of extrinsic dyes are described below.
Figure 1 gives a schematic overview of electronic transitions relevant for the properties of extrinsic fluorescent dyes. Excitation by absorption of light lifts electrons of the dye molecules within femtoseconds from the ground state S0 to higher singlet excited states. There are several processes competing with fluorescence, which can result in energy loss and thus a red shift of fluorescence emission with respect to absorption (Stokes shift). These processes are vibrational relaxation and/or internal conversion, solvent relaxation, and intramolecular charge transfer (ICT) or twisted intramolecular charge transfer (TICT; Fig. 1). Once the dye molecules have reached the lowest vibrational level of S1, or in case of charge transfer processes the lowest vibrational level of S(T)ICT, the molecules can relax to the ground state S0 by fluorescence emission or radiationless by internal conversion. Furthermore, intersystem crossing to the triplet state (not shown in Fig. 1) is facilitated from S(T)ICT as compared to S1. Vibrational relaxation refers to the relaxation of the excited molecules from higher vibrational levels within an excited level (e.g. within S1), whereas internal conversion is the relaxation from higher electronic excited state levels to lower ones (e.g., from S2 to S1; from S1 to S0). Both processes occur within picoseconds.
Fig. 1
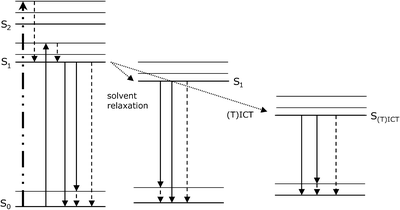
Simplified scheme showing the energy levels of the ground state So and various excited states, as well as electronic transitions taking place for fluorescent dyes. After light absorption, where the electrons are lifted from the ground state S0 to higher energetic excited levels (bold dashed dotted arrow; for sake of clarity, absorption to only one vibrational level of the S2 excited state level is shown), radiationless vibrational relaxation and internal conversion processes (dashed arrows), solvent relaxation and (twisted) intramolecular charge transfer (T)ICT (dotted arrows), as well as fluorescence (solid arrows) can occur. For sake of clarity the conversion to the triplet state, which can occur from S(T)ICT is not included in this figure.
The electronic transitions due to excitation are typically accompanied by a change of the dipole moment of the dye. If the solvent molecules possess a dipole moment as well, reorientation of solvent molecules surrounding the excited dye molecules to energetically more favorable positions can occur. Thereby, the energy level of the S1 state is lowered and the energy of the S0 state is raised, which increases the Stokes shift (Fig. 1). This process is referred to as solvent relaxation. Since solvent relaxation requires the presence of a dipole moment in the solvent molecules themselves, it is dependent on solvent polarity (37).
Other common mechanisms governing the fluorescence properties of extrinsic dyes are intramolecular charge transfer (ICT) including twisted intramolecular charge transfer (TICT). In ICT and TICT an electron is transferred from an electron donor group (e.g. an amino group) to an electron withdrawing group (e.g. an aromatic system) in the excited dye molecules. For TICT a change in the dye’s conformation, e.g. rotation or twist, is a prerequisite for the electron transfer to take place. The charge separation induced by (T)ICT gives rise to an increased dipole moment of the excited state S(T)ICT compared to S1. Formation of (T)ICT is favored in polar solvents and results in enhanced solvent relaxation processes and a more pronounced Stokes shift. Additionally, for fluorescent dyes (T)ICT states are in most cases more likely to relax by nonradiative processes than by fluorescence emission, resulting in low fluorescence intensities of the dyes in polar environments (39–41).
In addition to solvent polarity, solvent viscosity and temperature can affect the processes of solvent relaxation and (T)ICT. With increasing viscosity or decreasing temperature solvent relaxation is generally decelerated and (T)ICT processes are delayed, resulting in smaller Stokes shifts. Moreover, solvent viscosity and temperature can affect fluorescence intensity and lifetime.
Hydrogen bonding between solvent and dye molecules is another parameter capable to alter the fluorescence properties of a dye. Typically, the addition of a low amount (<0.1%) of a hydrogen-bonding solvent, e.g. methanol, to a solvent like hexane is sufficient to change the emission spectrum of a dye (42).
Interactions between dye molecules and solutes like proteins can lead to changes of fluorescence emission by a change of the preferred relaxation pathway (Fig. 1). This is the basis for the use of extrinsic fluorescent dyes for protein characterization. The spectral properties of the most frequently used dyes are explained below, with focus on how their fluorescence in presence of proteins can be explained. An overview of extrinsic fluorescent dyes for protein characterization is given in Table II.
Table II Overview of Extrinsic Fluorescent Dyes Applied for Protein Characterization
ANS and Bis-ANS
ANS and Bis-ANS (Fig. 2A and B) fluorescence is sensitive to the dyes’ environment with respect to polarity, viscosity and temperature. A blue shift of the emission maximum and an increase of quantum yield occurs with decreasing dielectric constant of the solvent (19). Figure 3A and B show the blue shift and intensity increase of ANS and Bis-ANS fluorescence when changing the solvent from water to methanol, ethanol and DMSO. ANS and Bis-ANS, the dimeric analogon of ANS, exhibit similar intrinsic fluorescence properties with respect to the underlying mechanisms, but the UV absorption maximum and fluorescence emission of Bis-ANS are shifted to longer wavelengths (20). In the excited state, ANS can undergo TICT with a charge transfer from the electron-donating amino-aryl group to the sulfonated naphthalene system, with a subsequent twist of the molecules (40,41). The higher polarity of STICT as compared to the S1 state favors its formation in polar environments. The relaxation of STICT to S0 generally occurs via rapid nonradiative decay, which is responsible for the low quantum yield of fluorescence in polar solvents (40,41). The effect of temperature and viscosity on the fluorescence properties of ANS and Bis-ANS can be explained by their impact on solvent relaxation processes and TICT (cf. Fig. 1 and the section Mechanisms of Dye Fluorescence). The deceleration of solvent relaxation and TICT in media of high viscosity results in blue shifted emission and increased fluorescence intensity (43,44). The photophysical mechanisms of ANS fluorescence were recently described by Klimtchuk et al., Kirk et al., Kirk and Klimtchuk, and Kirk and Wessels (45–49).
Fig. 2
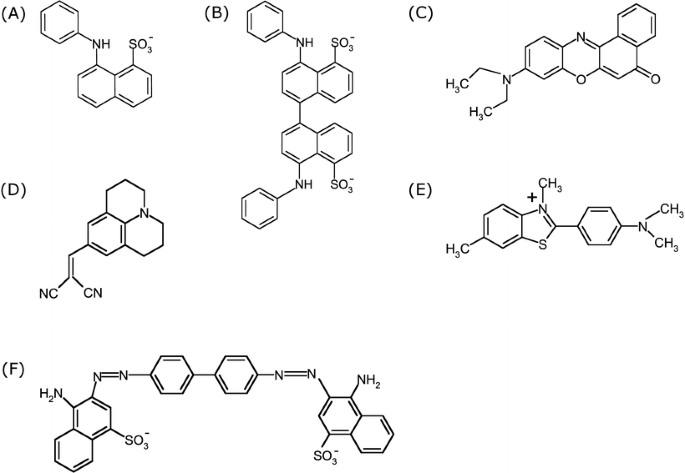
Chemical structures of commonly used fluorescent dyes: ANS (A), Bis-ANS (B), Nile Red (C), DCVJ (D), Thioflavin T (E) and Congo Red (F).
Fig. 3
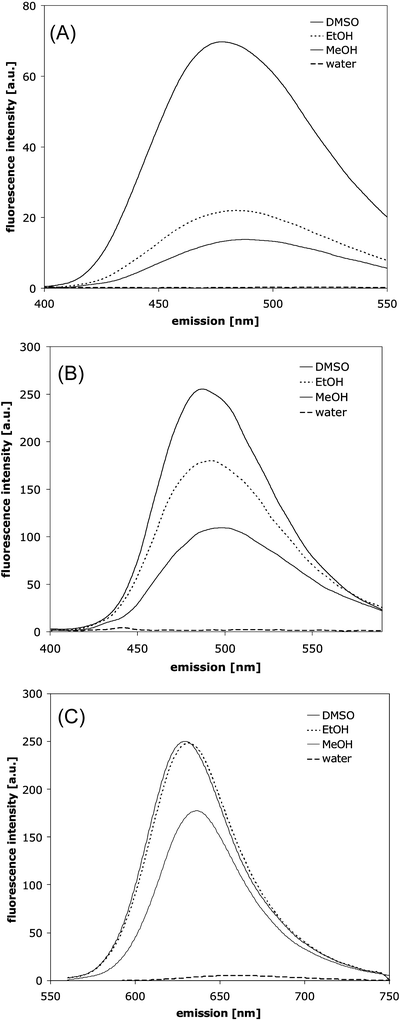
Polarity dependent fluorescence emission of 1 μM ANS excited at 350 nm (A), 1 μM Bis-ANS excited at 385 nm (B) and 1 μM Nile Red excited at 550 nm (C) in different solvents.
The fluorescence properties of ANS and Bis-ANS strongly depend on their interaction with protein molecules, which results in changes of polarity and viscosity of the environment. Hydrophobic interactions (19) and electrostatic interactions (50) have been discussed as binding mechanisms of ANS to proteins. More precisely, ion pairing between the negatively charged sulfonate groups of ANS with positively charged amino acids e.g. histidine, lysine or arginine is the predominant interaction (51). However, complementary interactions such as van der Waals interactions are required to stabilize the ion pairs (52). Interestingly, it was found that Bis-ANS binding to proteins is dominated by hydrophobic interactions (53). Furthermore, the different size of ANS and Bis-ANS may lead to differences in binding affinities and number of binding sites on proteins. For several proteins an increased affinity of Bis-ANS to the protein and a stronger fluorescence enhancement as compared to ANS has been described (20,54).
Nile Red
TICT, with an electron transfer from the diethylamino group to the electron-withdrawing aromatic system, governs the fluorescence properties of Nile Red (Fig. 2C). The formation of the TICT state and nonradiative relaxation occur in polar solvents. In apolar environments, the TICT process is thermodynamically unfavorable, with the consequence that both fluorescence lifetime and quantum yield increase significantly (55). In contrast, hydrogen bonding between the excited dye and solvent molecules was described to lower the fluorescence lifetime and quantum yield of Nile Red (56).
Figure 3C shows the blue shift and intensity increase of Nile Red fluorescence emission in methanol, ethanol and DMSO compared to water. Dutta et al. demonstrated that the fluorescence lifetime of Nile Red decreases in more polar solvents, and that at the same time the nonradiative constant increases (39). As Nile Red fluorescence is sensitive to the polarity of its environment, it can be employed to probe changes in protein conformation that are related to the formation of hydrophobic surfaces, e.g. during aggregation or unfolding of proteins, as first shown by Sackett and Wolff (22).
DCVJ
DCVJ (Fig. 2D) belongs to the group of TICT-forming fluorescent dyes, with an electron transfer from the nitrogen in the ring system to the nitrile groups. Contrary to the so far described fluorescent dyes, the fluorescence of DCVJ and the similar molecule 9-(2-carboxy-2-cyanovinyl)-julolidine (CCVJ) is mainly sensitive to the viscosity of the environment and to a minor extent to solvent polarity (23,57). In highly viscous media such as glycerol the TICT state is partially inhibited, which leads to an increase in quantum yield. This viscosity sensitive fluorescence behavior has been used to investigate the viscosity of blood plasma (58). When bound to proteins, the rigidity of the microenvironment inhibits the intramolecular rotation with the consequence that TICT is suppressed, which leads to an increase in the probes’ quantum yield (59).
Thioflavin T
Thioflavin T absorption maxima are affected by solvent polarity (60). However, the fluorescence properties of Thioflavin T (Fig. 2E), especially its quantum yield, are more affected by the solvent viscosity and the rigidity of the microenvironment than by polarity (61). The formation of a nonfluorescent TICT state accompanied by a change in the angle between benzothiazole and the benzene ring from 37° to 90° was proposed as prevailing mechanism of low Thioflavin T fluorescence in solvents of low viscosity (62).
Originally, Thioflavin T has been used as histochemical dye to stain amyloid-like deposits in tissues (63,64) and later for quantification of amyloid fibrils in vitro in presence of amyloid precursor proteins and amorphous aggregates (65). Amyloid fibrils are filamentous protein aggregates of about 10 nm width and 0.1 to 10 μm length. Typically, cross beta sheets are the predominant secondary structure of amyloid fibrils (66). In the presence of amyloids, Thioflavin T exhibits an additional absorption peak at 450 nm and becomes highly fluorescent with an emission maximum at 480 nm (Table II), pointing at an impact of interactions between Thioflavin T and amyloid fibrils on fluorescence. Several groups proposed mechanisms for these interactions. It has been proposed that Thioflavin T interacts with β-sheet structures, as these are a predominant structural element in amyloid fibrils (67). However, the binding of Thioflavin T is not strictly linked to β-sheet structures, as fluorescence can be induced by non-β-sheet cavities, e.g. in acetylcholinesterase or γ-cyclodextrin, while not all β-sheet rich structures, e.g. transthyretin, induce fluorescence (68). Khurana et al. attributed the fluorescence increase in presence of amyloid fibrils to the formation of Thioflavin T micelles binding to the grooves of the twisted protofibrils or fibrils as analyzed by atomic force microscopy (69). Krebs et al. suggested an about 6.5 to 6.9 Å wide channel within the β-sheets of amyloid fibrils, with the definite channel volume depending on the side chains of the protein, as site where Thioflavin T binds with its long axis parallel to that of the fibrils. When bound to the channel the dye remains in a flat conformation in the excited state, which results in increased quantum yields due to an inhibition of STICT. As these steric constraints of the channel are to a certain extent typical for amyloid fibrils, Thioflavin T fluorescence is suitable to probe amyloid fibrils (70). Fluorescence anisotropy measurements combined with isothermal titration calorimetry and molecular modeling revealed that fluorescence is induced to a greater extent when Thioflavin T binds to cavities of 8 to 9 Å, which are capable to host a Thioflavin T dimer. It was concluded that the restriction of the molecular rotation around the bond is not sufficient to induce fluorescence, but that probably an excimer formation within the dimeric Thioflavin T leads to the increase in fluorescence (68).
Congo Red
Congo Red (Fig. 2F) is extensively used in the field of amyloid fibril analysis, frequently in combination with Thioflavin T. Similar to Thioflavin T, Congo Red has been used for the staining of amyloids within tissues predominantly via nonionic interactions using alkaline ethanolic solutions (25). It differs from the so far described dyes as the analytical focus is less on the increase of fluorescence intensity but more on shifts of UV absorbance and other optical effects (71–73). When Congo Red interacts with amyloid fibrils, a shift in the UV absorbance from about 490 to 540 nm occurs. Furthermore, an apple green birefringence in polarized light and an induction of a circular dichroism between 300 and 600 nm can be observed for Congo Red in presence of amyloid fibrils. Congo Red is prone to self-assembly in water and has been suggested to bind to amyloids mainly as a supramolecular ligand (74,75). Various models for the binding of Congo Red to amyloids have been proposed, which are summarized in the review of Frid et al. (26).
PRACTICAL CONSIDERATIONS FOR THE APPLICATION OF EXTRINSIC DYES FOR PROTEIN CHARACTERIZATION
Besides general practicalities, such as avoidance of inner filter effects and photobleaching, as well as taking proper blanks and controls (16), the experimentalist should be aware of a few specific potential pitfalls when working with fluorescent dyes.
Depending on the solubility of the dyes, an adequate solvent needs to be employed to prepare the (typically 10- to 100-fold concentrated) stock solutions (Table II). Aqueous stock solutions can be prepared with the positively charged Thioflavin T (Fig. 2C) and with ANS/Bis-ANS (Fig. 2A and B), which are negatively charged over a wide pH range, because of the pKa <2 of the sulfonate group. Nile Red is poorly soluble in aqueous media (solubility in water below 1 µg/ml) and requires organic solvents such as DMSO or ethanol for preparing stock solutions (22,76). When organic solvents are to be used, it should be verified that they do not affect the integrity of the protein.
In many cases, interaction of the fluorescent dye with native proteins accompanied by an increase of fluorescence intensity occurs. For example, Nile Red and ANS exhibit high fluorescence intensities in the presence of native human serum albumin (HSA), due to binding of dye molecules to hydrophobic binding sites on the native protein (77–79). However, as long as the dye clearly changes its fluorescence properties upon a structural alternation of the protein it can nonetheless be employed for characterization.
Furthermore, the dye itself can promote or inhibit aggregation or fibrillation when it interacts with the protein molecules. ANS was shown to facilitate the refolding of cytochrome c at low pH due to electrostatic interactions (80) and to inhibit heat-induced aggregation of carbonic anhydrase (81). Bis-ANS was capable to suppress heat-induced deactivation of porcine citrate synthase, aggregation of insulin B chain and alcohol dehydrogenase (82), as well as aggregation of lipase from Bacillus subtilis (28). This was ascribed to an interaction of Bis-ANS with solvent exposed hydrophobic surfaces on partially unfolded proteins. Dimerization of human growth hormone was induced by Congo Red leading to a shift of the dye’s absorption spectrum (83).
APPLICATIONS OF FLUORESCENT DYES IN PROTEIN CHARACTERIZATION
Assessment of Protein Denaturation, Folding and Molten Globular Intermediates
Correct protein folding is of fundamental importance for the quality of protein pharmaceuticals (84). The transition from a native to an unfolded state (and back) can occur via two-state kinetics (85) or via folding intermediates (86) with aggregation being a competing reaction to the folding process (84). Partially folded intermediates, often referred to as molten globules, exhibit a compact globular shape and still retain a certain amount of secondary structure, but lack a defined tertiary structure (29,87). They can be stable under acidic conditions, high temperatures, as well as intermediate concentrations of strong denaturants like urea or guanidine hydrochloride (GndHCl) (88).
Extrinsic fluorescent dyes can provide information about folding and unfolding processes, and can be particularly valuable to evidence the presence or absence of molten globule intermediates. Semisotnov et al. revealed a strong affinity of ANS to the solvated hydrophobic core of molten globular intermediates of carbonic anhydrase B and α-lactalbumins (89). By monitoring the folding curve of interleukin-1 receptor antagonist by intrinsic fluorescence and circular dichroism (CD) spectroscopy, a clear transition folding midpoint was determined at approximately 5.5 M GndHCl (90). The absence of substantial ANS fluorescence indicated the nonexistence of molten globular intermediates during folding (90). Studying the acid denaturation behavior of 20 monomeric proteins, Fink et al. used ANS fluorescence to confirm the presence of molten globular intermediates, which showed a substantial increase in ANS fluorescence intensity, whereas ANS fluorescence was negligible for the native and unfolded states of the studied proteins (91). Furthermore, ANS has been employed to probe molten globular states of recombinant human growth hormone (92), stem bromelain (93), β-lactamase (29) and HSA (94).
Besides fluorescence intensity, the position of the emission maximum of a dye and changes in fluorescence lifetime can provide further information on the hydrophobicity of molten globules. A blue shift in ANS fluorescence emission maximum, in addition to an intensity increase demonstrated the exposure of hydrophobic surfaces on molten globules of β-lactamase at pH 1.7 and 12.2. The largest blue shift from 540 to 470 nm occurred in presence of potassium chloride at pH 1.7, indicated a stabilizing effect of the salt on the molten globular structure of β-lactamase (29). For bovine serum albumin (BSA) the fluorescence lifetime of ANS in BSA-ANS complexes was reduced in the presence of GndHCl. However, the lifetimes in presence of high denaturant concentrations were still considerably larger than those of free ANS in water, indicating that hydrophobic binding sites for ANS remain present on unfolded BSA (95).
Different fluorescent dyes can vary in their interaction with the molten globular states of a certain protein. When using Nile Red to monitor the GndHCl induced denaturation of HSA, fluorescence intensity of the dye was highest between 0.25 and 1.5 M GndHCl and dropped above 1.5 M GndHCl, accompanied by a red shift of the emission maximum from 620 to 645 nm (77). ANS on the other hand revealed the highest fluorescence intensity for HSA at 1.8 M GndHCl (94), which points to different binding properties of Nile Red and ANS. In another study, ANS was found to bind to dimeric unfolding intermediates of BSA (identified at pH 4.2 by CD spectroscopy, light scattering and analytical ultracentrifugation), as reflected by blue shifted fluorescence emission from 520 nm to 480 nm with a 20-fold intensity increase, whereas pyrene fluorescence emission in presence of BSA was equal at pH 7.0 and pH 4.2 (96). These studies reveal that interactions and associated fluorescence properties depend on both the properties of the particular dye and the conformational state of the protein, and parallel use of different dyes may provide complementary information.
ANS fluorescence emission has furthermore been used to elucidate complex unfolding/dissociation processes of multimeric proteins. For human prostatic acid phosphatase, a dimeric enzyme, the maximum ANS fluorescence intensity in presence of 0.8 to 1.2 M GndHCl was attributed to an exposure of hydrophobic monomer-monomer interfaces and an onset of monomer unfolding. This was confirmed by a determination of the activity of the enzyme and an evaluation of the quaternary structure by size exclusion chromatography (97). Other examples for the characterization of unfolding processes of multimeric proteins by ANS are the unfolding of pyridoxyl 5’-phosphate C–S lyase MalY with urea (98), of alkaline phosphatase with urea (99) and of streptomycin adenylyltransferase with GndHCl (100).
Detection of Protein Aggregation
ANS and Bis-ANS are particularly suitable for the detection of protein aggregation at early stages, which is a challenging task, as aggregated species are often present in very low concentrations and may exhibit a short life span, e.g. when formed during refolding of proteins. Finke and Jennings characterized the presence of aggregated states of Interleukin-1ß (IL-1ß) at early stages during folding by ANS and tryptophan fluorescence, as well as by CD spectroscopy in conjugation with stopped-flow mixing. ANS was most sensitive for small reversible IL-1ß aggregates formed during folding, which were shown to irreversibly form larger aggregates when the concentration is high enough to thermodynamically drive the aggregation to sizes larger than a critical nucleus size (101).
Recombinant human factor VIII forms soluble aggregates at 37°C after a lag time of about 6 h, which points at a slow nucleation phenomenon during the aggregation process. Within thermal scans, Bis-ANS fluorescence revealed aggregation induced changes of the protein already at 40°C, while DSC, intrinsic fluorescence, CD and FTIR spectroscopy did not show any changes below 45°C (102). Similarly, ANS fluorescence detected changes related to thermally induced aggregation of the dimeric ß-lactoglobulin (103) and bovine carbonic anhydrase (104).
The various fluorescent dyes can exhibit different specificity and selectivity for certain aggregate types and sizes. Heat-induced amorphous aggregates of concanavalin A formed at pH 5 led to an increase of ANS fluorescence but not Thioflavin T fluorescence. However, fibrillation at pH 9 could be probed by both ANS and Thioflavin T, revealing that Thioflavin T has a higher selectivity for fibril structures, whereas ANS interacts with hydrophobic areas present in both amorphous aggregates and amyloid fibrils (32). Lindgren et al. demonstrated by steady-state and time-resolved fluorescence that ANS, Bis-ANS, Thioflavin T and DCVJ exhibited different selectivity to diverse types of aggregates, prefibrillar amyloidogenic oligomers and protofibrils of transthyretin (33). ANS fluorescence intensity increased with increasing aggregate size, whereas Bis-ANS interacted strongest with the monomeric A-state and the unfolded monomers of the protein. The decrease in intensity of Bis-ANS fluorescence upon aggregation of the A-state of transthyretin was attributed to a steric hindrance of binding caused by the larger size of Bis-ANS. DCVJ was the most suitable dye for detecting early oligomers with a size of 300 to 500 kDa; moreover, it detected mature amyloid fibrils and spherical aggregates. Thioflavin T was most appropriate for the detection of mature fibrils, but it bound to early oligomers of transthyretin as well (33). Carrotta et al. revealed that chromatographically separated dimers and trimers of thermally stressed ß-lactoglobulin exhibited molten globular features as detected by ANS fluorescence and CD spectroscopy. Contrary to native ß-lactoglobulin, an increase in Thioflavin T fluorescence was measured for isolated dimers, trimers and larger aggregates, indicating that Thioflavin T binding is not restricted to complete amyloid fibrils (105).
With fluorescent dyes the detection of small amounts of large aggregates, which are typically not detected by size exclusion chromatography, is feasible. Sutter et al. showed that Nile Red was capable to detect amounts as low as 1 nM of large heat denatured aggregates of ß-galactosidase, which were not detected by size exclusion chromatography. These large aggregates resulted in a blue shift of the emission maximum from 660 to 611 nm and an increase in fluorescence intensity. Combination of Nile Red fluorescence with time-resolved anisotropy and fluorescence correlation spectroscopy (FCS) confirmed the presence of few large aggregates and FCS furthermore allowed a size estimation of about 130 nm for the aggregates (106). Tiny amounts of visible aggregates of a human multi-domain glycoprotein could also be detected by ANS fluorescence using steady-state fluorescence spectroscopy (107). Fluorescence microscopy can alternatively be employed to visualize small amounts of large aggregates in protein formulations. Demeule et al. used Nile Red and Thioflavin T to visualize human IgG aggregates in the size range from 0.5 µm to millimeters (11,108).
Detection of Amyloid Fibrils
For the analytical characterization of amyloid fibrils, as well as for studying the fibrillation mechanism and kinetics, extrinsic fluorescent dyes have proven to be highly sensitive. Generally, fibrillation can be detected after a certain lag time that is amongst other factors dependent on the sensitivity of the applied method. It was possible to detect calcitonin fibrils by Nile Red fluorescence microscopy already at a very early time-point, at which UV spectroscopy and steady-state fluorescence spectroscopy did not show changes in signal (108). Thioflavin T fluorescence was more sensitive than turbidity measurements in the detection of aggregates within transthyretin amyloidogenesis, as Thioflavin T fluorescence intensity increased immediately when the fibrillation conditions were set, whereas turbidity increased at first after a lag time of about 200 s (109).
Human insulin is known to form amyloid fibrils in vitro under certain conditions, e.g. below pH 2 at 60°C. Insulin oligomers present the nucleation sites for fibril formation, which is followed by a growth of the fibrils and finally by precipitation of the fibrils as floccules (110,111). Mauro et al. observed differences in aggregation kinetics of insulin at low pH, at different temperatures and concentrations depending on the used method (static and dynamic light scattering techniques, AFM and Thioflavin T fluorescence), with Thioflavin T fluorescence being the most sensitive method for detecting the formation of insulin fibrils (14). In addition, Thioflavin T fluorescence revealed that fibril nucleation and elongation of insulin was inhibited by glycerol, whereas low concentrations of ethanol and trifluorethanol favored fibril formation (112). Other examples are the application of Thioflavin T to characterize fibrillation of the prion-forming protein Sup35p (113), hen egg-white lysozyme (114) and protein L (15). In the latter two studies Congo Red was applied as second dye for fibril characterization. A red shift of the Congo Red UV absorption maximum from about 490 nm to 500 nm with a shoulder at 540 nm was observed in the presence of protein L and hen egg-white lysozyme fibrils (15, 114). The shoulder can be resolved by absorption difference spectroscopy, where a distinct peak at 540 nm was observed in the presence of HSA amyloid fibrils (115) and of cystatin B amyloid fibrils (116).
Assessment of Surface Hydrophobicity
The surface hydrophobicity of a protein affects its physical stability, solubility, aggregation tendency, as well as adsorption behavior, which are important parameters for formulation development. The use of hydrophobic fluorescent dyes offers a spectroscopic approach to measure surface hydrophobicity of native and denatured proteins. However, other binding mechanisms next to hydrophobic interactions, e.g. electrostatic interactions which may occur between the dye and the protein need to be taken in consideration when interpreting the results. Compared to other methods like theoretical calculations based on the amino acid sequence, chromatography (117), differential scanning calorimetry, or precision densimetry (118), the use of fluorescent dyes is relatively rapid and simple and can also be applied to proteins with unknown amino acid sequences. Cardamone and Puri established a method to quantitatively assess the relative surface hydrophobicity by calculating the association constant of ANS-protein binding from Klotz plots (119) and compared the results with RP-HPLC and a theoretical hydrophobicity index based on the amino acid sequence (30). According to the ANS fluorescence results, the studied proteins could be ranked in the order bacteriorhodopsin > BSA > ovalbumin > porcine somatotrophin > lysozyme > RNase, which reflects the number of ANS binding sites and indirectly the surface hydrophobicity. Except for porcine somatotrophin and ovalbumin, a good correlation of the ANS fluorescence results with RP-HPLC and the theoretical hydrophobicity index could be achieved. The exceptions were attributed to the fact that ANS probes the molecules at physiological conditions, whereas denatured proteins are analyzed in RP-HPLC and the theoretical approach neglects the tertiary structure of the proteins (30).
Febbraio et al. employed Nile Red fluorescence to study the effect of in-vivo post translational N-ɛ-methylation of Sulfolobus solfataricus β-glycosidase on the surface hydrophobicity, thermal stability and aggregation behavior. Nile Red was capable to distinguish between the methylated wildtype and the recombinantly produced unmethylated form of the enzyme. The higher Nile Red fluorescence intensity measured for the native methylated enzyme indicated a higher surface hydrophobicity due to the apolar nature of the methyl groups (120). Nile Red was used to identify surface properties of tubulin dimer consisting of α- and β-tubulin. Nile Red interaction with tubulin caused an emission blue shift of about 40 nm and fluorescence intensity enhancement. When tubulin polymerized in glutamate buffer, Nile Red fluorescence emission was red-shifted. The red shift of emission could be attributed to an increase in the mean environment polarity of the Nile Red binding site, as the steady state intensity was reduced and lifetime was decreased (121). Other examples are the application of Nile Red fluorescence for probing protein surface hydrophobicity of L-lactate dehydrogenase from Bacillus stearothermophilus (122) and photoactive yellow protein from Halorhodospira halophila (123).
Monitoring of Interactions between Proteins and other Molecules
Extrinsic fluorescent dyes have been employed to monitor interactions between proteins and other molecules by measuring the displacement of the dyes by particular molecules competing for the binding sites, e.g. surfactants, small drug molecules or ligands. This allows gaining structural information of the protein and in particular of its binding sites. Three ANS binding sites for ANS on BSA were determined by De et al. (35). ANS was displaced from the BSA binding sites upon the addition of the anionic surfactant SDS, the neutral surfactant Triton X-100 or the cationic surfactant cetyl trimethyl ammonium bromide. As the highest decrease in ANS fluorescence was observed upon the addition of the anionic SDS, it was concluded that ANS is predominantly bound by ion pairing with cationic residues of the protein. Fluorescence lifetime studies revealed a bi-exponential decay of ANS in the presence of surfactants; the shorter lifetime could be ascribed to micellar ANS and the longer one to BSA-bound ANS (35). Similarly, displacement of ANS from albumins by medium and long chained fatty acids was accompanied by a decline in ANS fluorescence intensity (124). Brown et al. performed a dual-probe study with ANS and Nile Red to characterize the impact of various ligands on the displacement of the two dyes from BSA, which was monitored by the fluorescence decrease of the dyes in presence of the ligands (78). It could be demonstrated that some ligands displaced either ANS (e.g. ethacrynic acid) or Nile Red (e.g. stearic acid) from BSA, whereas others (e.g. flufenamic acid and L-tryptophan) displaced both dyes equally. These results revealed that ANS and Nile Red occupy different binding sites on albumin, suggesting that Nile Red interacts with the relatively less polar ones (78).
Probing of Protein Structure in Ice
The structural characterization of proteins in the frozen state is of relevance for formulation development, as protein bulk solutions and formulations are sometimes frozen to achieve a longer shelf life, inadvertent freezing of refrigerated protein drugs may induce structural changes, and freezing is an important process step for developing lyophilized products. ANS fluorescence was used to probe irreversible changes in the tertiary structure of proteins in the frozen state (125,126). Because of the high optical density of ice, high ANS concentrations up to 500 μM were employed. The samples were measured in short-pathlength cells to reduce the inner filter effect and under rotation to correct for inhomogeneity of the sample within the ice. For azurin, a 13-fold increase in ANS fluorescence accompanied by a blue shift was determined when the samples were frozen to −13°C. This was attributed to an irreversible change in tertiary structure in the frozen state, which could be inhibited by the addition of glycerol as cryoprotector (125). In a follow-up study seven monomeric, dimeric and tetrameric proteins, some of which bind ANS only in the frozen state and some also in the liquid state, were compared for their ANS fluorescence after freezing. All of them exhibited enhanced ANS fluorescence in ice as compared to the liquid formulations. The studied tetrameric proteins exhibited the largest increase in intensity, proportional to the number of ANS binding sites (126).
Fluorescent Dyes for Protein Visualization
Fluorescent dyes can be employed to stain SDS-PAGE gels and Western blots, as well as to visualize protein aggregates and protein crystals in microscopic techniques. Daban reviewed the application of Nile Red for staining of SDS-PAGE gels and the covalent dye 2-methoxy-2,4-diphenyl-3(2H)-furanone (MDPF) for Western blots. Nile Red can be employed as sensitive staining approach, detecting about 5 ng protein per band. As compared to other staining protocols, Nile Red staining has the advantage of being a fast one-step procedure (127). Alternatively, the dyes Sypro Orange (excitation maximum 472 nm, emission maximum 569 nm) and Sypro Red (excitation maximum 547 nm, emission maximum 635 nm) can be used for staining of SDS-PAGE gels and Western blots with a sensitivity of 1 to 10 ng protein per band. The stained bands can be visualized by CDD cameras or laser scanners (128,129).
The approach to visualize protein aggregates and fibrils by extrinsic dyes using fluorescence microscopy was already discussed earlier in this review. Moreover, ANS has the ability to make protein crystals fluorescent, which has been utilized for the visualization and identification of protein crystals with fluorescence microscopy (36). Within a wide concentration range of ANS it was possible to discriminate between protein crystals and salt crystals, which was shown for several proteins (36).
New Trends in Protein Characterization with Extrinsic Fluorescent Dyes
The high sensitivity of fluorescent dyes and the possibility to measure in small volumes, e.g. in 1536-well plates, makes these dyes suitable for high throughput screening studies to detect conformational changes and aggregation during formulation development (130). A high throughput screening method using commercially available real-time PCR instruments, which is based on the increase in fluorescence intensity of the dye Sypro Orange when bound to hydrophobic areas of thermally unfolded proteins, was used to determine the melting point of proteins during a thermal scan. The goal of the screening was to identify optimum formulation conditions for crystallization and purification of several proteins, with respect to pH, buffer, excipients and ligands (131–133).
Another novel approach is the application of ANS fluorescence for kinetic measurements of urea and GndHCl induced denaturation and refolding processes within microchips using very low sample volumes. It was feasible to monitor the unfolding and refolding of HSA and bovine carbonic anhydrase in volumes of 20 μl within the microfluidic system, which was validated against fluorescence measurements on well plates (134).
The design of new fluorescent dyes to characterize proteins is ongoing (135,136). Such dyes are designed using combinatorial approaches, as it is difficult to predict the properties of the fluorophores a priori (137). For instance, new dyes with improved selectivity for amyloid fibrils have been evaluated. Volkova et al. evaluated the ability of several cyanine dyes based on benzothiazole and benzimidazole compounds, as well as benzothiazole squaraine to detect fibrillar β-lactoglobulin. Several of these dyes turned out to be more sensitive and more selective for fibrillar β-lactoglobulin than Thioflavin T. Detailed mechanistic explanations for the fluorescence properties of these new dyes have not yet been described (138).
CONCLUSIONS
Noncovalent extrinsic fluorescent dyes can be of use for protein characterization in several stages of pharmaceutical development. Especially the high sensitivity of extrinsic dyes and their suitability for high throughput approaches can facilitate screening studies in early stages of formulation development. The ability of extrinsic dyes to assess surface hydrophobicity or posttranslational modifications is particularly beneficial for the characterization of hydrophobic recombinant proteins, as surface hydrophobicity can be relevant for the activity, aggregation and adsorption properties of the protein. Extrinsic fluorescent dyes allow the detection of aggregation, fibrillation, folding intermediates, and other structural variants without chromatographic separation. Additionally, size information about aggregates or fibrils can be obtained when employing extrinsic fluorescent dyes in FCS or fluorescence microscopy. Extrinsic dyes are a valuable addition to intrinsic protein fluorescence, as they are independent of the presence and position of aromatic amino acids within the protein. Furthermore, they monitor aspects e.g. minor changes in surface hydrophobicity, which are not necessarily probed by tryptophan and tyrosine fluorescence. In conclusion, extrinsic fluorescent dyes are highly sensitive and versatile tools for protein characterization. Their application for probing protein structure can provide valuable information about protein structure, unfolding and aggregation and thereby contributes to the safety of protein pharmaceuticals.
Abbreviations
ANS:
8-Anilino-1-naphthalenesulfonate
Bis-ANS:
4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonate
BSA:
Bovine serum albumin
CD:
Circular dichroism
DCVJ:
9-(Dicyanovinyl)-julolidine
DMSO:
Dimethyl sulfoxide
FCS:
Fluorescence correlation spectroscopy
FTIR:
Fourier transform infrared spectroscopy
GndHCl:
Guanidine hydrochloride
HSA:
Human serum albumin
ICT:
Intramolecular charge transfer
TICT:
Twisted intramolecular charge transfer
References
- G. Walsh. Biopharmaceutical benchmarks 2006. Nature Biotechnol. 24:769–779 (2006).
CAS Google Scholar - M. C. Manning, K. Patel, and R. T. Borchardt. Stability of protein pharmaceuticals. Pharm. Res. 6:903–918 (1989).
PubMed CAS Google Scholar - E. Y. Chi, S. Krishnan, T. W. Randolph, and J. F. Carpenter. Physical stability of proteins in aqueous solutions: mechanisms and driving forces in nonnative protein Aggregation. Pharm. Res. 20:1325–1336 (2003).
PubMed CAS Google Scholar - W. Wang, S. Singh, D. Zeng, K. King, and S. Nema. Antibody structure, instability, and formulation. J. Pharm. Sci. 96:1–26 (2007).
PubMed CAS Google Scholar - W. Wang. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 289:1–30 (2005).
PubMed CAS Google Scholar - M. E. M. Cromwell, E. Hilario, and F. Jacobson. Protein aggregation and bioprocessing. AAPS J. 8:E572–E579 (2006).
PubMed CAS Google Scholar - S. Hermeling, D. J. A. Crommelin, H. Schellekens, and W. Jiskoot. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 21:897–903 (2004).
PubMed CAS Google Scholar - S. Hermeling, L. Aranha, J. M. A. Damen, M. Slijper, H. Schellekens, D. J. A. Crommelin, and W. Jiskoot. Structural characterization and immunogenicity in wild-type and immune tolerant mice of degraded recombinant human interferon alpha2b. Pharm. Res. 22:1997–2006 (2005).
PubMed CAS Google Scholar - A. S. Rosenberg. Effects of protein aggregates: an immunologic perspective. AAPS J. 8:501–507 (2006).
Google Scholar - H.-C. Mahler, R. Müller, W. Friess, A. Delille, and S. Matheus. Induction and analysis of aggregates in liquid IgG1-antibody formulations. Eur. J. Pharm. Biopharm. 59:407–417 (2005).
PubMed CAS Google Scholar - B. Demeule, M. J. Lawrence, A. F. Drake, R. Gurny, and T. Avinte. Characterization of protein aggregation: the case of a therapeutic immunoglobulin. Biochim. Biophys. Acta. 1774:146–153 (2007).
PubMed CAS Google Scholar - J. S. Philo. Is any measurement method optimal for all aggregate sizes and types. AAPS J. 5:E564–E571 (2006).
Google Scholar - L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky, and A. L. Fink. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40:6036–6046 (2001).
PubMed CAS Google Scholar - M. Mauro, E. F. Craparo, A. Podesta, D. Bulone, R. Carotta, V. Martorana, G. Tianaa, and P. L. San Biagio. Kinetics of different processes in human insulin amyloid formation. J. Mol. Biol. 366:258–274 (2007).
PubMed CAS Google Scholar - T. Cellmer, R. Douma, A. Huebner, J. Prausnitz, and H. Blanch. Kinetic studies of protein L aggregation and disaggregation. Biophys. Chem. 125:350–359 (2007).
PubMed CAS Google Scholar - W. Jiskoot, A. J. W. G. Visser, J. N. Herron, and M. Sutter. Fluorescence spectroscopy. In W. Jiskoot, and D. J. A. Crommelin (eds.), Methods for Structural Analysis of Protein Pharmaceuticals, AAPS press, Arlington, 2005, pp. 27–82.
Google Scholar - A. S. Ladokhin. Fluorescence spectroscopy in peptide and protein analysis. In R. A. Meyers (ed.), Encyclopedia of Analytical Chemistry, Wiley, New York, 2000, pp. 5762–5779.
Google Scholar - G. Weber, and D. J. R. Lawerence. Fluorescent indicators of adsorption in aqueous solution and on the solid phase. Biochem. J. 56(325th Meeting):xxxi (1954).
- L. Stryer. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13:482–495 (1965).
PubMed CAS Google Scholar - C. G. Rosen, and G. Weber. Dimer formation from 1-anilino-8-naphthalenesulfonate catalyzed by bovine serum albumin. A new fluorescent molecule with exceptional binding properties. Biochemistry 8:3915–3920 (1969).
PubMed CAS Google Scholar - P. Greenspan, E. P. Mayer, and S. D. Fowler. Nile Red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 100:965–973 (1985).
PubMed CAS Google Scholar - D. Sackett, and J. Wolff. Nile Red as polarity-sensitive fluorescent probe of hydrophobic protein surfaces. Anal. Biochem. 167:228–234 (1987).
PubMed CAS Google Scholar - C. E. Kung, and J. K. Reed. Fluorescent Molecular Rotors: A new class of probes for tubulin structure and assembly. Biochemistry 28:6678–6686 (1989).
PubMed CAS Google Scholar - M. L. Viriot, M. C. Carré, C. Geoffroy-Chapotot, A. Brembilla, S. Muller, and J. F. Scholtz. Molecular rotors as fluorescent probes for biological studies. Clin. Hemorheol. Microcircul. 19:151–160 (1998).
CAS Google Scholar - H. Puchtler, F. Sweat, and M. Levine. On the binding of Congo Red by amyloid. J. Histochem. Cytochem. 9:553–539 (1961).
PubMed CAS Google Scholar - P. Frid, S. V. Anisimov, and N. Popovic. Congo Red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 53:135–160 (2007).
PubMed CAS Google Scholar - H. Naiki, K. Higuchi, M. Hosokawa, and T. Takeda. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye Thioflavine T. Anal. Biochem. 177:244–249 (1989).
PubMed CAS Google Scholar - P. Acharya, and N. Madhusudhana Rao. Stability studies on a lipase from Bacillus subtilis in guanidinium chloride. J. Prot. Chem. 22:51–60 (2003).
CAS Google Scholar - Y. Goto, and A. L. Fink. Conformational States of β-Lactamase: Molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28:945–952 (1989).
PubMed CAS Google Scholar - M. Cardamone, and N. K. Puri. Spectrofluorimetric assessment of the surface hydrophobicity of proteins. Biochem. J. 282:589–593 (1992).
PubMed CAS Google Scholar - R. Takashi, Y. Tonomura, and M. F. Morales. 4,4’Bis(1-anilinonaphthalene 8-sulfonate) (bis-ANS): A new probe of the active site myosin. Biochemistry 74:2334–2338 (1977).
CAS Google Scholar - V. Vetri, C. Canale, A. Relini, F. Librizzi, V. Militello, A. Gliozzi, and M. Leone. Amyloid fibrils formation and amorphous aggregation in concanavalin A. Biophys. Chem. 125:184–190 (2007).
PubMed CAS Google Scholar - M. Lindgren, K. Sörgjerd, and P. Hammarström. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 88:4200–4212 (2005).
PubMed CAS Google Scholar - M. Anraku, K. Yamasaki, T. Maruyama, U. Krangh-Hansen, and M. Otagiri. Effect of oxidative stress on the structure and function of human serum albumin. Pharm. Res. 18:632–639 (2001).
PubMed CAS Google Scholar - S. De, A. Girigoswami, and S. Das. Fluorescence probing of albumin-surfactant interaction. J. Coll. Interf. Sci. 285:562–573 (2005).
CAS Google Scholar - M. R. Groves, I. B. Müller, X. Kreplin, and J. Müller-Dieckmann. A method for the general identification of protein crystals in crystallization experiments using a noncovalent fluorescent dye. Acta Crystallogr. D. Biol. Crystallogr. 63:526–535 (2007).
PubMed Google Scholar - J. R. Lakowicz. Principles of fluorescence spectroscopy. 3rd edition, Springer, Berlin, 2006.
Google Scholar - B. Valeur. Molecular fluorescence. Principles and applications. Wiley-VCH Verlag GmbH, Weinheim, 2002.
Google Scholar - A. Dutta, K. Kamada, and K. Ohta. Spectroscopic studies on nile red in organic solvents and polymers. J. Photochem. Photobiol. A: Chem. 93:57–64 (1996).
CAS Google Scholar - K. Das, N. Sarkar, D. Nath, and K. Bhattacharyya. Non-radiative pathways of aniline-naphthalene sulphonates: twisted intramolecular charge transfer versus intersystem crossing. Spectrochim. Acta. 48A:1701–1705 (1992).
CAS Google Scholar - T. L. Changm, and H. C. Cheung. A model for molecules with twisted intramolecular charge transfer characteristics: solvent polarity effect on the nonradiative rates of dyes in a series of water-ethanol mixed solvents. Chem. Phys. Lett. 173:343–348 (1990).
Google Scholar - B. S. Neporent, and N. G. Bakhshiev. On the role of universal and specific intermolecular interactions in the influence of the solvent on the electronic spectra of molecules. Opt. Spectroscop. 8:408–413 (1960).
CAS Google Scholar - T. Azumi, K. Itoh, and H. Shiraishi. Shift of emission band upon the excitation at the long wavelength absorption edge. III. Temperature dependence of the shift and correlation with the time dependent spectral shift. J. Phys. Chem. 65:2550–2555 (1976).
CAS Google Scholar - J. Slavik. Anilinonaphthalene sulfonate as probe of membrane composition and function. Biochim. Biophys. Acta. 694:1–25 (1982).
PubMed CAS Google Scholar - E. Klimtchuk, S. Venyaminov, E. Kurian, W. Wessels, W. Kirk, and F. G. Prendergast. Photophysics of ANS. I. Protein-ANS complexes: Intestinal fatty acid binding protein and single-trp mutants. Biophys. Chem. 125:1–12 (2007).
PubMed CAS Google Scholar - W. Kirk. Photophysics of ANS. II. Charge transfer character of near-UV absorption and consequences for ANS spectroscopy. Biophys. Chem. 125:13–23 (2007).
PubMed CAS Google Scholar - W. Kirk, and E. Klimtchuk. Photophysics of ANS. III. Circular dichroism of ANS and anilinonaphthalene in I-FABP. Biophys. Chem. 125:24–31 (2007).
PubMed CAS Google Scholar - W. Kirk, and W. Wessels. Photophysics of ANS. IV. Electron transfer quenching of ANS in alcoholic solvents and mixtures. Biophys. Chem. 125:32–49 (2007).
PubMed CAS Google Scholar - W. Kirk, E. Kurian, and W. Wessels. Photophysics of ANS. V. Decay modes of ANS in proteins: the IFABP-ANS complex. Biophys. Chem. 125:50–58 (2007).
PubMed CAS Google Scholar - D. J. R. Laurence. A study of the adsorption of dyes on bovine serum albumin by the method of polarization of fluorescence. Biochem. J. 51:168–180 (1952).
PubMed CAS Google Scholar - D. Matulis, and R. Lovrien. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 74:422–429 (1998).
PubMed CAS Google Scholar - O. K. Gasymov, and B. J. Glasgow. ANS fluorescence: Potential to augment the identification of the external binding sites of proteins. Biochim. Biophys. Acta. 1774:403–411 (2007).
PubMed CAS Google Scholar - A. Bothra, A. Bhattacharyya, C. Mukhopadhyay, K. Bhattacharyya, and S. Roy. A fluorescence spectroscopic and molecular dynamics study of Bis-ANS/protein interaction. J. Biomol. Struct. Dyn. 15:959–966 (1998).
PubMed CAS Google Scholar - A. V. Pastukhov, and I. J. Ropson. Fluorescent Dyes as Probes to Study Lipid-Binding Proteins. Proteins: Struct. Funct. Genet. 53:607–615 (2003).
CAS Google Scholar - N. Sarkar, K. Das, D. N. Nath, and K. Bhattacharyya. Twisted charge transfer process of Nile Red in homogeneous solution and in faujasite zeolite. Langmuir 10:326–329 (1994).
CAS Google Scholar - A. Cser, K. Nagy, and L. Biczok. Fluorescence lifetime of Nile Red as a probe for the hydrogen bonding strength with its microenvironment. Chem. Phys. Lett 360:473–478 (2002).
CAS Google Scholar - M. A. Haidekker, T. P. Brady, D. Lichlyter, and E. A. Theodorakis. Effects of solvent polarity and solvent viscosity on the fluorescent properties of molecular rotors and related probes. Bioorg. Chem 33:415–425 (2005).
PubMed CAS Google Scholar - W. J. Akers, J. M. Cupps, and M. A. Haidekker. Interaction of fluorescent molecular rotors with blood plasma proteins. Biorheol. 42:335–344 (2005).
CAS Google Scholar - T. Iio, M. Itakura, S. Takahashi, and S. Sawada. 9-(Dicyanovinyl)julolidine binding to bovine brain calmodulin. J. Biochem. (Tokyo) 109:449–502 (1991).
Google Scholar - A. A. Maskevich, V. I. Stispura, V. A. Kuzmitsky, I. M. Kuznetsova, O. I. Povarova, V. N. Uversky, and K. K. Turoverov. Spectral properties of Thioflavin T in solvents with different dielectric properties and in a fibril-incorporated form. J. Proteom. Res. 6:1392–1401 (2007).
CAS Google Scholar - E. S. Voropai, M. P. Samtsov, K. N. Kaplevskii, A. A. Maskevich, V. I. Stepuro, O. I. Povarova, I. M. Kuznetsova, K. K. Turoverov, A. L. Fink, and V. N. Uverskii. Spectral properties of Thioflavin T and its complexes with amyloid fibrils. J. Appl. Spectros. 70:868–874 (2003).
CAS Google Scholar - V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, K. K. Turovervov, and I. M. Kuznetsova. Computational study of Thioflavin T torsional relaxation in the excited state. J. Phys. Chem. A. 111:4829–4835 (2007).
PubMed CAS Google Scholar - P. S. Vassar, and C. F. A. Culling. Fluorescent stains with special reference to amyloid and connective tissue. Achr. Pathol. 68:487–498 (1959).
CAS Google Scholar - G. Kelenyi. On the histochemistry of azo group-free thiazole dyes. J. Histochem. Cytochem. 15:172–180 (1967).
PubMed CAS Google Scholar - H. LeVine III. Quantification of the β-sheet amyloid fibril structures with Thioflavin T. Methods Enzymol. 309:274–284 (1999).
PubMed CAS Google Scholar - M. F. B. G. Gebbink, D. Claessen, B. Bouma, L. Dijkhuizen, and H. A. B. Wösten. Amyloids- A functional coat for microorganisms. Nature Reviews 3:333–341 (2005).
PubMed CAS Google Scholar - H. LeVine III. Thioflavin T interaction with synthetic Alzheimers’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Prot. Sci. 2:404–410 (1993).
Article CAS Google Scholar - M. Groenning, L. Olsen, M. van der Weert, J. M. Flink, S. Frokjaer, and F. S. Jørgensen. Study of the binding of Thioflavin T to β-sheet-rich and non-β-sheet-rich cavities. J. Struct. Biol. 158:358–369 (2007).
PubMed CAS Google Scholar - R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, R. K. Grover, R. Roy, and S. Singh. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151:229–238 (2005).
PubMed CAS Google Scholar - M. R. H. Krebs, E. H. C. Bromley, and A. M. Donald. The binding of Thioflavin T to amyloid fibrils: localisation and implications. J. Struct. Biol. 149:30–37 (2005).
PubMed CAS Google Scholar - J. P. Verbelen, and S. Kerstens. Polarization confocal microscopy and congo red fluorescence: a simple and rapid method to determine the mean cellulose fibril orientation in plants. J. Microsc. 198:101–107 (2000).
PubMed CAS Google Scholar - W. E. Klunk, R. F. Jacob, and R. Preston Mason. Quantifying amyloid by Congo Red spectral shift assay. Methods Enzymol. 309:285–305 (1999).
PubMed CAS Google Scholar - R. Khurana, V. N. Uversky, L. Nielsen, and A. L. Fink. Is Congo Red an amyloid-specific dye. J. Biol. Chem. 276:22715–22721 (2001).
PubMed CAS Google Scholar - B. Stopa, M. Gorny, L. Konieczny, B. Piekarska, J. Rybarska, M. Skowronek, and I. Roterman. Supramolecular ligands: Monomer structure and protein ligation capability. Biochim. 80:963–968 (1998).
CAS Google Scholar - B. Stopa, B. Piekarska, L. Konieczny, J. Rybarska, P. Spolnik, G. Zemanek, I. Roterman, and M. Krol. The structure and protein binding of amyloid-specific dye reagents. Acta Biochim. Polon. 50:1213–1227 (2003).
PubMed CAS Google Scholar - P. Greenspan, and S. Fowler. Spectrofluorometric studies of the lipid probe nile red. J. Lip. Res. 26:781–789 (1985).
CAS Google Scholar - M. K. Santra, A. Banerjee, S. S. Krishnakumar, O. Rahaman, and D. Panda. Multiple-probe analysis of folding and unfolding pathways of human serum albumin. Evidence for a framework mechanism of folding. Eur. J. Biochem. 271:1789–1497 (2004).
PubMed CAS Google Scholar - M. B. Brown, T. E. Edmonds, J. N. Miller, and N. J. Seare. Use of Nile Red as a long-wavelength fluorophore in dual-probe studies of ligand-protein interactions. J. Fluoresc. 3:129–130 (1993).
CAS Google Scholar - S. Muzzammil, Y. Kumar, and S. Tayyd. Molten globule-like state of human serum albumin at low pH. Eur. J. Biochem. 266:26–32 (1999).
Google Scholar - V. Ali, K. Prakash, S. Kulkarni, A. Ahmad, K. P. Madhusudan, and V. Bhakuni. 8-Anilino-1-naphthalene sulfonic acid (ANS) induces folding of acid unfolded Cytochrome c to molten globule state as a result of electrostatic interactions. Biochemistry 38:13635–13642 (1999).
PubMed CAS Google Scholar - B. Kundu, and P. Guptasarma. Hydrophobic dye inhibits interaction of molten carbonic anhydrase during thermal unfolding and refolding. Proteins: Struct. Funct. Genet. 37:321–324 (1999).
CAS Google Scholar - X. Fu, X. Zhang, and Y. Chang. 4,4′-Dianilino-1,1′-binaphthyl-5,5′-sulfonate, a novel molecule having chaperon-like activity. Biochem. Biophys. Res. Comm. 329:1087–1093 (2005).
PubMed CAS Google Scholar - J. Sereikaite, and V. Bumelis. Congo Red interaction with α-proteins. Acta Biochim. Polon. 53:87–91 (2006).
PubMed CAS Google Scholar - J. L. Cleland, and D. I. C. Wang. In vitro protein refolding. In H.-J. Rehm, and G. Reed (eds.), Biotechnology: a multi-volume comprehensive treatise Vol 3, Bioprocessing, VCH-Wiley, New York, 1993, pp. 527–555.
Google Scholar - S. E. Jackson. How do small single-domain proteins fold. Fold. Des. 3:R81–R91 (1998).
PubMed CAS Google Scholar - R. L. Baldwin, and G. D. Rose. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci. 24:77–83 (1999).
PubMed CAS Google Scholar - O. B. Ptitsyn, R. H. Pain, G. V. Semisotnov, E. Zerovnik, and O. I. Razgulyaev. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 262:20–24 (1990).
PubMed CAS Google Scholar - K. A. Dill, and D. Shortle. Denatured states of proteins. Annu. Rev. Biochem. 60:795–825 (1991).
PubMed CAS Google Scholar - G. V. Semisotnov, N. A. Rodionova, V. P. Kutyshenko, B. Ebert, J. Blanck, and O. B. Ptitsyn. Sequential mechanism of refolding of carbonic anhydrase B. FEBS Lett. 224:9–13 (1990).
Google Scholar - R. F. Latypov, T. S. Harvey, E. Liu, P.V. Bondarenko, T. Kohno, R. A. Fachini II, R. D. Rosenfeld, R. R. Ketchem, D. N. Brems, and A. A. Raibekas. Biophysical characterization of structural properties and folding of interleukin-1 receptor antagonist. J. Mol. Biol. 368:1187–1201 (2007).
PubMed CAS Google Scholar - A. L. Fink, L. J. Calciano, Y. Goto, T. Kurotsu, and D. R. Palleros. Classification of acid denaturation of proteins: intermediates and unfolded States. Biochemistry 33:12504–12511 (1994).
PubMed CAS Google Scholar - N. B. Bam, J. L. Cleland, and T. W. Randolph. Molten globule intermediate of recombinant human growth hormone: stabilization of surfactants. Biotechnol. Prog. 12:801–809 (1996).
PubMed CAS Google Scholar - B. Ahmad, and R. H. Khan. Studies on the acid unfolded and molten globule states of catalytically active stem bromelain: A comparison with catalytically inactive form. J. Biochem. 140:501–508 (2006).
PubMed CAS Google Scholar - B. Ahmad, M. Zulfazal, S. K. Haq, and R. H. Khan. Guanidine hydrochloride denaturation of human serum albumin originates by local unfolding of some stable loops in domain III. Biochim. Biophys. Acta 1750:93–102 (2005).
PubMed CAS Google Scholar - D. M. Togashi, and A. G. Ryder. Time-Resolved fluorescence studies on bovine serum albumin denaturation process. J. Fluoresc. 16:153–160 (2006).
PubMed CAS Google Scholar - A. Brahma, C. Mandal, and D. Bhattacharyya. Characterization of a dimeric unfolding intermediate of bovine serum albumin under mildly acidic conditions. Biochim. Biophys. Acta 1751:159–169 (2005).
PubMed CAS Google Scholar - P. Wojciak, A. Mazurkiewicz, A. Bakalova, and R. Kuciel. Equilibrium unfolding of dimeric human prostatic acid phosphatase involves an inactive monomeric intermediate. Int. J. Biol. Macromol. 32:43–54 (2003).
PubMed CAS Google Scholar - M. Bertoldi, B. Cellini, D. V. Laurents, and C. Borri Voltattorni. Folding pathway of the pyridoxal 5’-phosphate C-S lyase MalY from Escherichia coli. Biochem. J. 389:885–898 (2005).
PubMed CAS Google Scholar - B. Ásgeirsson, and K. Guðjónsdóttir. Reversible inactivation of alkaline phosphatase from Atlantic cod (Gadus morhua) in urea. Biochim. Biophys. Acta 1764:190–198 (2006).
PubMed Google Scholar - S. Jana, T. K. Chaudhuri, and J. K. Deb. Effects of guanidine hydrochloride on the confirmation and enzyme activity of Streptomycin Adenylyltransferase monitored by circular dichroism and fluorescence spectroscopy. Biochemistry (Mosc.) 71:1230–1237 (2006).
CAS Google Scholar - J. M. Finke, and P. A. Jennings. Early aggregated states in the folding of Interleukin-1ß. J. Biol. Phys. 27:119–131 (2001).
CAS Google Scholar - A. O. Grillo, K. L. T. Edwards, R. S. Kashi, K. M. Shipley, L. Hu, M. J. Besman, and C. R. Middaugh. Conformational origin of the aggregation of recombinant human Factor VIII. Biochemistry 40:586–595 (2001).
PubMed CAS Google Scholar - V. Vetri, and V. Militello. Thermal induced conformational changes involved in the aggregation pathway of beta-lactoglobulin. Biophys. Chem. 113:83–91 (2005).
PubMed CAS Google Scholar - B. Kundu, and P. Guptasarma. Use of a hydrophobic dye to indirectly probe the structural organization and conformational plasticity of molecules in amorphous aggregates of carbonic anhydrase. Biochem. Biophys. Res. Com. 293:572–577 (2002).
PubMed CAS Google Scholar - R. Carrotta, R. Bauer, R. Waninge, and C. Rischel. Conformational characterization of oligomeric intermediates and aggregates in ß-lactoglobulin heat aggregation. Prot. Sci. 10:1312–1318 (2001).
CAS Google Scholar - M. Sutter, S. Oliveira, N. N. Sanders, B. Lucas, A. van Hoek, M. A. Hink, A. J. W. G. Visser, S. C. De Smedt, W. E. Hennink, and W. Jiskoot. Sensitive spectroscopic detection of large and denatured protein aggregates in solution by use of the fluorescent dye Nile Red. J. Fluoresc. 17:181–192 (2007).
PubMed CAS Google Scholar - P. O. Souillac. Biophysical characterization of insoluble aggregates of a multi-domain protein: An insight into the role of various domains. J. Pharm. Sci. 94:2069–2083 (2005).
PubMed CAS Google Scholar - B. Demeule, R. Gurny, and T. Avinte. Detection and characterization of protein aggregates by fluorescence microscopy. Int. J. Pharm. 329:37–45 (2007).
PubMed CAS Google Scholar - A. R. Hurshman, J. T. White, E. T. Powers, and J. W. Kelly. Transthyretin aggregation under partially denaturating conditions is a downhill polymerization. Biochemistry 43:7365–7381 (2004).
PubMed CAS Google Scholar - D. F. Waugh, D. F. Wilhelmson, S. L. Commerford, and M. L. Sackler. Studies of the nucleation and growth reations of selected types of insulin fibrils. J. Am. Chem. Soc. 75:2592–2600 (1953).
CAS Google Scholar - D. F. Waugh. A mechanism for the formation of fibrils from protein molecules. J. Cell. Physiol. 49:145–164 (1957).
CAS Google Scholar - S. Grudzielanek, R. Jansen, and R. Winter. Solvational tuning of the unfolding, aggregation and amyloidogenesis of insulin. J. Mol. Biol. 351:879–894 (2005).
PubMed CAS Google Scholar - J. Krzewska, M. Tanaka, S. B. Burston, and R. Melki. Biochemical and functional analysis of the assembly of full-length Sup35p and its prion-forming domain. J. Biol. Chem. 282:1679–1686 (2007).
PubMed CAS Google Scholar - B. A. Vernaglia, J. Huang, and E. D. Clark. Guanidine hydrochloride can induce amyloid fibril formation from henn egg-white lysozyme. Biomacromol. 5:1362–1370 (2004).
CAS Google Scholar - P. Taboada, S. Barbosa, E. Castro, and V. Mosquera. Amyloid fibril formation and other aggregate species formed by Human Serum Albumin association. J. Phys. Chem. B. 110:20733–20736 (2006).
PubMed CAS Google Scholar - E. Žerovnic, M. Pompe-Novak, M. Škarabot, M. Ravnikar, I. Muševič, and V. Turk. Human stefin B readily forms amyloid fibrils in vitro. Biochim. Biophys. Acta 1594:1–5 (2002).
Google Scholar - T. Blunk, E. Mak, and R. H. Mueller. Characterization of colloidal drug carriers. Determination of surface hydrophobicity by hydrophobic-interaction chromatography. Pharmaz. Ind. 55:612–615 (1993).
CAS Google Scholar - A. N. Danilenko, V. T. Dianova, E. E. Braudo, T. Henning, R. Mothes, and K. D. Schwenke. A novel approach to the evaluation of hydrophobicity of food proteins. Nahrung 42:179–182 (1998).
CAS Google Scholar - I. M. Klotz, and D. L. Hunston. Properties of graphical representations of multiple classes of binding sites. Biochemistry 10:3065–3069 (1971).
PubMed CAS Google Scholar - F. Febbraio, A. Andolfo, F. Tanfani, R. Briante, F. Gentile, S. Formisano, C. Vaccaro, A. Scire, E. Bertoli, P. Pucci, and R. Nucci. Thermal stability and aggregation of sulfolobus solfataricus β-glycosidase are dependent upon the N-ɛ-methylation of specific Lysyl residues. J. Biol. Chem. 279:10185–10194 (2004).
PubMed CAS Google Scholar - D. L. Sackett, J. R. Knutson, and J. Wolff. Hydrophobic surfaces of tubulin probed by time-resolved and steady-state fluorescence of Nile Red. J. Biol. Chem. 25:14899–14906 (1990).
Google Scholar - M. Kotik, and H. Zuber. Evidence of temperature-dependent conformational changes in the L-Lactate dehydrogenase from bacillus stearothermophilus. Biochemistry 31:7787–7795 (1992).
PubMed CAS Google Scholar - J. Hendriks, T. Gensch, L. Hviid, M. A. van der Horst, K. J. Hellingwerf, and J. J. van Thor. Transient exposure of hydrophobic surface in the photoactive yellow protein monitored with Nile Red. Biophys. J. 82:1632–1643 (2002).
PubMed CAS Google Scholar - E. C. Santos, and A. A. Spector. Effect of fatty acids on the binding of 1-Anilino-8-naphtahlenesulfonate to bovine serum albumin. Biochemistry 11:2299–2302 (1972).
PubMed CAS Google Scholar - E. Gabellieri, and G. B. Strambini. Perturbation of protein tertiary structure in frozen solutions revealed by 1-Anilino-8-naphthalene sulfonate fluorescence. Biophys. J. 85:3214–3220 (2003).
Article PubMed CAS Google Scholar - E. Gabellieri, and G. B. Strambini. ANS fluorescence detects widespread perturbations of protein tertiary structure in ice. Biophys. J. 90:3239–3245 (2006).
PubMed CAS Google Scholar - J. R. Daban. Fluorescent labeling of proteins with Nile red and 2-methoxy-2,4-diphenyl-3(2H)-furanone: Physico-chemical basis and application to the rapid staining of sodium dodecyl sulfate polyacrylamide gels and Western blots. Electrophor. 22:874–880 (2001).
CAS Google Scholar - T. H. Steinberg, L. J. Jones, R. P. Haugland, and V. L. Singer. SYPRO orange and SYPRO red protein gel stains: one-step fluorescent staining of denaturing gels for detection of nanogram levels of protein. Anal. Biochem. 239:223–237 (1996).
PubMed CAS Google Scholar - T. H. Steinberg, R. P. Haugland, and V. L. Singer. Application of SYPRO orange and SYPRO red protein gel stains. Anal. Biochem. 239:238–245 (1996).
PubMed CAS Google Scholar - M. A. H. Capelle, R. Gurny, and T. Avinte. High throughput of protein formulation stability: Practical considerations. High Throughput screening of protein formulation stability: Practical considerations. Eur. J. Pharm. Biopharm. 65:131–148 (2007).
PubMed CAS Google Scholar - U. B. Ericsson, B. M. Hallberg, G. T. DeTitta, N. Dekker, and P. Nordlund. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357:289–298 (2006).
PubMed CAS Google Scholar - M. Vedadi, F. H. Niesen, A. Allali-Hassani, O. Y. Fedorov, P. J. Finerty Jr., G. A. Wasney, R. Yeung, C. Arrowsmith, L. J. Ball, H. Berglund, R. Hui, B. D. Marsden, P. Nordlund, M. Sundstrom, J. Weigelt, and A. M. Edwards. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad Sci. USA 103:15835–15840 (2006).
PubMed CAS Google Scholar - A. P. Yeh, A. McMillan, and M. H. B. Stowell. Rapid and simple protein-stability screens: application to membrane proteins. Acta Cryst. D: Biological Crystallography D62:451–457 (2006).
CAS Google Scholar - M. B. Kerby, J. Lee, J. Ziperstein, and A. Tripathi. Kinetic measurements of protein conformation in a microchip. Biotechnol. Prog 22:1416–1425 (2006).
PubMed CAS Google Scholar - B. N. G. Giepmans, S. R. Adams, M. H. Ellisman, and R. Y. Tsien. The fluorescent toolbox for assessing protein location and lunction. Science 312:217–224 (2006).
PubMed CAS Google Scholar - J. Zhang, R. E. Campbell, A. Y. Ting, and R. Y. Tsien. Creating new fluorescent probes for cell biology. Nature Reviews 3:906–918 (2002).
PubMed CAS Google Scholar - N. S. Finney. Combinatorial discovery of fluorophores and fluorescent probes. Curr. Opin. Chem. Biol. 10:238–245 (2006).
PubMed CAS Google Scholar - K. D. Volkova, V. B. Kovalska, O. A. Balanda, R. J. Vermeij, V. Subramaniam, Y. L. Slominskii, and S. M. Yarmoluk. Cyanine dye-protein interactions: Looking for fluorescent probes for amyloid structures. J. Biochem. Biophys. Methods 70:727–733 (2007).
PubMed CAS Google Scholar - G. Weber, and L. B. Young. Fragmentation of Bovine Serum Albumin by pepsin. I. The origin of the acid expansion of the albumin molecule. J. Biol. Chem. 239:1415–1423 (1964).
PubMed CAS Google Scholar - X. C. Yu, and W. Margolin. Inhibition of assembly of bacterial cell division protein FtsZ by the hydrophobic dye 5,5’′-bis-(8-anilino-1-naphthalenesulfonate). J. Biol. Chem. 2273:10216–10222 (1998).
Google Scholar - G. R. Castro, B. K. Larson, B. Panilaitis, and D. L. Kaplan. Emulsan quantification by Nile Red quenching fluorescence assay. Appl. Microbiol. Technol. 67:767–770 (2005).
Article CAS Google Scholar - J. Wall, C. L. Murphy, and A. Solomon. In vitro immunoglobulin light chain fibrillogenesis. Methods Enzymol. 304:204–217 (1999).
Google Scholar - Y. S. Kim, T. W. Randolph, M. C. Manning, F. J. Stevens, and J. F. Carpenter. Congo red populates partially unfolded states of an amyloidogenic protein to enhance aggregation and amyloid fibril formation. J. Biol. Chem. 21:10842–10850 (2003).
Google Scholar
Acknowledgments
We thank Dr. Mirka Poprawa-Smoluch for critically reviewing the manuscript and her helpful comments. We thank Prof. Dr. Ulla Mitzdorf and Dr. Margit Weber from Ludwig-Maximilians University Munich and her co-workers for providing the HWP-grant for advancement of women in research to Dr. Andrea Hawe. This research was supported in part by the Technology Foundation STW, the applied science division of NWO and technology program of the Dutch Ministry of Economic Affairs.
Author information
Author notes
- Marc Sutter
Present address: Novartis Pharma AG, WSJ-316.4.14, CH-4056, Basel, Switzerland
Authors and Affiliations
- Division of Drug Delivery Technology, Leiden/Amsterdam Center for Drug Research, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands
Andrea Hawe, Marc Sutter & Wim Jiskoot
Authors
- Andrea Hawe
You can also search for this author inPubMed Google Scholar - Marc Sutter
You can also search for this author inPubMed Google Scholar - Wim Jiskoot
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toWim Jiskoot.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hawe, A., Sutter, M. & Jiskoot, W. Extrinsic Fluorescent Dyes as Tools for Protein Characterization.Pharm Res 25, 1487–1499 (2008). https://doi.org/10.1007/s11095-007-9516-9
- Received: 17 October 2007
- Accepted: 05 December 2007
- Published: 03 January 2008
- Issue Date: July 2008
- DOI: https://doi.org/10.1007/s11095-007-9516-9