Genotype determination for polymorphisms in linkage disequilibrium (original) (raw)
Background
Recent advances in genotyping technologies have greatly improved genotype call rates and accuracy. This consequently enhances our understanding of genetic variations that are responsible for complex human traits. In particular, studies with SNPs show great promise to identify genetic determinants of complex disorders. Despite these achievements, the large number of SNPs in today's genome-wide studies poses a number of serious challenges. For example, one important issue is how to handle missing genotypes. Simply excluding subjects that have any missing genotypes is impractical in many situations, as most subjects usually have one or more missing genotypes in large genotyping efforts. Consequently, ignoring missing genotypes may lead to huge loss of genetic information.
Genotypes of SNPs are usually determined one at a time based on signal intensities of alleles. And depending on the availability of training data, the assignment of genotypes is often treated as a clustering or classification problem. When the allele signal intensities of a SNP are not well separated, currently available genotype calling algorithms make no calls (so called missing values) on data points that are not assigned to a genotype cluster with a high posterior probability. Many strategies have been exploited to reduce the impact of missing genotypes on association analyses. One of them is to take genotype uncertainties into account when conducting association tests. For example, Plagnol et al. [1] treated posterior probabilities as weights of genotype assignments and used a weighted score statistic, and Kang et al. [2] incorporated genotype uncertainties into their haplotype estimation algorithm. Another strategy is to impute missing genotypes from called genotypes. For example, Souverein et al. [3] modelled a SNP and markers that are in LD with the SNP using the polytomous logistic regression model, and Dai et al. [4] used a classification tree method. A detailed comparison of several of those imputation methods can be found in Yu and Schaid [5]. Imputation of missing genotypes can also be a by-product of haplotype reconstruction, which estimates missing genotypes and unknown haplotype phase simultaneously [6–18]. All those imputation strategies are based on the fact that when SNPs are in LD, the unobserved genotypes can be imputed accurately based on genotypes observed at other SNPs.
Essentially, the above described approaches try to use information from two different perspectives: incorporating genotype uncertainty into association tests focuses on making full use of information from the signal intensities of each SNP, and imputing missing genotypes based on LD focuses on borrowing information from neighbouring SNPs. As each of them uses only partial information from the data, we would expect that genotype determination can be greatly improved in an approach that takes advantage of both "fuzzy" call and imputation of missing values on called genotypes. Here, we propose a new method that uses both signal intensities and LD information, with the two parts connected by jointly estimating the underlying cluster parameters and haplotype frequencies for multiple markers.
To show better the motivation for combining signal intensities and LD information, let's examine the clustering results of the signal data from Illumina BeadArray platform for about 1500 individuals at a SNP. Figure 1 shows the normalized signal intensities of two alternative alleles: A and C. Using the genotype calling algorithm illuminus [19], most data points can be clustered into one of three genotype groups: AA, AC, or CC. However, there are several data points that are located between two clusters. These data points are usually treated as missing values and their genotypes have to be imputed by some statistical methods. Despite the genotype uncertainty, the positions of these data points nevertheless provide some information for genotyping. For example, if the posterior probability for a data point to have genotype AA is 0.8, then it will be treated as a missing value when the threshold to assign genotype calls is set to 0.95; however, in the case that it is in LD with a neighbouring SNP, this partial genotype information, together with the LD between the two SNPs, can help to identify its underlying genotype.
Figure 1
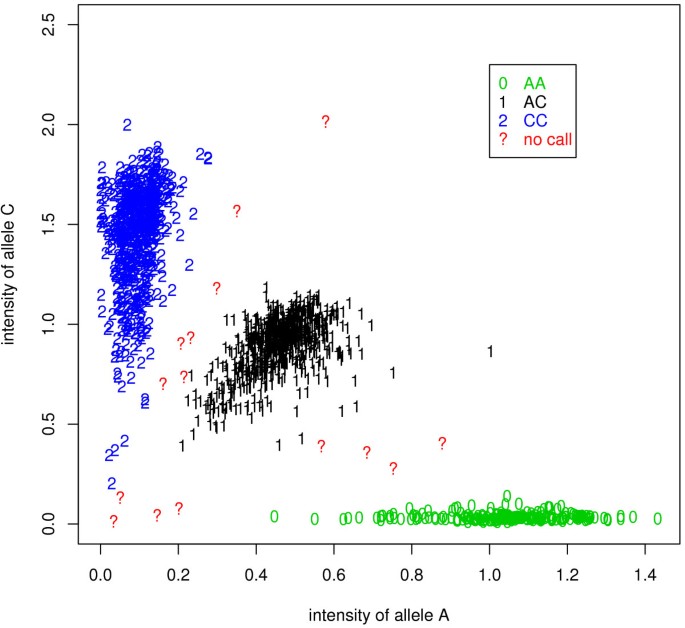
The clustering results based on a one-marker-at-a-time method. Values on the X-axis and Y-axis are normalized signal intensities of two alternative alleles (A and C). Estimated genotypes "AA", "AC", and "CC" are indicated by symbols "0", "1", and "2", respectively. Question marks represent missing values (i.e. no calls).
Methods
Notation
N: number of subjects.
M: number of markers.
H: number of distinguishable haplotypes.
X i = (X i1 , ..., X iM ): vector of signal intensities for subject i, i = 1, ..., N.
G i = (G i1 , ..., G iM ): genotype for subject i, i = 1, ..., N.
h j = (h j1 , ..., h jM ): the j th haplotype, j = 1,...,H. h jm is 1 for the rare allele and 0 for the common allele at SNP m.
Y i = (h i1/h i2): phased genotype (haplotype pairs) for subject i, where h _i1_∈ {h 1 ,...,h H } and h _i2_∈ {h 1 ,...,h H } are the haplotypes. i = 1, ..., N.
h j (Y i ): number of copies of haplotype h j in Y i .
θ S = (μ mt , Σ mt , π mt )m = 1,...,M: cluster parameters, where μ mt , Σ mt , π mt are the mean, covariance matrix (or variance for one-dimensional data), and probability of genotype cluster t at SNP m, respectively.
θ H = (θ h1 ,...,θ hH ): LD parameters, where θ hj is the frequency of haplotype h j .
In the notation, X i is observed, and G i and Y i are unobserved. X i = (X i1 , ..., X iM ) denotes signal intensities that are after proper normalization or transformation. Different normalization or transformation methods may be used for data generated by different platforms. For example, for signal data from Illumina BeadArray, X im is the contrast variable at SNP m. Similar to the algorithm illuminus by Teo _et al_[19], we define contrast as the ratio of the difference to the sum of the normalized signal intensities of two alternative alleles. For data from Affymetrix GeneChip 500 K arrays, X im is a two dimensional variable, with each dimension corresponding to the normalized signal of one allele, as in the Chiamo [20] software.
Our new method
In our method we partition parameters into two sets: the cluster parameters θ S , and the LD parameters θ H . Although they contain redundant information, i.e., all π mt are known when θ H is given, we adopt this parameterization to simplify the presentation of our algorithm. Our algorithm correlates the signal intensities of multiple markers by jointly estimating cluster parameters and LD parameters (i.e., haplotype frequencies). We assume signal intensities of different markers are independent, conditional on given genotypes. In addition, we make three reasonable assumptions:
- (1)
Conditional on phased genotypes and cluster parameters, the distribution of signal intensities does not rely on LD parameters θ H i.e., f(X i |Y i , θ S , θ H ) = f(X i |Y i , θ S ); - (2)
Conditional on LD parameters θ H the distribution of Y i does not depend on cluster parameters θ S , i.e., f(Y i |θ S , θ H ) = f(Y i |θ H ); - (3)
SNPs are in Hardy-Weinberg equilibrium (HWE).
The complete data likelihood is then:
f(X i , Y i | θ S , θ H ) = f(X i | Y i , θ S , θ H )f(Y i | θ S , θ H ) = f(X i | Y i , θ S )f(Y i | θ H )
We use the EM algorithm to estimate parameters in our method [21]. With some algebraic manipulation, we can show that the E step is equivalent to calculating the genotype posterior probability for the i th subject to have genotype t at the m th locus, given estimated parameters and observed signal intensities. Based on the relation between phased and unphased genotypes, the genotype posterior probability can be expressed as
w i m t = Pr ( G i m = t | X i , θ S , θ H ) = ∑ Y i : G i m = t f ( Y i | X i , θ S , θ H ) MathType@MTEF@5@5@+=feaagaart1ev2aaatCvAUfKttLearuWrP9MDH5MBPbIqV92AaeXatLxBI9gBaebbnrfifHhDYfgasaacPC6xNi=xI8qiVKYPFjYdHaVhbbf9v8qqaqFr0xc9vqFj0dXdbba91qpepeI8k8fiI+fsY=rqGqVepae9pg0db9vqaiVgFr0xfr=xfr=xc9adbaqaaeGaciGaaiaabeqaaeqabiWaaaGcbaGaem4DaC3aaSbaaSqaaiabdMgaPjabd2gaTjabdsha0bqabaGccqGH9aqpcyGGqbaucqGGYbGCcqGGOaakcqWGhbWrdaWgaaWcbaGaemyAaKMaemyBa0gabeaakiabg2da9iabdsha0jabcYha8jabdIfaynaaBaaaleaacqWGPbqAaeqaaOGaeiilaWIaeqiUde3aaSbaaSqaaiabdofatbqabaGccqGGSaalcqaH4oqCdaWgaaWcbaGaemisaGeabeaakiabcMcaPiabg2da9maaqafabaGaemOzayMaeiikaGIaemywaK1aaSbaaSqaaiabdMgaPbqabaGccqGG8baFcqWGybawdaWgaaWcbaGaemyAaKgabeaakiabcYcaSiabeI7aXnaaBaaaleaacqWGtbWuaeqaaOGaeiilaWIaeqiUde3aaSbaaSqaaiabdIeaibqabaGccqGGPaqkaSqaaiabdMfaznaaBaaameaacqWGPbqAaeqaaSGaeiOoaOJaem4raC0aaSbaaWqaaiabdMgaPjabd2gaTbqabaWccqGH9aqpcqWG0baDaeqaniabggHiLdaaaa@696D@
This is essentially a sum of probabilities of phased genotypes, conditional on estimated parameters and observed signal intensities. The probability of a phased genotype conditional on estimated parameters and observed signal intensities is
f ( Y i | X i , θ S , θ H ) = f ( X i | Y i , θ S ) f ( Y i | θ H ) ∑ Y i f ( X i | Y i , θ S ) f ( Y i | θ H ) MathType@MTEF@5@5@+=feaagaart1ev2aaatCvAUfKttLearuWrP9MDH5MBPbIqV92AaeXatLxBI9gBaebbnrfifHhDYfgasaacPC6xNi=xI8qiVKYPFjYdHaVhbbf9v8qqaqFr0xc9vqFj0dXdbba91qpepeI8k8fiI+fsY=rqGqVepae9pg0db9vqaiVgFr0xfr=xfr=xc9adbaqaaeGaciGaaiaabeqaaeqabiWaaaGcbaGaemOzayMaeiikaGIaemywaK1aaSbaaSqaaiabdMgaPbqabaGccqGG8baFcqWGybawdaWgaaWcbaGaemyAaKgabeaakiabcYcaSiabeI7aXnaaBaaaleaacqWGtbWuaeqaaOGaeiilaWIaeqiUde3aaSbaaSqaaiabdIeaibqabaGccqGGPaqkcqGH9aqpjuaGdaWcaaqaaiabdAgaMjabcIcaOiabdIfaynaaBaaabaGaemyAaKgabeaacqGG8baFcqWGzbqwdaWgaaqaaiabdMgaPbqabaGaeiilaWIaeqiUde3aaSbaaeaacqWGtbWuaeqaaiabcMcaPiabdAgaMjabcIcaOiabdMfaznaaBaaabaGaemyAaKgabeaacqGG8baFcqaH4oqCdaWgaaqaaiabdIeaibqabaGaeiykaKcabaWaaabuaeaacqWGMbGzcqGGOaakcqWGybawdaWgaaqaaiabdMgaPbqabaGaeiiFaWNaemywaK1aaSbaaeaacqWGPbqAaeqaaiabcYcaSiabeI7aXnaaBaaabaGaem4uamfabeaacqGGPaqkcqWGMbGzcqGGOaakcqWGzbqwdaWgaaqaaiabdMgaPbqabaGaeiiFaWNaeqiUde3aaSbaaeaacqWGibasaeqaaiabcMcaPaqaaiabdMfaznaaBaaabaGaemyAaKgabeaaaeqacqGHris5aaaaaaa@7494@
where
f ( X i | Y i , θ S ) = ∏ m = 1 M f ( X i m | Y i , θ S ) f ( Y i | θ H ) = θ h i 1 θ h i 2 ( 1 + I ( h i 1 = h i 2 ) ) MathType@MTEF@5@5@+=feaagaart1ev2aaatCvAUfKttLearuWrP9MDH5MBPbIqV92AaeXatLxBI9gBaebbnrfifHhDYfgasaacPC6xNi=xI8qiVKYPFjYdHaVhbbf9v8qqaqFr0xc9vqFj0dXdbba91qpepeI8k8fiI+fsY=rqGqVepae9pg0db9vqaiVgFr0xfr=xfr=xc9adbaqaaeGaciGaaiaabeqaaeqabiWaaaGceaqabeaacqWGMbGzcqGGOaakcqWGybawdaWgaaWcbaGaemyAaKgabeaakiabcYha8jabdMfaznaaBaaaleaacqWGPbqAaeqaaOGaeiilaWIaeqiUde3aaSbaaSqaaiabdofatbqabaGccqGGPaqkcqGH9aqpdaqeWbqaaiabdAgaMjabcIcaOiabdIfaynaaBaaaleaacqWGPbqAcqWGTbqBaeqaaOGaeiiFaWNaemywaK1aaSbaaSqaaiabdMgaPbqabaGccqGGSaalcqaH4oqCdaWgaaWcbaGaem4uamfabeaakiabcMcaPaWcbaGaemyBa0Maeyypa0JaeGymaedabaGaemyta0eaniabg+GivdaakeaacqWGMbGzcqGGOaakcqWGzbqwdaWgaaWcbaGaemyAaKgabeaakiabcYha8jabeI7aXnaaBaaaleaacqWGibasaeqaaOGaeiykaKIaeyypa0JaeqiUde3aaSbaaSqaaiabdIgaOnaaCaaameqabaGaemyAaKMaeGymaedaaaWcbeaakiabeI7aXnaaBaaaleaacqWGObaAdaahaaadbeqaaiabdMgaPjabikdaYaaaaSqabaGccqGGOaakcqaIXaqmcqGHRaWkcqWGjbqscqGGOaakcqWGObaAdaahaaWcbeqaaiabdMgaPjabigdaXaaakiabg2da9iabdIgaOnaaCaaaleqabaGaemyAaKMaeGOmaidaaOGaeiykaKIaeiykaKcaaaa@777B@
For given Y i and θ S , we assume that the signal intensities at different markers are independent, and they follow a distribution suitable to the normalized signal data, such as Gaussian distribution or _t_-distribution.
The M step in the EM algorithm calculates the set of θ S and θ H that maximizes the expected log-likelihood. The estimate for the frequency of haplotype h j is
θ h j = 1 2 n ∑ i = 1 n ∑ Y i f ( Y i | X i , θ S , θ H ) h j ( Y i ) MathType@MTEF@5@5@+=feaagaart1ev2aaatCvAUfKttLearuWrP9MDH5MBPbIqV92AaeXatLxBI9gBaebbnrfifHhDYfgasaacPC6xNi=xI8qiVKYPFjYdHaVhbbf9v8qqaqFr0xc9vqFj0dXdbba91qpepeI8k8fiI+fsY=rqGqVepae9pg0db9vqaiVgFr0xfr=xfr=xc9adbaqaaeGaciGaaiaabeqaaeqabiWaaaGcbaGaeqiUde3aaSbaaSqaaiabdIgaOnaaBaaabaGaemOAaOgabeaaaeqaaOGaeyypa0tcfa4aaSaaaeaacqaIXaqmaeaacqaIYaGmcqWGUbGBaaGcdaaeWbqaamaaqafabaGaemOzayMaeiikaGIaemywaK1aaSbaaSqaaiabdMgaPbqabaGccqGG8baFcqWGybawdaWgaaWcbaGaemyAaKgabeaakiabcYcaSiabeI7aXnaaBaaaleaacqWGtbWuaeqaaOGaeiilaWIaeqiUde3aaSbaaSqaaiabdIeaibqabaGccqGGPaqkaSqaaiabdMfaznaaBaaabaGaemyAaKgabeaaaeqaniabggHiLdGccqWGObaAdaWgaaWcbaGaemOAaOgabeaakiabcIcaOiabdMfaznaaBaaaleaacqWGPbqAaeqaaOGaeiykaKcaleaacqWGPbqAcqGH9aqpcqaIXaqmaeaacqWGUbGBa0GaeyyeIuoaaaa@5B1E@
Since θ S and θ H contain redundant information, there is no need to update the π mt 's in θ S once θ H is updated; as a result, we only need to estimate the mean and covariance matrix of each genotype cluster. In the M step of the conventional EM algorithm, the means and covariance matrices are updated by the set of values that maximizes the expected log-likelihood. For Illumina BeadArray data, our estimators for cluster parameters are based on data points with large genotype posterior probabilities (for example, greater than 0.95), since our study shows they are more robust than the estimators in the M step of the conventional EM algorithm. This strategy is also used in [19]. For data generated by the Affymetrix GeneChip 500 K arrays, we first obtained the regular estimates for the mean and covariance matrix of the t th cluster at SNP m, then estimated the cluster mean μ mt by using the (N × π mt /2) data points that have the smallest Mahalanobis distances, and finally updated the covariance matrix Σ mt by a weighted estimator based on estimated μ mt and all data points.
In our method, once the EM algorithm converges, we make genotype calls based on the genotype posterior probabilities and a predetermined threshold δ. Specifically, for a subject at a given SNP, if the posterior probability for the subject to have genotype t is greater than δ, we assign t to the corresponding data point; if the posterior probabilities for all of the three genotypes are less than δ, then the data point is labelled as missing and no call is assigned.
We emphasize that our method is different from treating signal intensities of different markers as a multi-dimensional variable. To show the difference, here we consider the case of two SNPs. The number of possible genotypes is nine for two SNPs. If we treat the contrast variable of the two SNPs as a bivariate _t_-distribution with known degrees of freedom, then we need to estimate 53 parameters, including nine two-dimensional means, nine two-by-two covariance matrices (three parameters in each covariance matrix), and eight cluster probabilities. By contrast, we only need to estimate 15 parameters in our method: six means, attributed to three genotype clusters for each SNP; six variances, attributed to three genotype clusters for each SNP; and three haplotype frequencies, attributed to four possible haplotypes. In addition, the cluster parameters in our method are estimated marker by marker; as a result, the difficulty brought by a SNP with a rare allele or low signal quality has little effect on the cluster parameters of other SNPs.
To assess the performance of our method (denoted as M3), we compared it with two other methods: the one-SNP-at-a-time method M1 and the LD based method M2. The method M1 models the cluster parameters θ S , and the method M2 models the LD parameters θ H . M1 and M2 use only part of the information from data: either the signal intensities of individual SNPs, or LD structure of multiple SNPs. By contrast, our method M3 uses both signal intensities of individual SNPs and LD structure between SNPs. In the method M1, we used the algorithm illuminus [19] for data from Illumina BeadArray and the algorithm Chiamo [20] for data from Affymetrix GeneChip 500 K arrays. Those two algorithms are used because they outperform other existing algorithms for their respective platforms [19, 20]. The illuminus algorithm assumes that the contrast and strength (the natural logarithm of the sum of normalized signal intensities) follow a _t_-mixture model and it uses the EM algorithm to estimate cluster parameters; the Chiamo algorithm uses a Bayesian hierarchical mixture model to call genotypes from normalized signal intensities. In the method M2, we first computed the posterior genotype probabilities for the missing genotypes based on the LD structure of genotypes assigned by illuminus or Chiamo, then assigned genotype calls to the missing ones based on whether the calculated posterior probabilities were greater than a threshold. The posterior genotype probabilities are calculated by using the EM algorithm [6] for our simulated data with two SNPs, and they are calculated by using the algorithm fastPHASE [11] for the two real data sets considered in the paper, due to the large number of SNPs in the real data sets.
Simulated data
To evaluate the performance of different methods, we simulated data by mimicking signals generated by Illumina BeadArray. In our simulation, the cluster parameters were based on SNPs in a real data set; the allele frequencies and LD levels were pre-determined to reflect different data structures. We considered two SNPs with the same allele frequencies. The minor allele frequency was chosen to be 0.05, 0.1, or 0.3, and the LD was chosen at a variety of levels, with the square of the Pearson's correlation (_r_2) being about 0, 0.3, 0.5, or 0.8. HWE was assumed for both SNPs. The threshold for assigning calls or no calls, δ, was chosen to be 0.85, 0.90, 0.95, or 0.975. The signal intensities (contrast and strength) for a given marker were assumed to have a three-component _t_-mixture distribution (bivariate), with six degrees of freedom. Contrast and strength were assumed to be independent from each other. To study the effect of signal quality, we considered three sets of cluster parameters:
- (1)
high quality, with means for contrast being -0.9, 0.2, 0.9, means for strength being -1.2, -1, -0.8, standard errors for contrast being 0.09, 0.09, 0.06, and standard errors for strength being 0.37, 0.22, 0.27; - (2)
low quality, with means for contrast being -0.5, 0.3, 0.9, means for strength being -1.4, -1.2, -0.8, standard errors for contrast being 0.22, 0.15, 0.06, and standard errors for strength being 0.56, 0.64, 0.48; - (3)
mixed quality, a situation with one SNP in high quality and the other one in low quality.
These parameters were taken from two SNPs in real data, representing SNPs with high or low genotype call rates. The sample size was 1500.
To quantify the performance of the methods, two quantitative metrics are computed: call rate and genotyping error rate. The call rate was the number of non-missing calls divided by the total number of calls, and the genotyping error rate was the proportion of incorrectly called genotypes among all assigned calls. For a set of chosen parameters, we used 1000 replicates to estimate the two metrics. As both the LD based method M2 and our method M3 estimate haplotype frequencies, it is interesting to investigate whether one method gives more accurate estimation for haplotype frequencies. And for two SNPs, the accuracy of haplotype frequencies of the two methods can be reflected by the square of the Pearson's correlation (_r_2) based on estimated haplotype frequencies.
Real data
We considered two sets of real data from two different genotyping platforms.
The first data set consists of 1599 unrelated case-control subjects (one third cases and two thirds controls) from a breast-cancer candidate-gene study conducted in the University of California at Irvine. For each subject, 1455 SNPs in 148 candidate genes were genotyped by Illumina BeadArray. Normalized signal data were obtained from BeadStudio. We used the algorithm illuminus to make genotype calls in the one-SNP-at-a-time method M1. In the LD based method M2, due to the large number of SNPs, we used the algorithm fastPHASE to compute the posterior probabilities for the missing genotypes (no calls) assigned by the algorithm illuminus. Jointly analyzing the signal data of all the 1455 SNPs is computationally prohibitive. To solve that problem, for each SNP, we searched all other SNPs to find the one with the maximum _r_2 with the given SNP, then analyzed the two SNPs jointly if their _r_2 is greater than 0.3. A _t_-mixture model with fixed degrees of freedom was then used to cluster the contrast of two alternative alleles for each SNP. In all the three methods (M1, M2, and M3), we used 0.95 as the threshold to assign genotype calls.
The second data set consists of signal data from Affymetrix GeneChip 500 K arrays for 1504 unrelated controls from the 1958 British Birth Cohort [20], obtained from the Wellcome Trust Case Control Consortium (WTCCC). We focused our study on the 6277 SNPs in chromosome 22. We chose the algorithm Chiamo as the one-SNP-at-a-time method M1, since it outperforms other genotype calling methods [20]. This method uses a Bayesian hierarchical mixture model to cluster quantile normalized data, and its default threshold of posterior probabilities for making a call is 0.9. For the no-calls assigned by Chiamo, we imputed them using the algorithm fastPHASE. The joint analysis of signal and LD was similar to that applied to the first real data set, except that here we used a Gaussian mixture model for the two-dimensional signal data. To compare the performance of different methods, we downloaded the Illumina Infinium 15 K data from the WTCCC. Among the 1504 subjects with Affymetirx 500 K data, 1457 of them were also genotyped by the Illumina Infinium 15 K. And the number of common SNPs on chromosome 22 in the two platforms was 31. For direct comparison, in each method, we computed the discordance rates between the most probable calls at the 31 common SNPs. The reason why we use the most probable call for each data point is that the thresholds for different methods and platforms are not comparable.
Results
Simulated data
The results for the simulated data are reported in Tables 1 to 4. They are mean values from 1000 replications. Tables 1, 2, and 3 show the call rates and genotyping error rates of the three methods when (1) both of the two SNPs were in high quality, (2) both of the two SNPs were in low quality, and (3) SNP1 was in high quality while SNP2 was in low quality, respectively. Table 4 reports the estimated _r_2 and mean square errors. Since the patterns of the results for different allele frequencies p were similar, here we only show the results for p = 0.1. Our study indicates that different thresholds δ led to very similar conclusions, thus only those of δ = 0.95 are described. And the results for _r_2 = 0 are not shown, because when _r_2 was close to 0, the performance of all the three methods were very similar, as expected.
Table 1 Call rate and genotyping error for two SNPs in high quality
Table 2 Call rate and genotyping error for two SNPs in low quality
Table 3 Call rate and genotyping error for two SNPs in mixed quality
Table 4 Comparison of M2 and our method M3 on the estimation of _r_2
The call rates of the two LD based methods (M2, and M3) depend significantly on the LD level between the two SNPs. As the method M1 (illuminus) calls one SNP at a time, the LD level does not affect its call rates, which can be seen from the almost identical call rates at all LD levels. When the two SNPs were independent from each other, as expected, the call rates of the three methods were very similar (data not shown). When the two SNPs were in LD, our method M3 consistently improved the call rates of M1, and with the increase of LD, its advantage became more considerable. When LD was not very strong, such as when _r_2 was below 0.5, the increment brought by M2 over M1 was very small; in contrast, our method still improved the call rates in the same situation.
Now we examine the effect of signal quality on the call rates. When the signal quality of both SNPs was high, all the three methods gave very high call rates and our method gave the highest, as illustrated in Table 1. When the signal quality of both SNPs was low, although the call rates of all methods were lower, as shown in Table 2, the benefit of using our method was more noticeable. To study if our method would improve the genotype calling of one SNP but deteriorate that of the other, we tested it on the simulated data of mixed quality, and the results are shown in Table 3. Here, because the two SNPs were assumed to have the same rare allele frequency, the signal intensities of the SNP in low quality were more difficult to cluster than those of the SNP in high quality. As a result, the call rates of M1 for SNP2 were much lower than those of SNP1 for all three methods. However, by jointly modelling signal intensities and LD, our method significantly increased the call rates of SNP2. In addition, the call rates of SNP1 were also increased. This might seem implausible at the first sight, but it can be explained by the fact that, although the signal intensities of SNP2 were noisy, they were nevertheless useful to help cluster the signal intensities of the associated marker SNP1. Consequently, jointly calling the two SNPs increased the call rates for both of them.
Now let's examine the other important metric – the genotyping error rate. For a given method with different thresholds, genotyping accuracy and call rates are often trade-off of each other. It is therefore important for an algorithm not to gain higher value of one at the cost of the other. The results in Tables 1, 2, and 3 illustrate that our method not only achieved greater call rates than M1, but also achieved higher genotyping accuracy, which is most obvious when LD was moderate or strong. By contrast, although the method M2 increased the call rates when LD was strong, this increase was gained at the sacrifice of genotyping accuracy. For example, when both SNPs were in low quality and _r_2 was about 0.8, both M2 and our method have higher call rates than M1; however, the genotyping error rate of M2 was 0.64%, which was 2 times of the error rate of our method (0.32%) and 1.2 times of that of M1 (0.53%).
When the signal intensities of a SNP are in low quality, the estimated genotypes are prone to genotyping errors. This error was amplified by the method M2 (Tables 2 and 3), because M2 is directly based on called genotypes. By contrast, our method achieved higher genotyping accuracy. This is because, for a SNP whose signal intensities are not well separated, its genotype assignments can be improved by the LD between itself and other SNPs. Strikingly, in the situation of mixed quality, when the two SNPs were in high LD, our method improved the genotyping accuracy for both SNPs over the method M1. Again, this can be attributed to the fact that although the signal of SNP2 was noisy, it still provided some clustering information for SNP1.
Table 4 shows the estimated _r_2 and mean square errors. With all LD levels and signal qualities, our method outperformed the method M2 by providing estimated correlations that were much closer to their true values, with smaller mean square errors. This is because our method gave better call rates and genotyping accuracy, and as a result, it provided more accurate estimation of _r_2 and haplotype frequencies.
Real data
For the data set of the breast-cancer candidate-gene study, we computed the call rates of the 1455 SNPs using all the three methods. The median call rates of M1 (illuminus), M2 (fastPHASE), and our method M3 were 98.75%, 98.87%, and 99.44%, respectively. Compared to the one-marker-at-a-time method illuminus, our method increased the call rates of 294 SNPs for at least 1%, and 13 SNPs for at least 5%. By contrast, fastPHASE increased the call rates of only 57 SNPs for at least 1%, and only 4 SNPs for at least 5%. Although there is no additional data to compare the genotype accuracy, the results here show convincingly that our method significantly improves the call rates of the other two methods.
In the WTCCC data, 6277 SNPs on chromosome 22 were genotyped for 1504 unrelated controls using the Affymetrix 500 K platform. Here we computed the discordance rates between the calls from the three methods and the calls from the Infinium 15 K. The discordance rates were 0.85% for Chiamo, 0.41% for fastPHASE, and 0.27% for our method. Those discordance rates were calculated based on the most probable calls. Among the data points with Chiamo scores below 0.9, i.e., the data points with "no calls" by Chiamo with its default threshold 0.9, the discordance rates of fastPHASE and our method were 27.74% and 14.23%, respectively. Note that the discordance rates for data points with low Chiamo scores were much higher than the overall discordance rates. We examined the pattern of Chiamo scores, and found that most of the data points with low scores were in a few SNPs that have relatively low call rates, which is likely an explanation for the high discordance rates. All those discordance rates indicate that our method increases the genotyping accuracy.
Discussion and Conclusion
In existing analyses, genotype calling and imputation of missing genotypes are usually considered as two separate problems. In this paper, using simulated data and real data, we have demonstrated that it is advantageous to treat the two problems simultaneously. Currently, approaches based on LD between SNPs have been widely used to impute missing genotypes. Those approaches, however, might decrease genotyping accuracy, when comparing to the traditional one-marker-at-a-time genotype calling method. By contrast, our method outperforms the traditional one-marker-at-a-time method in the respects of both call rate and genotyping accuracy. Besides genotype calls, our method also yields a weight matrix consisting of genotype probabilities. This weight matrix can be used in association analyses, in a similar way described by Plagnol et al [1].
Although in our method SNPs are assumed to be in Hardy-Weinberg equilibrium, we found our method still improves the results of call rate and genotyping accuracy for data with moderate departure from HWE (data not shown). Another assumption we made is that subjects are from a random sample of a population, which means subjects are unrelated and there is no population stratification. In the future, we plan to modify our method so that related subjects with known pedigree structure and population substructure can also be taken into account.
Our method does not put any constraint on the number of SNPs that can be called jointly. However, because the number of haplotypes increases exponentially with the number of jointly-analyzed SNPs, it is computationally demanding for the traditional EM algorithm to process a large number of SNPs. To solve this problem, what we have done in the application of our method to the real data is that, for each SNP, we made genotype calls by using another SNP that had the highest Pearson's correlation with the original SNP. Although this significantly mitigates the computation requirement, it might not be the best strategy. An alternative way is to use a sliding window method to fix the number of SNPs that are called together; however, here we decide not to do that, because LD between SNPs is usually a localized phenomenon and the number of SNPs that are analyzed together should be determined by local LD structure. Other alternatives include several recently proposed methods that can provide rapid and accurate estimation of haplotypes [11, 13–15].
What we considered here are signal intensities obtained from two different platforms: the Illumina BeadArray and the Affymetrix GeneChip 500 K arrays. There is no reason, however, to limit the method to those platforms. In fact, the idea of jointly analyzing signal intensities of multiple markers is quite general. It can be applied to other platforms, such as multiple inversion probe technology [22], and other DNA variations, such as copy number variants. The advantage of combining signal intensities and LD is confirmed by a few recent publications [23, 24], where the signal data and dependence between neighbouring markers are integrated to infer copy numbers and loss of heterozygosity.
References
- Plagnol V, Cooper J, Todd J, Clayton D: A method to address differential bias in genotyping in large-scale association studies. PLoS Genetics. 2007, 3: e74-
Article PubMed Central PubMed Google Scholar - Kang H, Qin ZS, Niu T, Liu JS: Incorporating genotyping uncertainty in haplotype inference for single-nucleotide polymorphisms. Am J Hum Genet. 2004, 74: 495-510.
Article PubMed Central CAS PubMed Google Scholar - Souverein O, Zwinderman A, Tanck M: Multiple imputation of missing genotype data for unrelated individuals. Ann Hum Genet. 2006, 70: 372-381.
Article CAS PubMed Google Scholar - Dai J, Ruczinski I, LeBlanc M, Kooperberg C: Imputation methods to improve inference in SNP association studies. Genet Epidemiol. 2006, 30: 690-702.
Article PubMed Google Scholar - Yu Z, Schaid DJ: Methods to impute missing genotypes for population data. Hum Genet. 2007, 122: 495-504.
Article PubMed Google Scholar - Excoffier L, Slatkin M: Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995, 12: 921-927.
CAS PubMed Google Scholar - Hawley M, Kidd K: HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered. 1995, 86: 409-411.
CAS PubMed Google Scholar - Long JC, Williams RC, Urbanek M: An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet. 1995, 56: 799-810.
PubMed Central CAS PubMed Google Scholar - Chiano MN, Clayton DG: Fine genetic mapping using haplotype analysis and the missing data problem. Ann Hum Genet. 1998, 62: 55-60.
Article CAS PubMed Google Scholar - Qin Z, Niu T, Liu J: Partition-Ligation EM Algorithm for Haplotype Inference with Single Nucleotide Polymorphisms. Am J Hum Genet. 2002, 71: 1242-1247.
Article PubMed Central CAS PubMed Google Scholar - Scheet P, Stephens M: A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006, 78 (4): 629-644.
Article PubMed Central CAS PubMed Google Scholar - Eronen L, Geerts F, Toivonen H: HaploRec: efficient and accurate large-scale reconstruction of haplotypes. BMC Bioinformatics. 2006, 7: 542-
Article PubMed Central PubMed Google Scholar - Browning SR, Browning BL: Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007, 81: 1084-1097.
Article PubMed Central CAS PubMed Google Scholar - Rastas P, Koivisto M, Mannila H, Ukkonen E: A hidden Markov technique for haplotype reconstruction. Lect Notes Comput Sci. 2005, 3692: 140-151.
Article Google Scholar - Kimmel G, Shamir R: A block-free hidden Markov model for genotypes and its application to disease association. J Comput Biol. 2005, 12: 1243-1260.
Article CAS PubMed Google Scholar - Niu T, Qin ZS, Xu X, Liu JS: Bayesian Haplotype Inference for Multiple Linked Single-Nucleotide Polymorphisms. Am J Hum Genet. 2002, 70: 157-169.
Article PubMed Central CAS PubMed Google Scholar - Stephens M, Donnelly P: A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73: 1162-1169.
Article PubMed Central CAS PubMed Google Scholar - Zhang Y, Niu T, Liu JS: A coalescent-guided hierarchical Bayesian method for haplotype inference. Am J Hum Genet. 2006, 79: 313-322.
Article PubMed Central CAS PubMed Google Scholar - Teo YY, Inouye M, Small KS, Gwilliam R, Deloukas P, Kwiatkowski DP, Clark TG: A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics. 2007, 23 (20): 2741-2746.
Article PubMed Central CAS PubMed Google Scholar - Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007, 447: 661-678.
Article Google Scholar - Dempster A, Laird N, Rubin D: Maximum likelihood from incomplete data via EM algorithm. J Roy Statist Soc Ser B. 1977, 39: 1-38.
Google Scholar - Moorhead M, Hardenbol P, Siddiqui F, Falkowski M, Bruckner C, Ireland J, Jones HB, Jain M, Willis TD, Faham M: Optimal genotype determination in highly multiplexed SNP data. Eur J Hum Genet. 2006, 14: 207-215.
Article CAS PubMed Google Scholar - Korn J, Kuruvilla F, McCarroll S, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins P, Darvishi K: Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008, 40: 1253-1260.
Article PubMed Central CAS PubMed Google Scholar - Scharpf R, Parmigiani G, Pevsner J, Ruczinski I: Hidden Markov Models for the assessment of chromosomal alterations using high-throughput SNP arrays. Ann Appl Stat. 2008, 2: 687-713.
Article PubMed Central PubMed Google Scholar