The Distributed Annotation System (original) (raw)
The basic system is composed of a genome server, one or more annotation servers, and an annotation viewer. The genome server is responsible for serving genome maps, sequences, and information related to the sequencing process. Annotation servers are responsible for responding to requests on a region and delivering annotations. The client, an annotation viewer, is a lightweight application whose behavior is analogous to a web browser. The viewer communicates with the genome and annotation servers using a well defined language specification.
At a fundamental level, all annotations can be reduced to their coordinates relative to a particular sequence landmark. The DAS viewer retrieves annotations from the various annotation servers and uses the sequence coordinates to generate an integrated index of what is on the genome. This integration is then presented to the user in tabular or graphical form. Annotation providers can provide a suggestion of how their annotations should be rendered in a graphical display, and can provide links back to their databases and web sites to allow the researcher to retrieve further information about the annotation.
Because it relies entirely on sequence coordinates to achieve integration, DAS does not attempt to resolve semantic contradictions between different data sources. The goal of the system is to provide indexing and visualization, thereby making contradictions between annotations visible.
Reference sequence
The distributed annotation system relies on there being a common "reference sequence" on which to base annotations. The reference server consists of a set of "entry points" into the sequence, and the lengths of each entry point. Entry points will vary from genome to genome. For some genome projects, entry points correspond to entire chromosomes. For others, entry points may be a series of contigs.
The entry points describe the top level items on the reference sequence map. It is possible for each entry point to have substructure, basically a series of subsequences (components) and their start and end points. This structure is recursive. Annotations take the form of a statement about a region of the reference sequence. Each annotation is unambiguously located by providing its position as the start and stop positions relative to a "reference sequence."
To give a concrete example, the C. elegans reference map consists of six top level entry points, one per chromosome. Each chromosome is formed from several contigs called "superlinks," and each superlink contains one or more smaller contigs called "links." Links in turn are composed of one or more fully-sequenced clones [11]. One could refer to an annotation by specifying its start or stop positions in clone, link, superlink, or chromosome coordinates.
The reference sequence server is responsible for providing the reference sequence map and the underlying DNA. The server can provide a list of sequence entry points or given a component of the map it can return its parent and children components. The reference server can provide arbitrarily long stretches of raw DNA sequence given a reference subsequence, start position, and stop position. Needless to say, bandwidth becomes a limiting factor for retrieving multi-megabase segments of DNA. However, in practice it is rare for users to retrieve more than a gene's worth of raw DNA at a time.
Annotation servers
Annotation servers are specialized for returning lists of annotations across defined regions of the genome. Each annotation is anchored to the genome map by way of a start and stop position relative to one of the entry points. Annotations have an identifier that is unique to the providing server and a structured description of its nature and attributes. The general description of an annotation follows loosely the general feature format (GFF) which intentionally aims for a basic lowest common denominator description http://www.sanger.ac.uk/Software/formats/GFF/. Annotations may also be associated with URLs where additional human or machine readable information about the annotation can be found.
The annotator is free to describe his annotations using any terms which he feels are appropriate, as DAS does not impose a controlled vocabulary. Annotations have categories, types, and methods defined by the annotator. The annotation type corresponds to a biologically significance description. In the Eddy Lab RNA track of the HGP three types are defined, "tRNA", "snoRNA", and "miscRNA". The annotation method is intended to describe how the annotated feature was discovered, and may include a reference to a software program. The annotation category is a broad functional category. "Homology", "variation" and "transcribed" are example categories. This structure allows researchers to add new annotation types if the existing list is inadequate without entirely losing all semantic value. It is intended that larger annotation servers provide URLs to human-readable information that describes its types, methods and categories in more detail.
Another optional feature of annotation servers is the ability to provide hints to clients on how the annotations should be rendered visually. This is done by returning a DAS "stylesheet." Stylesheets use the type and category information to associate each annotation with a particular graphical representation, a glyph.
Although the servers are conceptually divided between reference servers and annotation servers, there is in fact no key difference between them. A single server can provide both reference sequence information and annotation information. The main functional difference is that the reference sequence server is required to serve the coordinate map and the raw DNA, while annotation servers have no such requirement.
Specification
The main component of DAS is the XML specification, which defines all valid DAS communication. As with HTML, our goal is a language which is human readable, easily parsed, and extensible. The additional file [appendix.pdf] provides a summary of version 1.01 of the DAS specification.
While a client can query multiple servers simultaneously, the communication between the client and any single server follows a simple client server model. Clients query the reference and annotation servers by sending a formatted URL request to each server. Each URL has a site-specific prefix, followed by a standardized path and query string. The standardized path begins with the string /das. This is followed by URL components containing the data source name and a command. For example:
http://stein.cshl.org/das/elegans/features?segment=ZK154:1000,2000
In this case, the site-specific prefix is http://stein.cshl.org/. The request begins with the standardized path /das, and the data source, in this case /elegans. This is followed by the command /features, which requests a list of features relative to a given set of named arguments (?segment=ZK154:1000,2000). The data source component allows a single server to provide information on several genomes.
Servers process the request and return a response as defined by the DAS specification, typically a formatted XML document. The response from the server to the client consists of a standard HTTP header with DAS status information within that header followed optionally by an XML file that contains the answer to the query. The DAS status portion of the header consists of two lines. The first is X-DAS-Version and gives the current protocol version number, currently DAS/1.0. The second line is X-DAS-Status and contains a three digit status code which indicates the outcome of the request. The defined status codes are listed in Table 1.
Table 1 Server Status Codes Server status codes are modeled after the familiar status codes of the HTTP 1.0 protocol.
An example HTTP header: (provided by server)
HTTP/1.1 200 OK
Date: Sun, 12 Mar 2000 16:13:51 GMT
Server: Apache/1.3.6 (Unix) mod_perl/1.19
Last-Modified: Fri, 18 Feb 2000 20:57:52 GMT
Connection: close
Content-Type: text/plain
X-DAS-Version: DAS/1.0
X-DAS-Status: 200
DATA FOLLOWS ...
The specification outlines seven basic queries which a client can use to interrogate a DAS server. The valid queries are briefly summarized in Table 2. Two queries, "dsn" and "entry points", essentially provide information to the client about the structure of the server and the reference sequence. The "dna" query can be used to fetch a segment of DNA from a reference server. A client can request annotations, "features", or a summary of the annotations available, "types", from any DAS server. The main annotation content query, "features", basically follows the general feature format (GFF). The servers provide a "stylesheet" to suggest representations to the client's graphical display. When more information is desired about a particular annotation, the client makes a "link" request. The "link" request, the only query which does not return a structured XML document, returns HTML. It is anticipated that DAS clients will hand off the link requests to the local web browser or other web-accessible genome database.
Table 2 Queries Summary The basic seven queries of the DAS 1.01 specification.
Prototypes
A series of prototypes for both the client and server components were developed to test various versions of the DAS specification.
Servers
A server is expected to respond to the DAS specification's defined queries with the appropriate content, usually XML. The details of server implementation are left to the various annotation source providers. We provide a sample Perl script for converting ACeDB-based databases into DAS servers, and the Dazzle Java library does the same thing for annotation databases based on the Ensembl code base (T. Down, personal communication, 2001).
The first reference DAS server was written for WormBase [11] and piggybacks on the WormBase software architecture: an Apache/mod_perl web server communicating with an ACeDB database via the AcePerl database access library. The Perl DAS server accepts incoming DAS requests, translates them into the ACeDB query language, reformats the results as XML, and returns them. The WormBase DAS server is currently serving as the C. elegans reference server at http://www.wormbase.org/db/das/. A set of servers containing test data, one reference and four annotation, are available at http://skynet.wustl.edu/cgi-bin/das/.
Viewers
We have developed two prototype DAS client programs. One, called Geodesic, is a stand alone Java application. It connects to one or more DAS servers, retrieves annotations, and displays them in an integrated map, as seen in Figure 2. The other, called DasView, is a Perl application that runs as a server-side script. It connects to one or more DAS servers, constructs an integrated image, and serves the image to a web browser as a set of click-able image map, as seen in Figure 3.
Figure 2
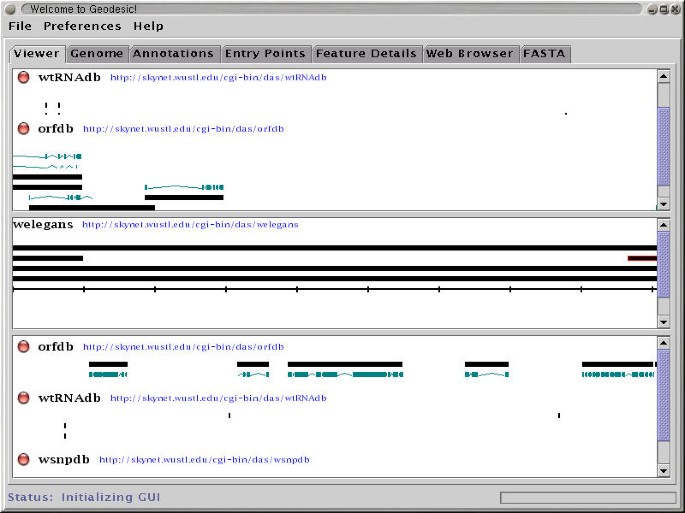
Geodesic A screen-shot of the current version of Geodesic. The view is on clone ZK154 using sources from the C. elegans test server set.
Figure 3
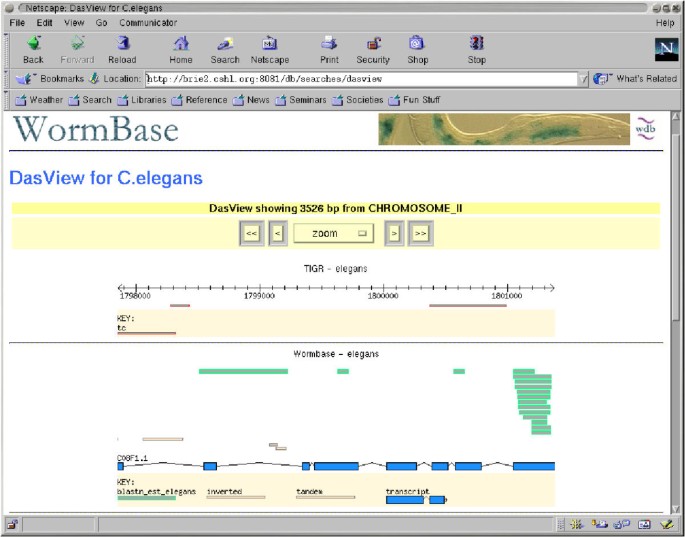
DasView A screen-shot of the current version of DasView. The view is on Chromosome II of WormBase.
Geodesic is mouse and menu driven. The user can choose which data sources to display. The user identifies a segment of the genome to view by browsing through entry points or entering a region name directly. By clicking on a feature, the user obtains additional information in the Feature Details tab and can optionally follow available links back to the original data source. The user can save displayed data as FASTA, GFF, or DAS XML. The user can, to a limited extent, customize the display within the preferences menu.
The DasView prototype implements an alternative mode of using DAS, browserless server side integration. A database can hook into trusted third party servers behind the scenes. The third party data are then integrated into the normal data displays of the database. In this scenario, no DAS client software would be needed.
Both viewers provide the user with one-click linking back the primary data sources where they can learn more about a selected annotation, and are sufficiently flexible to accept a wide range of annotation types and visualization styles. The stand alone Java viewer is appropriate for extensive, long-term use. The Perl implementation is suitable for casual use because it does not require the user to preinstall the software.