Sex genes for genomic analysis in human brain: internal controls for comparison of probe level data extraction. (original) (raw)
Background
Recent developments in DNA microarrays permit a systematic investigation of gene involvement in biological systems. The microarray technology relies on the quantification of relative changes in RNA abundance between samples, which are assumed a priori to represent changes in function or activity of the cell. Accordingly, efforts in genome sequencing and functional gene annotations are shifting the focus to a more global view of biological mechanisms. However, the large amount of data being generated represents a considerable analytical challenge. The typical structure of genomic datasets is complex and evolving rapidly as new microarray analytical tools are being developed and as genomic information gets periodically updated. Currently, a large proportion of the human genome can be surveyed on a single microarray (~22,000 genes and expressed sequenced tags [ESTs]). On Affymetrix GeneChip™ oligonucleotide DNA microarray [1], each gene is probed by 11 to 20 probe pairs (a probeset), consisting of 25 base pairs long oligonucleotides corresponding to different parts of the gene sequence. In a probe pair, a perfect match (PM) oligonucleotide corresponds to the exact gene sequence, while the mismatch (MM) oligonucleotide differs from the PM by a single base in the center of the sequence. The use of probe pair redundancy to assess the expression level of a specific transcript, improves the signal to noise ratio (efficiencies of hybridization are averaged over multiple probes), increases the accuracy of RNA quantification (removal of outlier data) and reduces the rate of false positives. The intensity information from these probes can be combined in many ways to get an overall intensity measurement for each gene, but there is currently no consensus as to which approach yields more reliable results.
Alternative algorithms have been recently described to extract and combine multiple probe level information, however comparative studies assessing the reliability of these different approaches have been limited to analysis based on few synthetic internal control genes [2]. Once gene expression levels have been determined, genomic studies are confronted with issues of multiple statistical testing of large number of genes (in the 10,000s) in much smaller number of samples (from two to less than a hundred in most cases). Typically, this issue has been circumvented by empirically setting statistical thresholds for expression level, fold change between samples and significance levels, based on a small number of internal controls that were added either during processing or before hybridization of the samples onto microarrays.
In the context of a wider study of brain dysfunction in psychiatric disorders, we have been collecting large-scale gene expression profiles in two areas of the brain prefrontal cortex from postmortem human samples, including male and female samples. Thus, as an approach to evaluate the specificity and sensitivity of microarray methods, we used sex-chromosome genes as biological internal controls for assessing microarray data extraction procedures and for developing improved statistical analysis. Sexual dimorphism originates in the differential expression of X- and Y-chromosome linked genes, mostly as a secondary consequence of male or female gonadal hormone secretion. However, not all Y-chromosome genes are restricted to expression in the testes. For instance, several Y-chromosome genes are expressed in the male rodent [3] and human [4] central nervous system. The function of these genes outside the testes is unknown, but a certain level of sexual dimorphism is manifested in the brain of male mice in the total absence of testes [5].
In the central nervous system, sex differences have been described in total brain size [6, 7], in areas controlling reproductive functions and sexual behavior [8], as well as in structure [9], information processing [10], serotonin concentration [11], synthesis [12] and receptor binding [13]. Y-chromosomal dosage also affects behavioral phenotype across mouse strains [14] and in humans [15]. Most sexual dimorphisms originate not as a primary effect of sex chromosome genes in individual tissues, but as a secondary consequence of male or female gonadal hormone secretion. However, evidence exists for cell autonomous realization of genetic sex in neurons, independently of hormonal environment [16, 17], and for direct contributing roles of Y-linked genes in structural features, such as vasopressin-immunoreactive fibers in the lateral septum [5].
Here, we compared three probe level data extraction algorithms: Microarray Suite 5.0 (MAS5.0) Statistical Algorithm from Affymetrix, Model Based Expression Index (MBEI) of Li and Wong [18] and Robust Multi-array Average (RMA) of Irizarry et al. [2]. The three methods were tested on our brain genomic dataset using transcripts from Y-chromosome genes as internal controls for reliability and sensitivity of signal detection. RMA-extracted gene expression values were determined to be less variable and more reliable than MAS5.0 and MBEI-derived values. Expression values for males and females were compared using t-tests with unequal variance, on a gene-by-gene basis and separately for the two brain areas. This means that 22,283 tests were performed for each area and method. The multiple testing problem was addressed by using the Benjamini-Hochberg method for adjusting the resulting p-values. This approach conserves the false discovery rate (the expected proportion of errors among the genes identified as differentially expressed) with no evidence for obvious false negative results. Fourteen probesets with significant sex effect were identified in both brain areas, representing nine Y- and two X-chromosome linked genes (including redundant probesets). These results provide supporting evidence for a putative direct role of sex-chromosome genes, in addition to gonadal hormones, in differentiation and maintenance of sexual dimorphism of the central nervous system.
Results
Probe level data extraction: MAS5.0, MBEI and RMA comparison
We used an oligonucleotide DNA microarray approach [1] to monitor large-scale gene expression profile in the human prefrontal cortex, using dissected samples from postmortem brains. Total RNA was extracted and processed for hybridization onto U133A oligonucleotide microarrays (22,283 genes, expressed sequenced tags [ESTs] and controls). Quality control parameters were based on MAS5.0 extracted information, using thresholds established across numerous microarray studies (see methods). Seventy-five arrays from two brain areas (Brodmann area 9 [BA9], 39 arrays; BA47, 36 arrays) were retained for further comparative analysis. Three different methods were applied to estimate gene expression intensities from the 11 to 16 probesets that represents each gene or EST on the microarrays: Microarray Suite 5.0 (MAS5.0) Statistical Algorithm from Affymetrix, Model Based Expression Index (MBEI [18]) and Robust Multi-array Average (RMA, [2]). MAS5.0 detected on average 53% of the genes (~11,800 transcripts) as expressed in the brain samples. MBEI systematically detected the presence of an additional 8% of genes, while RMA does not provide direct qualitative information about gene expression status.
To assess the reliability of the respective probe-level data analysis methods, we compared the variance in signal detection for each gene across all arrays for the three alternative methods (Fig. 1). Irizarry et al. [2] showed that RMA is less noisy at lower concentrations than the other two methods. The coefficient of variation for each gene (standard deviation as a percentage of the mean) was computed and plotted as a function of the gene expression level, measured by the percentage of samples in which the gene was detected as present. Ideally, this function should have a low constant value, because then the variability of the log-transformed intensity measurements is approximately constant for all expression levels. Variability in MAS5.0 signal intensity measurements was high for background detection (absent genes) and genes with low expression levels, and decreased as signal intensity increased, reaching a level of variation close to that of MBEI and RMA for highly expressed genes (Fig. 1, top curves). The RMA analysis detected gene expression intensities with low variability, regardless of expression levels (Fig. 1, bottom curves), while MBEI analysis results displayed intermediate variability (Fig. 1, Middle curves). Results from all three methods were highly reproducible between the two brain areas, as demonstrated by the closeness of the curves corresponding to the two brain areas for all three methods (Fig. 1).
Figure 1
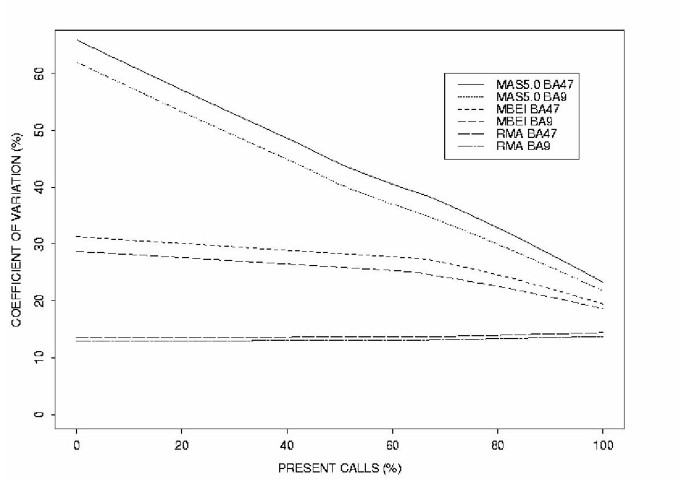
MAS5.0, MBEI and RMA signal variability. The variability in signal intensity measurement obtained with three different probe-level data extraction methods is represented by the lowess curves of the coefficient of variation. The X-axis represent increasing signal intensities, as measured by the percentage of arrays on which this gene is detected as present (% present calls). Presence calls were obtained with MBEI (BA9, n = 39; BA47, n = 36). Note that curves for the two brain areas are very close to each other for all three methods.
The coefficient of variation as a measure of dispersion is not robust to outliers; thus, two other measures were also considered. A popular robust measure of dispersion is the MAD (Median Absolute Deviation). We used MAD as a proportion of the median as an alternative to the coefficient of variation and reached similar conclusions for the comparison of the three methods (data not shown). A second alternative to the coefficient of variation was obtained by using trimmed means and variances (with extreme observations on both ends of the scale removed) and also lead to the conclusion that RMA extracted data were less variable than data extracted with MAS5.0 or MBEI (data not shown).
Next we investigated whether lower signal variability obtained with the RMA probe-level data extraction method was reflected in greater sensitivity to detect differentially expressed genes between experimental groups. To this end, array samples were divided into male (BA9, n = 29; BA47, n = 27) and female (BA9, n = 10; BA47, n = 9) sample groups and Log2-transformed gene expression levels (MAS5.0 and MBEI Log2 converted, RMA values) were compared by t-test between both groups. Y-chromosome-linked genes should only be detected in male samples and were therefore considered true biological internal controls for group comparisons. Out of 45 Y-chromosome probesets on the U133A array, eleven of them yielded consistent low p-values (less than 10E-7) with RMA extracted values, against nine probesets or less with MAS5.0 or MBEI (Table 1 and 2).
Table 1 MAS, MBEI and RMA detection sensitivity for Y-chromosome-linked genes.
Table 2 Male-Female differentially expressed genes.
T-tests rely on the assumption of normality in the two groups of the (log-transformed) gene expression. This assumption can fail for some genes, and the use of the t-tests can be especially questionable for low sample sizes. In our case, the sample sizes were large enough for the males, but there were relatively few females. Therefore, the rank-based Wilcoxon test has also been run on all genes. The performance of the MAS5.0 and MBEI methods on the Y-chromosome genes improved with the Wilcoxon method, although RMA still found the highest number of Y-linked probesets in BA9.
Thus, based on statistical results obtained for biological internal controls and on analysis of signal variability between alternate probe-level data extraction algorithms, RMA-extracted values were deemed superior to those obtained using MBEI and MAS5.0. Log2-transformed-RMA data was used exclusively for the rest of this study.
Testing for differences in sex-chromosome gene expression in prefrontal cortex
We used gene-by-gene t-tests with unequal variance to compare average expression levels between males and females. The results were then checked using the rank-based Wilcoxon test, to discover differentially expressed genes with non-normal distributions of the gene intensities. To adjust for multiple testing and establish the significance of the resulting p-values, we computed cut-off values using the Benjamini-Hochberg technique of controlling the false discovery rate. For independent test statistics, this method aims to guarantee that the proportion of genes with non-significant differences that are detected as being different (the false discovery rate), is below the pre-established experiment-wise error rate of 5%. It has also been shown to work well for gene microarray data, where the test statistics are not independent [19]. At the same time, the technique is somewhat less conservative than using the Bonferroni adjustment for multiple testing although the comparison is not well defined since the two methods aim to control different criteria. Thirteen probesets displayed significant differential expression in both brain areas between males and females (i.e. p-values below the false discovery rate threshold, Fig. 2A, Table 2), representing eight Y-chromosome and two X-chromosome-linked genes. The gene UTY was also detected as differentially expressed in BA47. The X-linked gene PCDH11X was upregulated in male BA47 samples. Twenty additional genes (including some autosomal genes) survived the false discovery rate screen in BA9 or BA47, but with overall low fold changes (1.20 ± 0.06, Mean ± SD) and higher p-values, probably representing a combination of weak effects and lower analytical limit under present conditions. Additional analysis of signal intensities for 30 estrogen-related probesets revealed no discernable trends towards male-female differential gene expression. Individual examination of 31 additional Y-linked probesets also indicated a complete absence of specific signal for these genes (Fig. 2.B), thus confirming that the false discovery rate threshold, as applied here, was not excessively conservative and detected all trends towards sex differences. In comparison, Bonferroni's correction confirmed 30% fewer comparisons in the top 15 X-Y-related probesets in both brain areas combined, and only 53% of all probesets that survived the false discovery rate screen. Assessing the distribution of the sex t-test p-values suggested that additional genes may show weak tendencies towards sex effect (Fig. 3). The p-values from the original groups show a trend toward smaller p-values than expected from a uniform distribution for the low p-values. The size of this shift indicates a possibility that additional genes may be weakly affected by sex, however our sample size was too low to detect them with the adjustment method used in this report. Alternatively, the shift in the p-value distribution could have been introduced by the dependence of the test statistics and effects of data normalization.
Figure 2
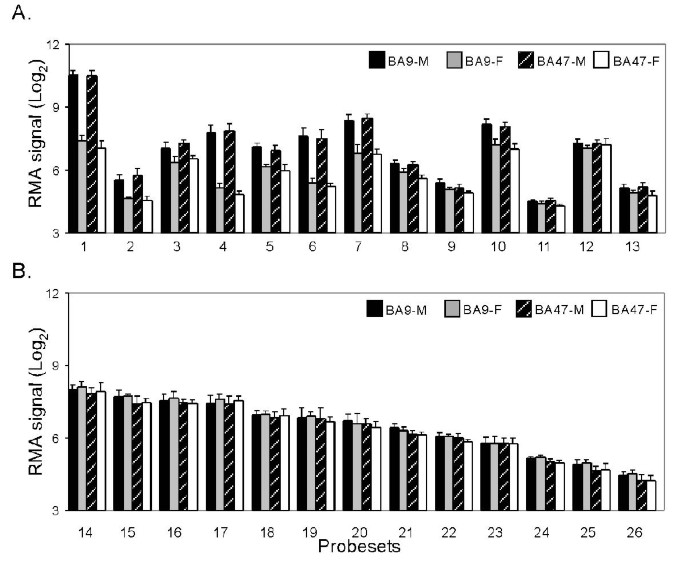
Y-chromosome-linked probesets: male-female expression comparisons. RMA-based averaged values (± STDEV) are displayed. A) Probesets with significant differences in expression levels for male and female samples in BA9 and/or BA47. All male-female comparisons were statistically significant with the exception of #11 in BA9 and # 12 and 13 in BA47 (See also Table 2). Probesets are organized according to order of y-linked genes in Table 2. B) Selected Y-linked probesets without sex-differences. All these genes were detected as ''absent'' by MAS5.0 or MBEI. Signal level represent background estimates. Probesets are: 1, 201909_at; 2, 204409_s_at; 3, 204410_at; 4, 205000_at; 5, 205001_s_at; 6, 206624_at; 7, 206700_s_at; 8, 207063_at; 9, 207246_at; 10, 214983_at; 11, 211149_at; 12, 208067_x_at, 13, 211227_s_at; 14, 214983_at; 15, 217261_at; 16, 217162_at; 17, 221179_at; 18, 211461_at; 19, 209596_at; 20, 210322_x_at; 21, 216376_x_at; 22, 216922_x_at; 23, 211462_s_at; 24, 207909_x_at; 25, 207918_s_at; 26, 207912_s_at.
Figure 3
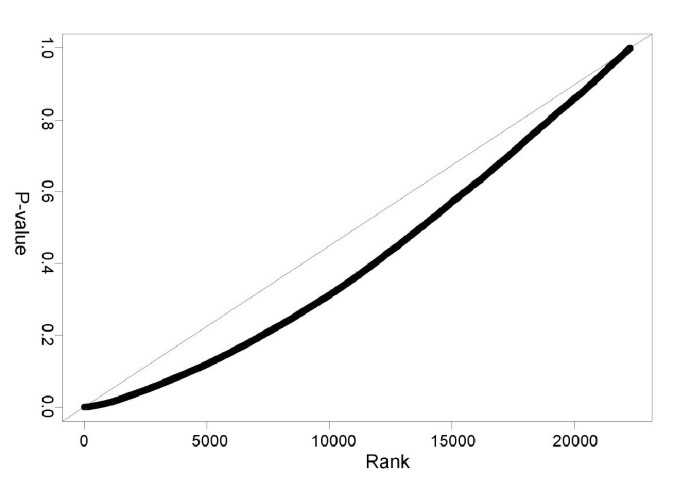
Distribution of the t-tests p-values for sex differences or random group labels. Distribution of the p-values from the t-tests comparing males and females. These p-values are slightly lower than expected from a uniform distribution, representing a mixture distribution of p-values from differentially expressed and not affected genes.
Changes in gene expression were confirmed by real-time quantitative PCR, as an alternative experimental platform to measure RNA levels. As expected, real-time PCR analysis for selected X- and Y-linked genes systematically generated much higher fold changes (Table 2, see Methods and Discussion).
Effect of other demographic and experimental variables on gene expression
The integrity of mRNA samples was assessed by gel electrophoresis and by the ratio of hybridization signal that is obtained between the 3' and 5' mRNA ends for control genes [3'/5' ratio for Actin and GAPDH on oligonucleotide microarrays (see Methods and Table 3)]. A ratio close to one indicates low or absent mRNA damage. Overall, the samples retained for analysis displayed low 3'/5' ratios (Table 3). No correlation between sample variability and brain pH or PMI was observed, indicating all together a high RNA integrity for postmortem brain samples.
Table 3 Microarray quality control parameters.
The cohort of subjects was racially diverse (see Methods). Gene-by-gene analysis of variance (ANOVA) was used to seek differences in gene expression for the respective racial groups (the sole Asian subject was excluded from this analysis). The significance of the resulting p-values was determined by using the Benjamini-Hochberg method described for the sex effect. None of the race p-values were significant at this threshold. To confirm that the negative finding was not a result of a breakdown in the distributional assumptions of the ANOVA, the non-parametric Mann-Whitney test was also applied to all genes. The resulting p-values were not significant. If any differences in gene expression were present between racial groups, they may be too weak to be detected with our current sample size.
Age is known to influence brain function and structure. We will present our findings on the complex effects of aging on gene expression in a separate report. For the purpose of studying sex-related differences in gene expression, no statistical interactions were noted between subject age and sex, therefore we do not address here the effect of age in our comparison of male and female groups.
Postmortem interval (PMI), representing the time elapsed between death and brain collection, may affect gene expression. As described in the Methods section, the effect of PMI was studied on a gene-by-gene basis using three different statistical models. The Pearson correlation coefficient tested for linear correlation between gene expression intensity and PMI, the rank-based Spearman correlation tested for any monotonic relationship, while the analysis of variance, based on a binned version of the PMI, tested for a more general relationship. The resulting p-values were adjusted using the Benjamini-Hochberg method. Only one gene was marginally correlated with PMI, and in one of the two brain areas only. Probeset 220675_s interrogates the expression level of a putative transcript coding for an unknown protein. Signal levels were at background levels, therefore likely representing the lower analytical threshold for PMI effect. No genes with significant PMI effect on expression level were found using the analysis of variance approach. Individual examination of genes that may be directly affected by PMI, such as early immediate genes or genes coding for heat shock proteins, did not reveal any trends or correlation with PMI.
The pH of brain tissue is affected by agonal and postmortem conditions, and in turn may affect RNA levels and integrity [20]. The effect of brain tissue pH on gene expression was studied in our samples by the Pearson correlation coefficient, the Spearman (rank-based) correlation and the analysis of variance approach. When comparing the resulting p-values to the Benjamini-Hochberg thresholds, no significant changes were found. No correlations were detected between pH and PMI (ρ = 0.13, p-value = 0.4), or pH and age (ρ = -0.14, p-value = 0.4), PMI, but age and PMI were positively correlated in this sample (ρ = 0.4, p-value= 0.01). When these variables were entered as covariates in an analysis of covariance model with gene expression as response and gender as the group effect, the resulting adjusted p-values for PMI and pH were not significant compared to the Benjamini-Hochberg thresholds.
Discussion
In this study, we assessed the reliability of probe-level data extraction methods for GeneChip™ oligonucleotide microarrays by comparing the variance in signal detection for each gene across all arrays for three alternative methods (MAS5.0, MBEI and RMA) and found that RMA extracted data displayed low and constant variability in expression values (as expressed by the coefficient of variation), regardless of overall signal intensities. In comparison, MAS5.0 and MBEI generated higher signal variability for low expressed genes and similar detection sensitivity at higher expression levels (Fig. 1). While it may seem intuitive that low-expressed genes have lower signal to background ratios, and should yield more variable intensity measurements, as seen with MAS5.0 (Fig. 1), it is important to remember that the numerical outputs for signal intensity summate complex patterns of hybridization across numerous oligonucleotides, representing both specific (PM) and non-specific (MM) signals, in addition to local background signal from non-probe surfaces. MBEI and RMA use probeset information across several microarrays to determine specific signal from background or non-specific hybridization, while MAS5.0 analyzes one array at a time. It is clear from these results, that the multiple array approach represents an improvement in lowering the variability in signal detection, to the point where background variability becomes constant, even in the case of negligible specific signal (see Fig. 1, 0% presence versus 100% in RMA analysis). However, while MBEI reduced the variability in probeset data extraction over multiple arrays, when compared to MAS5.0, it is not clear why its overall performance at detecting specific signals for Y-linked internal control genes decreased in comparison to both MAS5.0 and RMA algorithms (Table 1).
RMA data analysis seems to provide further benefit in reliability of specific signal detection by uncoupling MM probes from their respective PM, and by incorporating them in their estimates of local background signal. These results confirm reports of lower variability in signal detection by RMA, based on a few synthetic internal controls [2], and extends the approach by using true biological internal controls. While spiked-in genes would have the advantage of being added at known concentrations, the baseline expression of sex-chromosome genes in different tissues is not known. Nevertheless sex genes offer the advantage of being universal, systematic and practical, while allowing the comparison of detection sensitivity for analytical approaches across different experiments using similar biological material. The good performance of RMA in this particular dataset may also be due in part to the comparatively large number of arrays (39 and 36 arrays for the two brain areas). When the number of arrays is small, robust averaging may not work as well. Establishing the groups of arrays to be extracted together is an important step; the need to have a large number of arrays per group has to be balanced with the requirement that dramatically different arrays should not be averaged together. In this study probe-level data from BA9 and BA47 arrays were extracted separately.
The discussion of the variability of a measure, i.e. a signal intensity, has to be coupled with the discussion of its bias. Bias can be estimated in the case of spiked-in positive control transcripts that are added to one of the sample sets but not to the others as the averaged deviation from the true value. In our case, however, the true gene intensities are unknown. Measurements of transcript levels across genes by alternate approaches (Real-time PCR, Northern blot, in situ hybridization) are not absolute, as assays are linear for particular gene products between different samples, but not necessarily across different genes that have different sequences and hybridization efficiencies. Thus we will have to rely on the evidence of the studies with internal controls (2) to estimate that the RMA method is comparable in terms of bias with the other two data extraction methods.
Sensitivity of the three data extraction methods was evaluated on a low number of genes (45 Y-chromosome probesets, 31 were determined to be absent, see Figure 2, eleven were found to be differentially expressed using RMA and t-tests). In this case, false negatives are hard to define, since sex genes that are absent in most of the samples are not expected to be differentially expressed, while the three data extraction methods also differ in their present-absent calls (or lack of it, for RMA). False positives could only be defined under the assumption that only X and Y-chromosome genes are truly different for males and females. Thus our findings as to the sensitivity and specificity of the three methods are suggestive, but not a reliable proof of superiority.
Parametric tests like the t-test and ANOVA rely on the assumption of normality of the response variable, especially for low sample sizes. Testing the distribution of the intensities separately for ~22,000 genes is problematic. Visual inspection of normal quantile plots for a random sample of genes indicates that the assumption is violated for a relatively low proportion of the genes. The obvious solution is to use non-parametric tests (like the Wilcoxon test for comparing two groups, or Mann-Whitney for more than two groups). However, non-parametric tests lose power compared to the parametric tests when the assumptions hold. Thus we ran non-parametric tests in addition, not instead, of the parametric tests, and in questionable cases (when a difference was detected by only one method) inspected the distribution of the gene intensity.
Genomic studies are confronted with issues of multiple statistical testing of large number of genes in much smaller number of samples. Multiple testing is known to lead to high false rejection rates. Most studies involving microarray data analysis have dealt with this problem either by using arbitrary cut-off values for significance, or by using the Bonferroni adjustment method, which can be very conservative. Controlling the family-wise type I error rate (the probability that one error is committed in the family of hypotheses) is too stringent for the purpose of gene discovery and can result in a severe loss of power. Gene discovery can typically tolerate a small number of false positives among a larger pool of selected genes. One alternative is to conserve the expected proportion of false rejections out of all rejections, the false discovery rate. Controlling the false discovery rate has been investigated in other medical research areas involving similar very large datasets involving many comparisons, such as neuroimaging [21]. The Benjamini-Hochberg method that was applied here is a simple way to control the FDR for independent hypotheses, and has been used to address the issue of multiple testing in microarray data analysis [22, [23](/article/10.1186/1471-2105-4-37#ref-CR23 "Storey JD, Tibshirani R: Estimating False DiscoveryRates Under Dependence with Applications to DNA Microarrays. Technical report 2001. [ http://www-stat.stanford.edu/~tibs/research.html
]")\]. Theoretically, one of the underlying assumptions of this technique is that the test statistics used are statistically independent. More conservative methods can be used when the hypotheses are not independent \[[24](/article/10.1186/1471-2105-4-37#ref-CR24 "Benjamini Y, Yekutieli D: The control of the false discovery rate in multiple testing under dependency.
Annals of Statistics 2001, 29: 1165–1188.")\], however, it has been shown that the Benjamini-Hochberg technique will control the FDR when applied to microarray data \[[19](/article/10.1186/1471-2105-4-37#ref-CR19 "Reiner A, Yekutieli D, Benjamini Y: Identifying differentially expressed genes using false discovery rate controlling procedures.
Bioinformatics 2003, 19: 368–375. 10.1093/bioinformatics/btf877"), [23](/article/10.1186/1471-2105-4-37#ref-CR23 "Storey JD, Tibshirani R: Estimating False DiscoveryRates Under Dependence with Applications to DNA Microarrays.
Technical report 2001. [
http://www-stat.stanford.edu/~tibs/research.html
]")\] and our findings support that claim.Using RMA-extracted gene intensity measurements and controlling for multiple testing by the described method, we identified several X- and Y-chromosome linked genes that were differentially expressed between males and females. The statistical methods applied here provided no evidence for false negative results. There were no changes in gene expression that correlated with the different race subgroups, although the sample numbers were relatively small with limited analytical power to detect minor differences. Importantly, we found no evidence for correlation between transcript levels and/or integrity of RNA and two experimental variables that are often taken into consideration for pairing human brain samples: postmortem interval (PMI) and brain pH.
Differences in transcript levels for X- and Y-linked genes ranged between 1.2 and 15 fold, as opposed to a theoretical infinite fold (present in males and absent in females), reflecting the presence of background hybridization signal for genes that are not expressed (i.e. Y-linked genes in female samples). Real-time PCR analysis for selected genes systematically generated much higher fold changes (Table 2), (see Methods). These observations highlight technical issues relating to probeset selection, redundancy and hybridization efficiency on oligonucleotide microarrays. Typically, different probesets for the same gene can yield very different signal levels, reflecting either splice-variants, cross-hybridization or low hybridization efficiency for some oligonucleotides. Based on Y-chromosome internal controls for which biological differences between sexes are known, detection by any probeset seems to be sufficient to identify differential expression. Furthermore, while there is usually an overall good correlation between quantitative changes in expression levels obtained by microarray and real-time PCR (data not shown), these results indicate that background signal and/or less efficient probesets can greatly attenuate real fold changes between samples for genes that are either absent and/or expressed at very low levels in at least one sample. A clear analytical implication of these observations is that selecting differentially expressed genes based on arbitrary cut-off values for fold change will be less reliable than statistical thresholds.
Pre- and postmortem factors have been suggested to influence pH, mRNA quality and quantity in human brains [20]. The pH values that are reported in this study represent storage conditions and it is not known whether they may have varied at the time of brain collection. However, more systematic analyses by Harrison et al. [20] and Johnston et al. [25] indicated that the major factors affecting brain pH may be pre-morbid agonal conditions and longer PMI values than applicable to the samples used in this study. With this regard, it is important to note that all subjects in this study died rapidly, therefore precluding any effect of prolonged agonal state on pH values and overall gene expression. The pH values reported here are similar to values reported in studies of samples involving rapid death (less than 1 hour) and short PMI (less than 36 hours) [20, 25], and lower than values reported in other microarray studies [26]. The reason for differences in reported pH values may reflect methodological differences in assays. Importantly, there was no evidence for an influence of pH on either total RNA levels or individual gene transcripts, nor was there any correlation between pH and PMI or age. Furthermore, PMI did not have any significant effect on any signal intensities for the ~22,000 genes investigated in this study. This absence of PMI effect in our samples does not rule out changes in gene expression in the early period following death. It is possible that the timeframe of the study (PMI= 17.2 hours ± 1.2, Mean ± SD) masked any early effect of postmortem conditions on gene expression.
For X-chromosome genes, transcripts are maintained at male levels by inactivation of a copy on one of the two X-chromosomes in females. This function is directly mediated by the XIST gene RNA [27]. Accordingly, XIST transcript levels were significantly upregulated in female samples (Table 2), with background levels in male samples. Only one other X-chromosome gene was differentially expressed between sexes in our study, while detected changes in autosomal genes were sparse, of marginal amplitude and only in one area at a time. Only two to three genes (MAPK14, EIF4B and PCDH9, Table 2) demonstrated tendencies towards sex-related differential expression. The prefrontal cortex exhibits very little sexual dimorphism, therefore it is not known whether this paucity of detection of autosomal changes in the prefrontal cortex of human subjects is representative of other brain areas and non-gonadal tissue. It is also possible that changes restricted to few cells between sexes may be diluted and considerably reduced in the pooling of cellular subtypes in our gray matter samples.
The SRY gene encodes the master-switch for the development of the testis, but has also been detected at very low levels in human male brains, including frontal cortex [4]. In this study, SRY gene expression in male prefrontal cortex was either absent or below the detection level of DNA microarrays. Y-linked genes detected as expressed in the prefrontal cortex (Table 2) map to the part of the Y-chromosome that does not crossover with the X-chromosome [28]. Genes of this non-recombinant part of the Y-chromosome (NRY) are not present in females, but have functional homologues on the X-chromosome with similar but non-identical protein isoforms [28]. NRY genes identified in this study can be broadly divided in two functional groups: general cellular function (RPS4Y, USP9Y, UTY) or control of transcription-translation (DBY, SMCY, ZFY, EIF4Y). Several of these genes have been previously described as expressed in developing and adult brain tissue, either in rodents (SMCY, UTY, DBY [3]) or in humans (ZFY [4]). The fact that brain expression levels for most genes of the non-recombinant part of the Y-chromosome are independent of testicular secretions and levels of androgens [3] provides further support for a putative independent role for these genes products on brain function. However, an important consideration to keep in mind is that functional differences would have to rely on translation of these transcripts, for which no evidence is being provided. Y-linked genes are typically expressed at low levels and respective protein levels have often been difficult to monitor, even in testis [4].
Conclusions
An important issue in the analysis of gene expression data is the assessment of i) the data quality, ii) the performance of algorithms used for data extraction, and iii) the statistical methods to detect differentially expressed genes. In this study, we have demonstrated the use of sex genes as true biological internal controls to address some of these issues. Based on reliability of detection of sex-differences in sex chromosome gene expression, we have described analytical methods for testing differential gene expression in complex tissues, using robust RMA-extracted signal intensities from oligonucleotide microarrays and by correcting for multiple testing by controlling the false discovery rate. Our results also emphasized the importance of statistical threshold to detect differential expression, as opposed to arbitrary cut-off values. Under our experimental conditions, gene expression profiles in the brain were found to be robust and mostly independent of several demographic and experimental variables, such as race, brain pH and PMI. A consistent sex effect was identified on a set of genes corresponding mostly to sex-chromosomes. These results provide further evidence for a direct role of sex-chromosome genes in the differentiation and maintenance of brain sexual dimorphism. Taken together, our results suggest analytical guidelines for testing microarray data extraction and for adjusting multiple statistical analysis of differentially expressed genes. Importantly, these analytical approaches are applicable to all microarray studies that include male and female human or animal subjects.
Methods
Clinical samples
Samples were obtained from the Brain collection of the Human Neurobiology Core, Sylvio Conte Center for the Neuroscience of Mental Disorders, at the New York State Psychiatric Institute. There were 31 males and 10 females in the sample cohort. Caucasians represent 71%, African-Americans 20%, Hispanics 7% and Asians 2%. As a group, males did not differ significantly from females on age (44.9 ± 20.6 years vs. 46.5 ± 42.2 years, Mean ± STDEV), race (71% Caucasian vs. 72% Caucasian), postmortem delay (18.6 ± 6.2 hours vs. 16.6 ± 8.8 hours, Mean ± STDEV) or brain pH (6.56 ± 0.21; subjects, 6.49 ± 0.20, Mean ± STDEV). Therefore, for the purpose of this study, male and female samples were combined in two separate groups. All subjects were psychiatrically characterized by psychological autopsies and underwent a toxicological screen. 22 subjects committed suicide (psychological autopsies indicated that 19 of them had a lifetime diagnosis of major depression) and 19 died of causes other than suicide (psychological autopsies found them free of psychopathology). In this study probe-level data from BA9 and BA47 arrays were extracted separately, but arrays from the control and subject groups were extracted together. The validity of this step is based on the assumption that the psychiatric subject and the control groups differ at most in the expression level of a limited number of genes. The details of our finding on the comparison of these two groups will be published separately.
Microarray samples
Brodmann areas 9 (BA9) and 47 (BA47) were dissected from frozen brain sections that had been transferred from -80C to -20C for two hours. Brodmann areas were identified using gyral and sulcal landmarks, cytoarchitecture and a standardized coronal atlas (Robert Perry and Edward Bird, personal communication), as previously described [29]. Blocks were sectioned with a cryostat at 200 μm (-20°C). Meninges and white matter were removed as much as possible during sectioning, before collection into microtubes. 200 μm thick sections were collected in microtubes at -20C. Total RNA was extracted by the TRIZOL method (Invitrogen, Carlsbad, CA) and cleaned with Rneasy microcolumns (QIAGEN GmbH, Germany). The RNA purity and integrity were assessed by optical densitometry, gel electrophoresis and subsequent array parameters. Microarray samples were prepared according to the Affymetrix protocol http://www.affymetrix.com/support/. In brief, 10 μg of total RNA was reverse-transcribed and converted into double-stranded cDNA. A biotinylated complementary RNA (cRNA) was then transcribed in vivo, using an RNA polymerase T7 promoter site which had been introduced during the reverse-transcription of RNA into cDNA. After fragmentation in pieces of 50 to 200 bases long, 15 μg of labeled cRNA sample was hybridized onto oligonucleotide U133A microarrays, using standard protocols with the Affymetrix microarray oven and fluidics station at the Columbia University Genome Center. A high-resolution image of the hybridization pattern on the probe array was obtained by laser scanning, and fluorescence intensity data was automatically stored in a raw file. To reduce the influence of technical variability, samples were randomly distributed at all experimental steps to avoid any simultaneous processing of related samples. Microarray quality control parameters were as follows: noise (RawQ) less than 5, background signal less than 100 (250 targeted intensity for array scaling), consistent number of genes detected as present across arrays, consistent scale factors, Actin and GAPDH 3'/5' signal ratios less than 3 and consistent detection of BioB and BioC hybridization spiked controls. Based on these criteria, 39 arrays were retained for further analysis in BA9 and 36 arrays in BA47 (Table 3).
Real-time PCR
Small PCR products (100–200 base-pairs) were amplified in quadruplets on an Opticon real-time PCR machine (MJ Research, Waltham, MA), using universal PCR conditions (65C to 59C touch-down, followed by 35 cycles [15"at 95C, 10" at 59C and 10" at 72C]). 150 pg of cDNA was amplified in 20 μl reactions [0.3X Sybr-green, 3 mM MgCl2, 200 μM dNTPs, 200 μM primers, 0.5 unit Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA)]. Primer-dimers were assessed by amplifying primers without cDNA. Primers were retained if they produced no primer-dimers or non-specific signal only after 35 cycles. Results were calculated as relative intensity compared to actin. The last cycle was retained as baseline for comparison with "absent" genes.
Gene intensity measures (Probe level data extraction)
MAS5.0, MBEI and RMA represent different ways to combine probe-level data from oligonucleotide microarrays. Software for all three methods can be found in the R package affy that can be downloaded from the Bioconductor project website http://www.bioconductor.org. All three methods model the gene intensity as a measure of the "specific binding": the difference between the binding intensity of the perfect match probe and a measure of the non-specific and/or background binding. MAS5.0 and MBEI use mismatch (MM) probe intensities as a relative measure of non-specific binding while RMA uses a local background signal computed using summaries of these mismatch values as non-specific binding. All three methods are based on an explicit statistical model of gene intensity as a function of probe-level intensities and include some sort of mechanism for removing or downweighting outlier probes and/or arrays, so that a few outstanding values will not exercise a disproportionate influence on the gene intensity measure. Hence, all three can be called robust statistical methods. However, there are important differences in the form of the model and the underlying assumptions that are worth mentioning here.
Let us denote the intensities for each probe by PMijn and MMijn, where n represents the probe sets or genes, i represents the array or sample, and j the probe pair number.
For probe set n on array i, the intensity measure (signal) as given by the MAS5.0 software is computed on the log scale as the weighted average of the probe-level signals
log(Signalijn) = Tukey Biweight{log(PMijn - CTijn)}, j = 1, ..., J, [1]
where the CTijn are the mismatch MMijn values wherever they are smaller than the corresponding perfect match intensities and a modified background value otherwise. The weighting function used is the Tukey Biweight [30] which removes/downweights outlier probe intensities so they will not distort the signal. MAS5.0 extracts data from each array separately and independently, so data from different arrays do not influence each other. The other two methods use data from multiple arrays to estimate the signal.
The MBEI, described in [18], for each probe set n, from 1 to N, on array i is the maximum likelihood estimate of the θin (after outlier removal) from the equation
PMijn - MMijn = θin * φjn + εijn, i = 1, ..., I; j = 1, ..., J [2]
where φjn represent probe-specific affinities and the εijns are assumed to be independent normally distributed errors for each n. The model uses data from different arrays (denoted by i in the above formula) for the same probe set and iteratively estimates the array-specific probe-set intensity and the probe-affinity parameters in subsequent steps, until convergence of the parameter estimates is achieved. It assumes a multiplicative model on the original scale for the two sets of parameters, with normal errors. Outlier arrays or probe sets are tagged and discarded from the analysis.
The RMA log scale expression level for probe set n, n = 1, ..., N, is the estimate of μin from the linear additive model
Yijn = μin + αjn + εijn, i = 1, ..., I; j = 1, ..., J, [3]
where αjn are probe affinity effects that sum up to 0 for each probe set n, εijns are assumed to be independent normally distributed errors, and Yijn are the background adjusted, normalized and log transformed PM intensities. See [2] for details. As in the case of the MBEI method, this equation uses data from I microarrays and J probes for each probe set. Note that the probe-level MM intensities in this approach are used only indirectly, for calculating the background intensity. Instead of maximum likelihood, a robust method (median polish, see [30]) is used to obtain the parameter estimates, to diminish the influence of outlier probes/arrays.
From a theoretical point of view the MAS5.0 method is very different from the other two since it uses data from a single array. The other two methods differ mainly in the scale they use and the estimation method. For datasets with a small number of arrays, "averaging" over arrays is generally not going to lead to much improvement in the gene intensity measure, but for larger datasets, theoretically at least, the MBEI and RMA methods might yield more reliable results. The quality of the intensity measure in this case will depend on the estimation method, but also on the heterogeneity of the samples. Using arrays with very different gene expression levels in the same model might lead to biased intensity measures. To avoid this pitfall, arrays with samples from BA9 and BA47 were submitted separately to the MBEI and RMA probe-level extraction programs.
Statistical Testing
After obtaining the gene intensity measures, the next step was to study the effect of sex, race, postmortem delay and pH on gene expression levels. Since the co-regulation between genes is a very complex issue, we used a gene-by-gene statistical testing instead of a multivariate analysis. The log-transformed values were used instead of the raw intensities because of the variance stabilizing effect of this transformation. The most widely used statistical method for detecting differential gene expression between two groups is gene-by-gene t-test. The standard two-sample t-test assumes that the intensities in the two groups are normally distributed and have equal variances. In case of the Y chromosome genes, which are not present in one of the groups, the assumption of equal variances is clearly violated. Thus, a modification, the t-test for unequal variances was used for all genes. This t-test still relies on the normal distribution of the (log transformed) gene expression. That assumption can be violated for some genes. Thus a more prudent approach is to use a rank-based method like the Wilcoxon test for comparing the two gender groups. When the data is normally distributed, this can lead to a loss in power compared to the t-test.
There are three racial groups among the subject in the study (Caucasians, African Americans and Hispanics, the sole Asian subject was removed from this part of the analysis). To test for the effect of race on gene expression, gene-by-gene analysis of variance (ANOVA) with three groups was used. This method tests for any difference between the group averages. If a significant p-value is found, so called post-hoc tests (Scheffe's, Tukey's etc.) can be computed to ascertain the nature of this difference, without affecting the experiment-wise Type I error level. Non-parametric analysis of variance tests (Mann-Whitney) have also been performed to ascertain that the negative results were not due to a breakdown in the normal distribution assumption.
There are several ways to test for the effect of a continuous variable, like postmortem delay, on gene expression, depending on the form of the hypothesized relationship. Linear correlation was tested using the Pearson correlation coefficient. To test for a relationship of a more general form, the continuous variable can be binned into three or more categories and an analysis of variance, as described above, can be performed. For example, postmortem delay values were divided into 4 bins (less than 15 hours, 15–18, 18–22 and above 22 hours), and then ANOVA was used to detect differences among the four groups. Spearman's rank-based correlation coefficient and the non-parametric Mann-Whitney test for the comparison of the four groups have also been computed.
Finally, all of the continuous covariates (age, PMI, pH) were included in an analysis of covariance model with gene expression as the response variable and gender as the group effect, to compute adjusted p-values for the covariates. When the covariates are correlated, the adjusted p-values can differ from the unadjusted values.
Performing a separate test for each gene raises the problem of adjusting the resulting p-values for multiple testing. A conservative adjustment method (Bonferroni's adjustment) is to divide the experiment-wise Type I error level (α) by the number of hypotheses tested (N = 22,283) and use that as the cut-off for significant p-values. This method will usually result in very few, if any, significant p-values. Another alternative is to use cut-offs given by a linear step-up technique first proposed by Benjamini and Hochberg [31] for the control of the False Discovery Rate.
When testing N independent null hypotheses H1, H2, ..., HN with p-values P1, P2, ..., PN the false discovery rate rejection rule Rα guarantees that the expected proportion of rejected null hypotheses that are actually true (denoted by FDR) stays at or below α.
Let iα = max{i: P(i) ≤ (i/N) * α }, where P(i) are the ordered p-values. Then, under independence of the hypotheses Hi, the rejection rule given by
Rα = {Reject all Hi with Pi ≤ P(iα)} satisfies FDR(Rα) ≤ α [4]
The cut-off value for the lowest p-value is the Bonferroni cut-off, but for the subsequent p-values it is higher by a multiple equal to the order of the p-value.
Test statistics for gene microarray data are unlikely to be independent from each other because of gene coregulation. For positively dependent test statistics, a modified version of the technique was developed [20]. Measurement error of microarray data tends to have a positive dependence structure, but gene coregulation need not result in positive dependency, thus it is not clear if the second approach is more valid than the first one in the present case. However, in [19] the "naïve" Benjamini-Hochberg approach is compared on simulated gene microarray data to three other, more complicated techniques designed to control the FDR, and the authors conclude that the Benjamini-Hochberg approach does control the FDR on the desired level. Their second conclusion is that it retains more power than the traditional adjusting procedures, although it has less power than the more complicated, resampling-based techniques the paper describes. As it often happens in statistical testing, more power can be obtained at the expense of computational simplicity.
Author's contributions
HCG designed and carried out all statistical analysis and drafted the manuscript. LEB and PS designed and performed the real-time PCR quantifications and sequence annotations. PP helped with bioinformatic approaches and data extraction algorithms. SPE provided theoretical statistical support. JJM and VA participated in the conception of the study. VA provided and carried out the anatomical dissection of the brain experimental samples. ES designed the study and performed the microarray experiments and analysis. All authors read and approved the final manuscript.
References
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL: Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 1996, 14: 1675–1680.
Article CAS PubMed Google Scholar - Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP: Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31: e15. 10.1093/nar/gng015
Article PubMed Central PubMed Google Scholar - Xu J, Burgoyne PS, Arnold AP: Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet 2002, 11: 1409–1419. 10.1093/hmg/11.12.1409
Article CAS PubMed Google Scholar - Mayer A, Lahr G, Swaab DF, Pilgrim C, Reisert I: The Y-chromosomal genes SRY and ZFY are transcribed in adult human brain. Neurogenetics 1998, 1: 281–288. 10.1007/s100480050042
Article CAS PubMed Google Scholar - De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, Swain A, Lovell-Badge R, Burgoyne PS, Arnold AP: A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci 2002, 22: 9005–9014.
CAS PubMed Google Scholar - Luders E, Steinmetz H, Jancke L: Brain size and grey matter volume in the healthy human brain. Neuroreport 2002, 13: 2371–2374. 10.1097/00001756-200212030-00040
Article PubMed Google Scholar - Swaab DF, Hofman MA: Sexual differentiation of the human brain. A historical perspective. Prog Brain Res 1984, 61: 361–374.
Article CAS PubMed Google Scholar - Swaab DF, Hofman MA: Sexual differentiation of the human hypothalamus in relation to gender and sexual orientation. Trends Neurosci 1995, 18: 264–270. 10.1016/0166-2236(95)93913-I
Article CAS PubMed Google Scholar - Rabinowicz T, Petetot JM, Gartside PS, Sheyn D, Sheyn T, de CM: Structure of the cerebral cortex in men and women. J Neuropathol Exp Neurol 2002, 61: 46–57.
PubMed Google Scholar - Shaywitz BA, Shaywitz SE, Pugh KR, Constable RT, Skudlarski P, Fulbright RK, Bronen RA, Fletcher JM, Shankweiler DP, Katz L: Sex differences in the functional organization of the brain for language. Nature 1995, 373: 607–609. 10.1038/373607a0
Article CAS PubMed Google Scholar - Tordjman S, Roubertoux PL, Carlier M, Moutier R, Anderson G, Launay M, Degrelle H: Linkage between brain serotonin concentration and the sex-specific part of the Y-chromosome in mice. Neurosci Lett 1995, 183: 190–192. 10.1016/0304-3940(94)11148-C
Article CAS PubMed Google Scholar - Nishizawa S, Benkelfat C, Young SN, Leyton M, Mzengeza S, de Montigny C, Blier P, Diksic M: Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci U S A 1997, 94: 5308–5313. 10.1073/pnas.94.10.5308
Article PubMed Central CAS PubMed Google Scholar - Mann JJ, Huang YY, Underwood MD, Kassir SA, Oppenheim S, Kelly TM, Dwork AJ, Arango V: A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry 2000, 57: 729–738. 10.1001/archpsyc.57.8.729
Article CAS PubMed Google Scholar - Guillot PV, Carlier M, Maxson SC, Roubertoux PL: Intermale aggression tested in two procedures, using four inbred strains of mice and their reciprocal congenics: Y chromosomal implications. Behav Genet 1995, 25: 357–360.
Article CAS PubMed Google Scholar - Ratcliffe SG, Butler GE, Jones M: Edinburgh study of growth and development of children with sex chromosome abnormalities. IV. Birth Defects Orig Artic Ser 1990, 26: 1–44.
CAS PubMed Google Scholar - Beyer C, Pilgrim C, Reisert I: Dopamine content and metabolism in mesencephalic and diencephalic cell cultures: sex differences and effects of sex steroids. J Neurosci 1991, 11: 1325–1333.
CAS PubMed Google Scholar - Carruth LL, Reisert I, Arnold AP: Sex chromosome genes directly affect brain sexual differentiation. Nat Neurosci 2002, 5: 933–934. 10.1038/nn922
Article CAS PubMed Google Scholar - Li C, Wong WH: Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc Natl Acad Sci U S A 2001, 98: 31–36. 10.1073/pnas.011404098
Article PubMed Central CAS PubMed Google Scholar - Reiner A, Yekutieli D, Benjamini Y: Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19: 368–375. 10.1093/bioinformatics/btf877
Article CAS PubMed Google Scholar - Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC: The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett 1995, 200: 151–154. 10.1016/0304-3940(95)12102-A
Article CAS PubMed Google Scholar - Ellis SP, Underwood MD, Arango V, Mann JJ: Mixed models and multiple comparisons in analysis of human neurochemical maps. Psychiatry Res 2000, 99: 111–119. 10.1016/S0925-4927(00)00051-2
Article CAS PubMed Google Scholar - Efron B, Tibshirani R, Storey JD, Tusher V: Empirical Bayes analysis of a microarray experiment. Journal of the American Statistical Association 2001, 96: 1151–1160. 10.1198/016214501753382129
Article Google Scholar - Storey JD, Tibshirani R: Estimating False DiscoveryRates Under Dependence with Applications to DNA Microarrays. Technical report 2001. [http://www-stat.stanford.edu/~tibs/research.html]
Google Scholar - Benjamini Y, Yekutieli D: The control of the false discovery rate in multiple testing under dependency. Annals of Statistics 2001, 29: 1165–1188.
Article Google Scholar - Johnston NL, Cervenak J, Shore AD, Torrey EF, Yolken RH, Cerevnak J: Multivariate analysis of RNA levels from postmortem human brains as measured by three different methods of RT-PCR. Stanley Neuropathology Consortium. J Neurosci Methods 1997, 77: 83–92. 10.1016/S0165-0270(97)00115-5
Article CAS PubMed Google Scholar - Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P: Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 2000, 28: 53–67.
Article CAS PubMed Google Scholar - Willard HF: X chromosome inactivation, XIST, and pursuit of the X-inactivation center. Cell 1996, 86: 5–7.
Article CAS PubMed Google Scholar - Lahn BT, Page DC: Functional coherence of the human Y chromosome. Science 1997, 278: 675–680. 10.1126/science.278.5338.675
Article CAS PubMed Google Scholar - Arango V, Underwood MD, Gubbi AV, Mann JJ: Localized alterations in pre- and postsynaptic serotonin binding sites in the ventrolateral prefrontal cortex of suicide victims. Brain Res 1995, 688: 121–133. 10.1016/0006-8993(95)00523-S
Article CAS PubMed Google Scholar - Mosteller F, Tukey JW: Data analysis and regression: A secondary course in statistics. Reading MA : Addison-Wesley Publishing Co. Inc 1977.
Google Scholar - Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B, Methodological 1995, 57: 289–300.
Google Scholar