GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies (original) (raw)
Background
Microarray and proteome technologies are producing sets of genes and proteins that are differentially regulated under varying conditions. Other studies such as quantitative trait analysis, large-scale mutagenesis studies, and other large-scale genetic studies are also producing sets of interesting genes. The number of genes in the gene sets may be large. The functional data that can be associated with each gene is quite complex. However, the in-depth knowledge of gene function possessed by individual biologists is limited to relatively narrow research fields. Searching for patterns and evaluating the functional significance of those patterns from large groups of genes constitutes a big challenge for biologists. Most resources that are available for retrieving functional information are displayed in a one-gene-at-a-time format. Bioinformatics tools are needed for assisting the functional profiling of large sets of genes.
Gene nomenclature has been used frequently to describe gene products [1]. While the goal for gene nomenclature is to create a unique designation for gene names, gene name is often not unique even within a species. Trying to attach significant biological information to the name can be problematic. In fact, many revisions in nomenclature have occurred as the knowledge of the function of the gene product has developed [2]. The information about gene function is primarily contained in the articles indexed in the Medline database. In this form, it is readable by scientists but not easily interpreted by computers on a large scale. Tools based on literature profiling have been developed by a few groups to assist biologists in the interpretation of sets of interesting genes [3–5]. However, these methods depend on the identification of gene-reference relationships and have problems such as ambiguous gene names and symbols, context of categories etc. [3].
The use of ontological methods to structure biological knowledge is an active area of research and development [2]. Ontologies provide a mechanism for capturing a community's view of a domain in a shareable form. One of the most important ontologies in molecular biology is the Gene Ontology (GO) [2, 6]. GO is beginning to produce a structured, precisely defined, common, controlled vocabulary for describing the roles of genes and gene products in different species. It comprises three major categories that describe the attributes of biological process, molecular function and cellular component for a gene product. As of August 2003, GO contains about 14000 phrases, representing categories of concepts held within a Directed Acyclic Graph (DAG). Categories can have multiple parents and multiple children along a branch. As they form a standard vocabulary across many biological resources, this shared understanding provides a valuable, computationally accessible form of the community's knowledge about these attributes. Several programs have been developed for profiling gene expression based on GO, and demonstrated to be very useful in translating sets of differentially regulated genes into functional profiles [7–[12](/article/10.1186/1471-2105-5-16#ref-CR12 " GoSurfer[ http://biosun1.harvard.edu/complab/gosurfer/
]")\]. GoMiner\[[10](/article/10.1186/1471-2105-5-16#ref-CR10 "Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN: GoMiner: a resource for biological interpretation of genomic and proteomic data.
Genome Biol 2003, 4: R28. 10.1186/gb-2003-4-4-r28")\], MAPPFinder\[[11](/article/10.1186/1471-2105-5-16#ref-CR11 "Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR: MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data.
Genome Biol 2003, 4: R7. 10.1186/gb-2003-4-1-r7")\] and GoSurfer\[[12](/article/10.1186/1471-2105-5-16#ref-CR12 "
GoSurfer[
http://biosun1.harvard.edu/complab/gosurfer/
]")\] are standalone software packages while FatiGO\[[7](/article/10.1186/1471-2105-5-16#ref-CR7 "Herrero J, Al-Shahrour F, Diaz-Uriarte R, Mateos A, Vaquerizas JM, Santoyo J, Dopazo J: GEPAS: A web-based resource for microarray gene expression data analysis.
Nucleic Acids Res 2003, 31: 3461–3467. 10.1093/nar/gkg591")\] and Onto-Express\[[8](/article/10.1186/1471-2105-5-16#ref-CR8 "Draghici S, Khatri P, Martins RP, Oscategoryeier GC, Krawetz SA: Global functional profiling of gene expression.
Genomics 2003, 81: 98–104. 10.1016/S0888-7543(02)00021-6"), [9](/article/10.1186/1471-2105-5-16#ref-CR9 "Khatri P, Draghici S, Ostermeier GC, Krawetz SA: Profiling gene expression using Onto-Express.
Genomics 2002, 79: 266–270. 10.1006/geno.2002.6698")\] are web-based software. Web-based service provides experimental biologists easy access to tools by avoiding problems in installing software locally. However, the two web-based software packages did not visualize the data with the GO hierarchical structure – the fundamental defining feature of GO. The current implementation (as of August, 2003) of FatiGO is restrictive in that the user must specify ahead of time one particular level of the GO hierarchy that is to be used for analysis of the data. Although Onto-Express allows multilevel analysis, it visualizes the classification in flat view tables and the significantly enriched GO categories are presented as bar charts \[[8](/article/10.1186/1471-2105-5-16#ref-CR8 "Draghici S, Khatri P, Martins RP, Oscategoryeier GC, Krawetz SA: Global functional profiling of gene expression.
Genomics 2003, 81: 98–104. 10.1016/S0888-7543(02)00021-6"), [9](/article/10.1186/1471-2105-5-16#ref-CR9 "Khatri P, Draghici S, Ostermeier GC, Krawetz SA: Profiling gene expression using Onto-Express.
Genomics 2002, 79: 266–270. 10.1006/geno.2002.6698")\].As the GO categories are held within a DAG and have a natural hierarchical structure, we believe that the tree structure is more intuitive and representative. To create a web-based and tree-based data mining environment for gene sets, we have developed GOTree Machine (GOTM).
Implementation
Schematic overview of GOTM
GOTM is implemented in PHP. It is accessible through IE5.0 or higher and Netscape 7 or higher from multiple platforms. GOTM can be accessed from the website http://genereg.ornl.gov/gotm/. Figure 1 shows the schematic overview of GOTM. After reading the input parameters and data files from the user, GOTM interacts with the local database GeneKeyDB (S.K. et al., manuscript in preparation) to convert gene symbols, Affymetrix probe set IDs, Unigene IDs, Swiss-Prot IDs or Ensembl IDs to LocusIDs. The hierarchical GOTree structure is then generated using the PHP Layers Menu System [[13](/article/10.1186/1471-2105-5-16#ref-CR13 " PHP Layers Menu System[ http://phplayersmenu.sourceforge.net/
]")\] and sent back to the user. It is based on the GO annotation for LocusIDs as recorded in GeneKeyDB. The user can browse or query the GOTree for desired GO categories. The GOTree can be exported and stored locally in html format. Bar charts for GO categories at different annotation levels can be generated for publication. The bar chart is created using ChartDirector \[[14](/article/10.1186/1471-2105-5-16#ref-CR14 "
ChartDirector[
http://www.advsofteng.com/index.html
]")\]. Statistical analysis compares the interesting gene set and the reference gene set and provides the user with GO categories with enriched gene numbers. The enriched GO categories are presented in flat view format, sub-tree view format and DAG view format. The DAG is created using Graphviz \[[15](/article/10.1186/1471-2105-5-16#ref-CR15 "
Graphviz[
http://www.research.att.com/sw/tools/graphviz/
]")\]. Subsets of genes in each GO category can be displayed and additional information for each gene can be further retrieved from GeneKeyDB.Figure 1
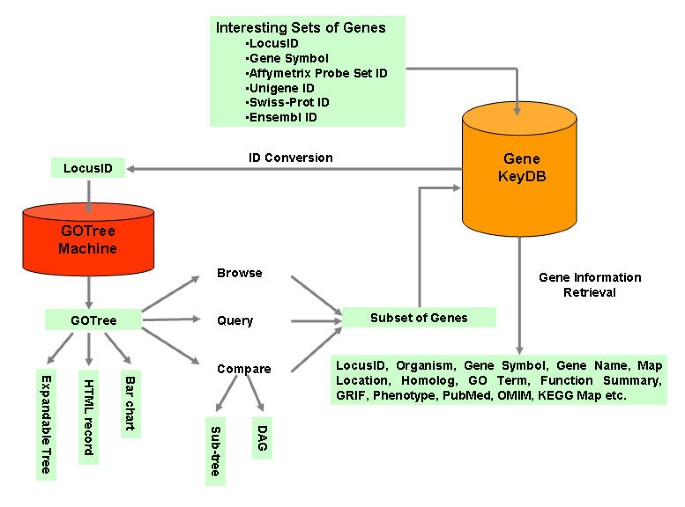
Schemetic overview of the GOTM GOTM is flexible in the input identifier (LocusID, gene symbol, Affymetrix Probe Set ID, Unigene ID, Swiss-Prot ID and Ensembl ID). GOTM produces different kinds of visualizations for different purposes, including 1) an expandable GOTree for online browsing 2) HTML output for an archivable record and 3) a bar chart for publication. Statistical analysis is used to compare gene sets. Sub-tree and DAG (Direct Acyclic Graph) can be generated for enriched GO categories.
Database: GeneKeyDB
The ORACLE relational database GeneKeyDB was initially built from the NCBI LocusLink database [[16](/article/10.1186/1471-2105-5-16#ref-CR16 " NCBI LocusLink database[ http://www.ncbi.nlm.nih.gov/LocusLink/
]")\]. It has adopted a strong gene-centric viewpoint rather than a sequence entry-centric view. Gene information was further taken from Ensembl, Swiss-Prot, HomoloGene, Unigene, Gene Ontology Consortium and Affymetrix etc. and was integrated into GeneKeyDB. The GO annotation for genes is based on the LocusLink data. However, the GO annotation for genes in the LocusLink data only provides the most detailed information available. Genes are annotated to the most granular GO category(s) possible. For example, the GO biological process annotation for the mouse _Birc4_ (LocusID 11798) gene is "apoptosis", so _Birc4_ is directly related to "apoptosis". However, because of the hierarchical relationship between the parent and the child, "apoptosis" is a "programmed cell death"; "program cell death" is, in turn, a "cell death", and so on. This continues until we reach the most general annotation "biological process". Thus, _Birc4_ is also indirectly related to "programmed cell death", "cell death" etc. If we are interested in all genes involved in "programmed cell death", by using only the annotation provided by the LocusLink data, we will miss the _Birc4_ gene. Moreover, if we want to find GO categories with enriched gene numbers, failing to implement the parent-child relationship will miss known information. In order to map the granular annotations such as "apoptosis" to general categories like "cell death", GO files for the 3 main categories were downloaded from the current ontologies section from the Gene Ontology consortium website \[[17](/article/10.1186/1471-2105-5-16#ref-CR17 "
Gene Ontolgy consortium[
http://www.geneontology.org
]")\] as flat text files and parsed by a Perl script. The relationships between genes and all their directly or indirectly related GO categories are created and stored in tables in GeneKeyDB.GeneKeyDB is updated periodically. It comprises several independent sub-modules, such as LocusLink and GOTree Machine. Each of the modules is updated independently during the updating. The process is automated by pre-prepared scripts. More detailed information on GeneKeyDB will be presented in a separate paper (S.K. et al., manuscript in preparation).
Statistical analysis
Identifying GO categories with significantly enriched gene numbers in the interesting gene set compared to a reference gene set will allow the user to focus on biological areas that are most important for the interesting gene set. In order to identify GO categories with significantly enriched gene numbers, we need to compare the distribution of genes in the interesting gene set in each GO category to those in the reference gene set. A reference gene set could be all genes in a genome or another appropriate reference gene set (e.g. the list of genes on the array). We need to mention that an inappropriate reference gene set will lead to possibly false positives and negatives. Unless the user can find the right reference gene set from our stored data, uploading an appropriate reference gene set for the analysis is always suggested. Suppose n genes were identified as interesting genes based on a microarray experiment (such as responsive, up-regulated or down-regulated genes) using an array with N genes. For a given GO category X, a gene is either in the category or not in the category. Suppose further that K out of the N reference genes and k out of the n interesting genes are in category X. If the n interesting genes were effectively a random sample uniformly selected from the reference gene set, the expected value of k would be k e = (n/N)K. If, on the other hand, k exceeds the above expected value, category X is said to be enriched, with a ratio of enrichment (R) given by R = k/k e . Statistical tests that have been used for the assessment of enrichment by related published software include Fisher's exact test, χ2 test, T test and binomial test [8–[12](/article/10.1186/1471-2105-5-16#ref-CR12 " GoSurfer[ http://biosun1.harvard.edu/complab/gosurfer/
]")\]. As genes can be selected only once, this is sampling without replacement and can be appropriately modelled by the hypergeometric distribution \[[8](/article/10.1186/1471-2105-5-16#ref-CR8 "Draghici S, Khatri P, Martins RP, Oscategoryeier GC, Krawetz SA: Global functional profiling of gene expression.
Genomics 2003, 81: 98–104. 10.1016/S0888-7543(02)00021-6")\]. GOTM reports only those enrichments that are statistically significant as determined by the hypergeometric test. The significance of enrichment (_P_) for a given category is determined by 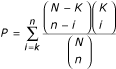. GO is organized on the basis of the three relatively independent categories: biological process, molecular function and cellular component. The Ns used for each category: biological process, molecular function and cellular component, represent the number of genes having GO annotation in that category.Results and Discussion
Input
Figure 2 shows the input user interface of GOTM. The input identifiers for GOTM can be LocusIDs, Gene Symbols, Affymetrix probe set IDs, Unigene IDs, Swiss-Prot IDs or Ensembl IDs. GOTM currently supports Gene Symbols from human, mouse, rat and fly, and Affymetrix probe set IDs from 8 human arrays and 6 mouse arrays. The user can choose either single gene set analysis or interesting gene set vs. reference gene set analysis. For single gene set analysis, only the file of the interesting gene set is needed, and the result will be a GOTree for the gene set. For interesting gene set vs. reference gene set analysis, the user needs to upload the file of the interesting gene set, and choose an existing reference gene set from our pre-stored gene sets, including all genes in the mouse genome, all genes in the human genome and gene sets from 14 Affymetrix arrays, or upload the file of the reference gene set. The result will be a GOTree for the interesting gene set, and identified GO categories with relatively enriched gene numbers in the interesting gene set compared to the reference gene set. The user can browse his local machine for the input files. The input file should be a plain text file, including the appropriate ID (required) and corresponding microarray ratio (optional), separated by tabs in the format of one ID per row. A unique analysis name is assigned and can be used to retrieve the results for a subsequent user session. Stored results can be accessed through the RETRIEVE TREE button and deleted through the DELETE TREE button at the top of input user interface. The results will be stored until the next periodical upgrading of GeneKeyDB. An email notice will be sent to the users after the updating.
Figure 2
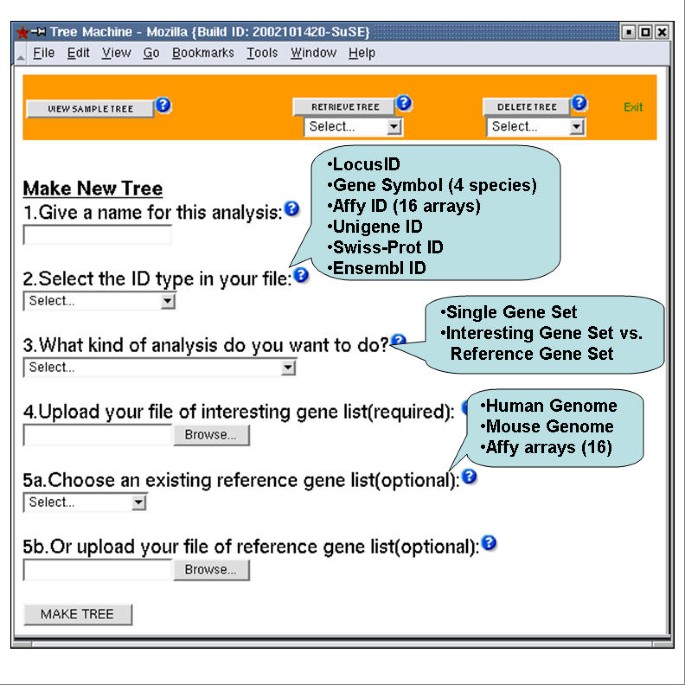
Input user interface of the GOTM Input interface for uploading analysis parameters (analysis name, ID type and analysis type) and data (interesting gene list and reference gene list).
Output
Figure 3 shows the output user interface of GOTM. The output view is divided into 3 windows. The upper-left window is the GOTree window, the upper-right window is the gene/category list window and the bottom window is the gene information window. The expandable GOTree will be shown in the GOTree window. The user can browse the tree by clicking the "+" symbol. For single gene set analysis, the number of genes in each GO category will be given. If the interesting gene set vs. reference gene set analysis is selected, three parameters will be given for each GO category: O (observed gene number in the category), E (expected gene number in the category), R (ratio of enrichment for the category). For those GO categories with R > 1, the fourth parameter P indicating significance of enrichment will be given. GO categories with significantly enriched gene numbers (P < 0.01) will be colored red. By clicking on individual GO categories, the genes in the category will be shown in the gene/category list window. It might be sometimes difficult for a user to browse and find the GO category in which the user is interested. In this case, the user can do an exact search for a GO category using "GO Term Search" or a fuzzy key word search using "Keyword Search" at the top of the GOTree window. The returned GO categories and genes inside each category will be shown in the gene/category list window. The number of GO categories with enriched gene numbers will also be shown in the GOTree window. By clicking on the number, the names of enriched GO categories will be shown in the gene/category list window. GOTree provides comprehensive classification of the genes in a hierarchical structure, however, due to the complex structure, it's not easily publishable. After browsing the GOTree, the user may pick appropriate annotation levels and get corresponding bar charts for publication using the Bar Chart button (for an example, see http://genereg.ornl.gov/gotm/paper/testis_bar.png). GOTree can also be exported and locally stored in html format using the Export GOTree button. Enriched GO categories are colored red, and genes in each category are also included in the exported GOTree (for an example, see http://genereg.ornl.gov/gotm/paper/testis_output.html).
{kind=link}
Figure 3
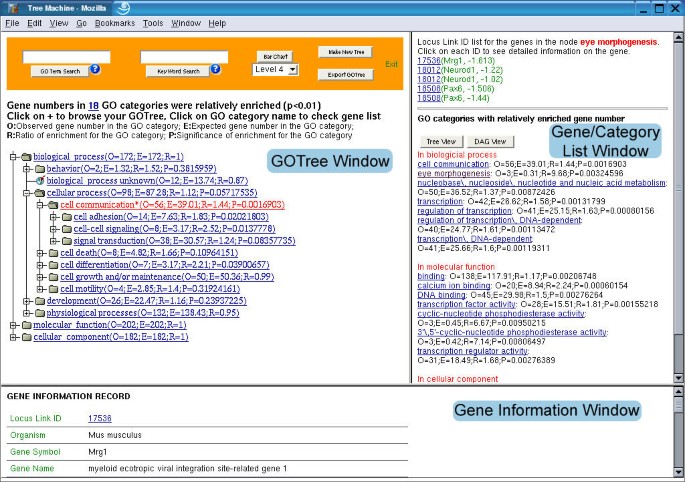
Output user interface of the GOTM The GOTree window displays the expandable tree structure of the GO categories. Each GO category is followed by three parameters: O (Observed gene number in the category); E (Expected gene number in the category) and R (Ratio of enrichment for the category). The fourth parameter P (p value calculated from the hypergeometric test) is given for the categories with R > 1 to indicate the significance of enrichment. Categories with P < 0.01 are colored red. The gene/category list window displays genes in selected GO categories ("eye morphogenesis" in this case) and the names of enriched GO categories followed by the parameters O, E, R and P. The genes are represented by LocusIDs followed by gene symbols and ratios in the microarray experiment. The gene information window displays the gene information record for the selected gene.
The gene/category list window shows the genes in a selected GO category, and enriched GO categories in the three main GO categories, biological process, molecular function and cellular component respectively. Each gene is represented by a LocusID, followed by the input ID. In addition, the ratio in the microarray experiment is shown if that information was included in the input file. Up-regulated genes are colored red while down-regulated genes are colored green. A flat view of enriched GO categories doesn't reveal the relationship among the GO categories. When tens or hundreds of GO categories are identified as significantly enriched, it becomes difficult for users to interpret the results. In this case, the user can press the TREE VIEW button to get a sub-tree (Figure 4) or press the DAG VIEW button to get a DAG (Figure 5) for the enriched GO categories in a new window. The GO categories in red in the sub-tree or the DAG are the enriched GO categories while the black ones are their non-enriched parents. The sub-tree and the DAG assemble related enriched GO categories together indicating important biological areas that are worth further study. By clicking on individual LocusIDs in the gene/category list window, related information for the genes will be queried from GeneKeyDB and shown in the gene information window.
Figure 4
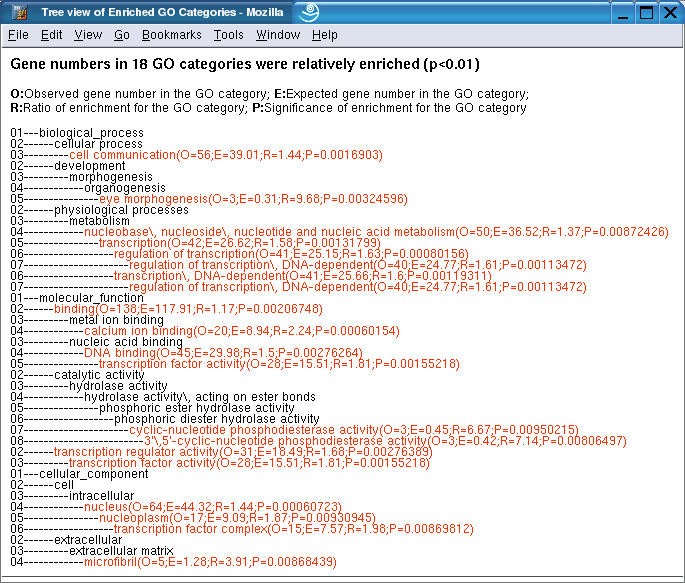
Sub-tree view of enriched GO categories The enriched GO categories are brought together and visualized as a sub-tree. Categories in red are enriched ones while those in black are non-enriched parents. Enriched categories are followed by four parameters, O (Observed gene number in the category); E (Expected gene number in the category), R (Ratio of enrichment for the category) and P (p value calculated from the hypergeometric test). Numbers at the left indicate the Gene Ontology annotation level.
Figure 5
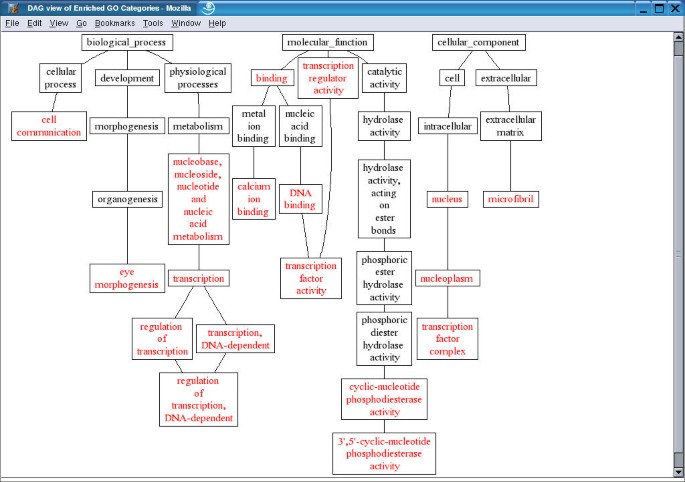
DAG view of enriched GO categories The enriched GO categories are brought together and visualized as a Directed Acyclic Graph (DAG). Categories in red are enriched ones while those in black are non-enriched parents. The list of genes in each category can be retrieved by click on the name of the categories.
The gene information window shows the gene information record for the selected gene, which includes LocusLink ID, organism, gene symbol, gene name, map location, homolog, GO terms, function summary, GRIF (Gene Reference Into Function), phenotype, PubMed record, OMIM (Online Mendelian Inheritance in Man) record, KEGG (Kyoto Encyclopaedia of Genes and Genomes) Map etc. A link is given to external databases such as PubMed, OMIM, KEGG etc when available.
Application
High-throughput gene expression profiling has become an important tool for investigating transcriptional activity in a variety of biological samples. Data from the published, large-scale expression analysis of Su et al is used here to illustrate the use of this tool [18]. They profiled gene expression from 91 human and mouse samples across a diverse array of tissues, organs, and cell lines and showed a preliminary description of the normal mammalian transcriptome. 311 human and 155 mouse tissue-restricted genes with known function were identified by examining gene expression across a panel of tissues. These genes were hypothesized to perform specific cellular and physiological functions in each tissue. Among the 85 human genes restricted to the testis, the authors only mentioned three genes which were known to be involved in testis function (SOX5, TEKT2 and ZPBP). It would be interesting to show the functional profiles and identify the important functional categories from the tissue restricted gene sets. To do this analysis, 85 human genes restricted to the testis and the 58 human genes restricted to the liver were downloaded from the supporting information on the PNAS web site for the paper. As the HG_U95A array was used for the experiment, all the genes on the HG_U95A array were used as the reference gene set. GOTM was used to identify GO categories with significantly enriched gene numbers (P < 0.01) in the testis gene set and the liver gene set. This analysis was carried out in August 2003 based on GeneKeyDB version GKDB200307.1. LocusLink data used for this version was downloaded on July 18, 2003 from NCBI. The versions for the GO files were 2.378, 2.747 and 2.857 for biological process, molecular function and cellular component respectively. GOTrees were generated for the two gene sets. GO annotation was found for 58, 53 and 50 genes respectively in the biological process, molecular function and cellular component categories for the testis gene set (see text output at http://genereg.ornl.gov/gotm/paper/testis_output.html), while 47, 48 and 39 respectively for the liver gene set http://genereg.ornl.gov/gotm/paper/liver_output.html. 59 and 79 enriched GO categories were identified in the testis and the liver gene set respectively. Examples can be seen from http://genereg.ornl.gov/gotm/paper/. These examples include bar charts under biological process (at the 4th level from the root), sub-trees, and DAGs of the enriched GO categories.
For the testis gene set, the statistics suggested 36 enriched GO categories in the biological process part of GO. As shown in the DAG http://genereg.ornl.gov/gotm/paper/testis_dag.png and the sub-tree http://genereg.ornl.gov/gotm/paper/testis_subtree.html, these GO categories comprise mainly four groups. The largest group of enriched GO categories includes those related to cell proliferation, cell cycle, mitosis and meiosis. The gametogenic function of the testis is to produce the male gametes or spermatozoa. Formation of the male gamete occurs in sequential mitotic, meiotic and postmeiotic phases. As reviewed by Eddy et al, many germ cell-specific transcripts are produced during this process [19]. The second group contains GO categories that are related to testis specific development, such as sex differentiation and reproduction. The third group of GO categories are those related to protein phosphorylation. Spermatozoa undergo a series of changes before and during egg binding to acquire the ability to fuse with the oocyte. These priming events are regulated by the activation of compartmentalized intracellular signalling pathways, which control the phosphorylation status of sperm proteins. Increased protein tyrosine phosphorylation is associated with capacitation, hyperactivated motility, zona pellucida binding, acrosome reaction and sperm-oocyte binding and fusion [20]. The fourth group consists of GO categories related to glycerolipid metabolism. Some glycerolipids were reported to be responsible for the unique fusogenic potential of sperm plasma membrane domains [21, 22].
{kind=link}
For the liver gene set, there are 35 enriched GO categories in the biological process part of GO. As shown in the DAG http://genereg.ornl.gov/gotm/paper/liver_dag.png and the sub-tree http://genereg.ornl.gov/gotm/paper/liver_subtree.html, there are mainly two groups of enriched GO categories. One group includes those related to different kinds of metabolism, which is consistent with the key role of liver in the metabolism. The other group includes those related to response to external stimuli and stress, which may be consistent with the roles that liver cells play in response to a variety of physiological states (e.g. production of acute phase proteins [23]). The GO categories, homeostasis and blood coagulation, are also enriched, which may be consistent with the ability of liver to synthesize various protein molecules that are responsible for clotting of blood.
{kind=link}
These two examples demonstrate that besides organizing interesting gene sets using GO hierarchies, based on statistical analysis, GOTM can help transfer these expression profiles into functional profiles. This transformation may be useful in helping biologists interpret high-throughput data. GOTM was applied to interpret tissue restricted gene sets identified by microarray experiment in this paper. In fact, it can be applied to any other interesting gene sets.
Related software comparison
Several GO based functional profiling software packages have been published recently. A complete list of GO Tools can be found at http://www.geneontology.org/GO.tools.html. Zeeberg et al did an extensive comparison of some of these software packages [10]. Table 1 compares GOTM to related software for the main features. It is based on the information available from individual websites as of August, 2003 when the comparison was done. GoMiner and GoSurfer are standalone software packages while FatiGO, Onto-Express and GOTM are web-based software. Different kinds of IDs have been used as input identifiers. The LocusLink Database is one of the most comprehensive resources for gene related information. Using LocusID as the primary identifier enables GOTM to access the abundant gene information resources in LocusLink database. GOSurfer is the only one among the others that includes LocusID as an input identifier. Gene symbol, Unigene ID, Swiss-Prot ID, Ensembl ID, and Affymetrix probe set IDs can also be used in GOTM owing to their broad adoption by end-users. FatiGO is also very flexible in the input identifiers. FatiGo, however, requires the user to specify ahead of time one particular level of the GO hierarchy that is to be used for analysis of the data. Although Onto-Express allows multilevel analysis, the classification information is presented in bar charts and flat view tables. Both of these web-based software packages do not, in our opinion, visualize well the fundamental hierarchical nature of GO. GO was originally organized in DAG, thus GoMiner's use of a DAG as the visual output format seems appropriate; however, visualization becomes difficult when the gene set is significantly large. The same visualization problem exists for the fixed tree as used in GOSurfer. The expandable tree in GOTM and GOMiner is very similar to the widely used GO browser, AmiGO [[24](/article/10.1186/1471-2105-5-16#ref-CR24 " AmiGO[ http://www.godatabase.org/cgi-bin/go.cgi/
]")\], and is suitable for the visualization of the GOTree structure. All of the software packages provide statistical analysis for identifying important GO categories. GOTM uses the hypergeometric test for assessing significance of enrichment. Since repeated tests are conducted to determine the significantly enriched GO categories, a correction for multiple tests is necessary. FatiGO and the commercial version of Onto-Express have implemented the correction. However, as stated on the webpage of FatiGO, the cost for the correction is the slow speed. This slowness is not desirable for a web based service. Correction for multiple tests is not implemented in GOTM. As a result, the _P_ values can be considered as a relative measure for indicating possible statistical significance. It is not very difficult for an experienced biologist to identify truly interesting areas from the enriched GO categories given by GOTM. Moreover, in GOTM, the unique visualization of the enriched GO categories as sub-trees or DAGs (Figure [4](/article/10.1186/1471-2105-5-16#Fig4), [5](/article/10.1186/1471-2105-5-16#Fig5)) brings functionally related GO categories together, which can guide users to find interesting biological areas. Although there are usually tens of enriched GO categories, the sub-tree or DAG of enriched GO categories actually focuses on several biological areas. In contrast, tables and bar charts of enriched GO categories in FatiGO and Onto-Express can't reveal such information. GOSurfer and GoMiner highlight the enriched GO categories in the whole GOTree or DAG. Owing to the complex structure of the GO hierarchy, they may not be as intuitive as the visualization of sub-tree or DAG of enriched GO categories in GOTM.Table 1 Comparison of GOTM with related software*
Conclusions
As a web-based platform for interpreting sets of interesting genes using GO hierarchies, GOTM provides user friendly data visualization and statistical analysis for comparing gene sets. GOTM complements and extends the functionality of similar data mining tools. Statistical analysis helps users to identify the most important GO categories for the gene sets of interest and suggests biological areas that warrant further study. GOTM should have a broad application in functional genomic, proteomic and large scale genetic studies from which high-throughput data are continuously generated. The application of GOTM is limited by the number of genes that have GO annotation. However, with the bioinformatics effort in automatic prediction of protein functions based on literature, gene expression data and protein sequence information [25–29], rapid growth in GO is expected, and GOTM will become more useful with the improvement of GO.
Availability and requirements
Project Name: GOTM (GOTree Machine)
Project Homepage: http://genereg.ornl.gov/gotm/
Operating System: Platform independent
Programming Language: PHP
Other Requirements: IE5.0 or higher, or Netscape 7 or higher
License: GNU GPL
Any Restrictions to use by non-academics: License needed
References
- Wain HM, Bruford EA, Lovering RC, Lush MJ, Wright MW, Povey S: Guidelines for Human Gene Nomenclature. Genomics 2002, 79: 464–470. 10.1006/geno.2002.6748
Article CAS PubMed Google Scholar - The Gene Ontology Consortium: Creating the Gene Ontology resource: design and implementation. Genome Res 2001, 11: 1425–1433. 10.1101/gr.180801
Article Google Scholar - Jenssen T-K, Legreid A, Komorowski J, Hovig E: A literature network of human genes for high-throughput analysis of gene expression. Nat Genet 2001, 28: 21–28. 10.1038/88213
CAS PubMed Google Scholar - Masys DR, Welsh JB, Fink JL, Gribskov M, Klacansky I, Corbeil J: Use of keyword hierarchies to interpret gene expression patterns. Bioinformatics 2001, 17: 319–326. 10.1093/bioinformatics/17.4.319
Article CAS PubMed Google Scholar - Chaussabel D, Sher A: Mining microarray expression data by literature profiling. Genome Biol 2002, 3: R55. 10.1186/gb-2002-3-10-research0055
Article Google Scholar - The Gene Ontology Consortium: Gene Ontology: tool for the unification of biology. Nat Genet 2000, 25: 25–29. 10.1038/75556
Article PubMed Central Google Scholar - Herrero J, Al-Shahrour F, Diaz-Uriarte R, Mateos A, Vaquerizas JM, Santoyo J, Dopazo J: GEPAS: A web-based resource for microarray gene expression data analysis. Nucleic Acids Res 2003, 31: 3461–3467. 10.1093/nar/gkg591
Article PubMed Central CAS PubMed Google Scholar - Draghici S, Khatri P, Martins RP, Oscategoryeier GC, Krawetz SA: Global functional profiling of gene expression. Genomics 2003, 81: 98–104. 10.1016/S0888-7543(02)00021-6
Article CAS PubMed Google Scholar - Khatri P, Draghici S, Ostermeier GC, Krawetz SA: Profiling gene expression using Onto-Express. Genomics 2002, 79: 266–270. 10.1006/geno.2002.6698
Article CAS PubMed Google Scholar - Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN: GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol 2003, 4: R28. 10.1186/gb-2003-4-4-r28
Article PubMed Central PubMed Google Scholar - Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR: MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol 2003, 4: R7. 10.1186/gb-2003-4-1-r7
Article PubMed Central PubMed Google Scholar - GoSurfer[http://biosun1.harvard.edu/complab/gosurfer/]
- PHP Layers Menu System[http://phplayersmenu.sourceforge.net/]
- ChartDirector[http://www.advsofteng.com/index.html]
- Graphviz[http://www.research.att.com/sw/tools/graphviz/]
- NCBI LocusLink database[http://www.ncbi.nlm.nih.gov/LocusLink/]
- Gene Ontolgy consortium[http://www.geneontology.org]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB: Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 2002, 99: 4465–4470. 10.1073/pnas.012025199
Article PubMed Central CAS PubMed Google Scholar - Eddy EM: Male germ cell gene expression. Recent Prog Horm Res 2002, 57: 103–28. 10.1210/rp.57.1.103
Article CAS PubMed Google Scholar - Urner F, Sakkas D: Protein phosphorylation in mammalian spermatozoa. Reproduction 2003, 125: 17–26. 10.1530/reprod/125.1.17
Article CAS PubMed Google Scholar - Nolan JP, Hammerstedt RH: Regulation of membrane stability and the acrosome reaction in mammalian sperm. FASEB J 1997, 11: 670–682.
CAS PubMed Google Scholar - Reisse S, Rothardt G, Völkl A, Beier K: Peroxisomes and ether lipid biosynthesis in rat testis and epididymis. Biol Reprod 2001, 64: 1689–1694.
Article CAS PubMed Google Scholar - Kmiec Z: Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol 2001, 161: 1–151.
Article Google Scholar - AmiGO[http://www.godatabase.org/cgi-bin/go.cgi/]
- Blaschke C, Valencia A: Automatic classification of protein functions from the literature. Compar Funct Genom 2003, 4: 75–79. 10.1002/cfg.241
Article Google Scholar - Raychaudhuri S, Chang JT, Sutphin PD, Altman RB: Associating genes with gene ontology codes using a maximum entropy analysis of biomedical literature. Genome Res 2002, 12: 203–214. 10.1101/gr.199701
Article PubMed Central CAS PubMed Google Scholar - Lagreid A, Hvidsten TR, Midelfart H, Komorowski J, Sandvik AK: Predicting Gene Ontology biological process from temporal gene expression patterns. Genome Res 2003, 13: 965–979. 10.1101/gr.1144503
Article PubMed Google Scholar - Hvidsten TR, Komorowski J, Sandvik AK, Laegreid A: Predicting gene function from gene expressions and ontologies. Pac Symp Biocomput 2001, 299–310.
Google Scholar - Schug J, Diskin S, Mazzarelli J, Brunk BP, Stoeckert CJ: Predicting Gene Ontology functions from ProDom and CDD protein domains. Genome Res 2002, 2: 648–655. 10.1101/gr.222902
Article Google Scholar