Inborn errors in the metabolism of glutathione (original) (raw)
Abstract
Glutathione is a tripeptide composed of glutamate, cysteine and glycine. Glutathione is present in millimolar concentrations in most mammalian cells and it is involved in several fundamental biological functions, including free radical scavenging, detoxification of xenobiotics and carcinogens, redox reactions, biosynthesis of DNA, proteins and leukotrienes, as well as neurotransmission/neuromodulation. Glutathione is metabolised via the gamma-glutamyl cycle, which is catalyzed by six enzymes. In man, hereditary deficiencies have been found in five of the six enzymes. Glutathione synthetase deficiency is the most frequently recognized disorder and, in its severe form, it is associated with hemolytic anemia, metabolic acidosis, 5-oxoprolinuria, central nervous system (CNS) damage and recurrent bacterial infections. Gamma-glutamylcysteine synthetase deficiency is also associated with hemolytic anemia, and some patients with this disorder show defects of neuromuscular function and generalized aminoaciduria. Gamma-glutamyl transpeptidase deficiency has been found in patients with CNS involvement and glutathionuria. 5-Oxoprolinase deficiency is associated with 5-oxoprolinuria but without a clear association with other symptoms. Dipeptidase deficiency has been described in one patient. All disorders are very rare and inherited in an autosomal recessive manner. Most of the mutations are leaky so that many patients have residual enzyme activity. Diagnosis is made by measuring the concentration of different metabolites in the gamma-glutamyl cycle, enzyme activity and in glutathione synthetase and gamma-glutamylcysteine synthetase deficiency, also by mutation analysis. Prenatal diagnosis has been preformed in glutathione synthetase deficiency. The prognosis is difficult to predict, as few patients are known, but seems to vary significantly between different patients. The aims of the treatment of glutathione synthesis defects are to avoid hemolytic crises and to increase the defense against reactive oxygen species. No treatment has been recommended for gamma-glutamyl transpeptidase, 5-oxoprolinase and dipeptidase deficiency.
Similar content being viewed by others



Amino acid synthesis deficiencies
Article Open access 26 June 2017
A. Gamma-glutamylcysteine synthetase deficiency
Disease name and synonyms
Gamma-glutamylcysteine synthetase deficiency (OMIM #230450)
Glutamate-cysteine ligase deficiency
Definition and diagnostic criteria
Gamma-glutamylcysteine synthetase deficiency is a very rare autosomal recessive disease characterized by hemolytic anemia, and, in some cases, by neurological symptoms. The diagnosis is established by:
• Low activity of gamma-glutamylcysteine synthetase in red blood cells, leukocytes and/or cultured skin fibroblasts.
• Low levels of glutathione and gamma-glutamylcysteine in red blood cells and/or cultured skin fibroblasts.
• Presence of mutation(s) in the gamma-glutamylcysteine synthetase genes. The patients whose results have been published have had homozygous mutations in the gene encoding the heavy subunit of the enzyme.
In red blood cells heterozygous carriers have an enzyme activity of about 50% of the normal mean and normal levels of glutathione [1].
Epidemiology
Gamma-glutamylcysteine synthetase deficiency is very rare disease. Nine patients in seven families have been reported worldwide (USA, Germany, Japan, The Netherlands, Poland, and Spain).
Clinical description
All patients with gamma-glutamylcysteine synthetase deficiency have had hemolytic anemia, usually rather mild [1–8]. In addition, two siblings also had spinocerebellar degeneration, peripheral neuropathy, myopathy and aminoaciduria [3, 8]. Treatment with sulfonamide precipitated psychosis and pronounced hemolytic anemia in one of these siblings. One patient was reported to have learning disability with dyslexia and was also thought to be mentally retarded [5], and another had delayed psychomotor development and progressive sensory neuropathy of lower extremities, ataxia, hyperreflexia, dysarthria, and a peculiar gait suggestive of spinocerebellar degeneration [2]. Other symptoms found in patients with γ-glutamylcysteine synthetase deficiency are transient jaundice, reticulocytosis, and hepatosplenomegaly.
Etiology
Gamma-glutamylcysteine synthetase catalyzes the first and rate-limiting step in the synthesis of glutathione (GSH) (Figure 1) [9]. Hereditary gamma-glutamylcysteine synthetase deficiency is transmitted as an autosomal recessive trait. The human gamma-glutamylcysteine synthetase enzyme is a dimer consisting of a heavy (catalytic) and a light (regulatory) subunit. The human gene for the heavy subunit has been localized to chromosome 6p12 [10] and the gene for the light subunit to chromosome 1p21 [11]. The heavy subunit (molecular weight about 73 kDa) exhibits the catalytic activity of the native enzyme and is also responsible for the feedback inhibition by GSH. The light subunit (molecular weight about 28 kDa) is catalytically inactive but plays an important regulatory role [12].
Figure 1
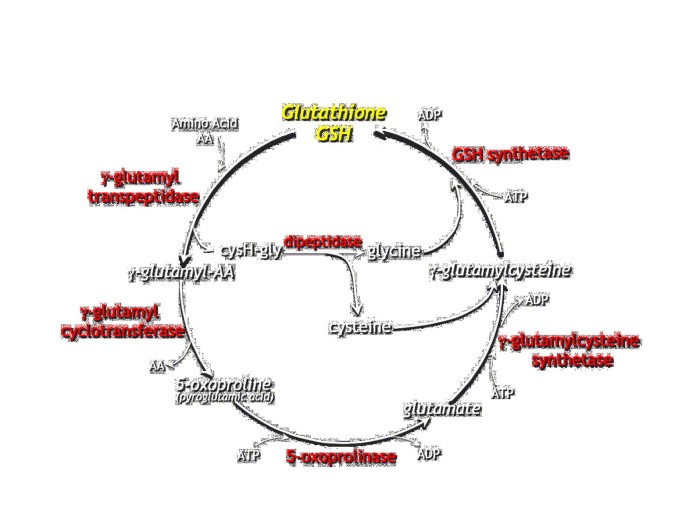
The gamma-glutamyl cycle.
Four different mutations in the heavy subunit have been identified in four families affected by gamma-glutamylcysteine synthetase deficiency [1, 2, 4, 5].
Gammaglutamylcysteine synthetase knock-out mice have been developed for both the heavy and the light subunits [13–15]. Homozygous mice embryos with knockout of the heavy subunit fail to gastrulate and die before day 8.5 of gestation [13]. The knockout mice of the light subunit are viable and fertile and have no overt phenotype [15].
Diagnostic methods
Glutathione and sulphydryl compunds in erythrocytes can be measured using 5,5'-dithiobis (2-nitrobenzoic acid), while glutathione in fibroblasts can be measured with the 5,5'-dithiobis (2-nitrobenzoic acid) glutathione recycling assay [16], or using high performance liquid chromatography (HPLC) [17].
Gamma-glutamylcysteine synthetase in erythrocytes and/or cultured fibroblasts can be measured as described elsewhere [1].
Sequence analysis can be made by polymerase chain reaction (PCR) and sequence analysis of the γ-glutamylcysteine synthetase genes (GLCLC, GLCLR) [1].
Differential diagnosis
Low levels of GSH can also be due to glutathione synthetase deficiency.
Genetic counseling
Families with gamma-glutamylcysteine synthetase deficiency can be tested in order to analyze the enzyme activity as well as their mutations. Limited mutation analysis data are available but it is essential to determine the underlying mutations to learn more about the functional properties of the mutant enzyme. Families should also be referred for genetic counseling.
Antenatal diagnosis
Antenatal diagnosis has not been reported. However, it should be possible if needed by measurement of gamma-glutamylcysteine synthetase activity or mutational analysis (if the mutation in the family is known) of chorionic villi or cultured amniocytes.
Management including treatment
Patients with gamma-glutamylcysteine synthetase deficiency should avoid drugs known to precipitate hemolytic crises in patients with glucose-6-phosphate dehydrogenase deficiency, e.g. phenobarbital, acetylsalicylic acid, sulfonamides. It is possible that patients would benefit from treatment with anti-oxidants but no studies have been made.
B. Glutathione synthetase deficiency
Disease name and synonyms
Glutathione synthetase deficiency (OMIM #266130).
5-oxoprolinuria (pyroglutamic aciduria) is sometimes used when referring to glutathione synthetase deficiency; it should be borne in mind however that 5-oxoprolinuria may have other causes (see 'Differential diagnosis').
Definition and diagnostic criteria
Hereditary glutathione synthetase deficiency is a rare autosomal recessive disease characterized by hemolytic anemia, metabolic acidosis, 5-oxoprolinuria, progressive neurological symptoms and recurrent bacterial infections. The diagnosis usually involves the following: clinical findings, the finding of 5-oxoprolinuria, low levels of glutathione, low activity of glutathione synthetase, and mutation analysis of the glutathione synthetase gene. The diagnosis of glutathione synthetase deficiency is established by:
• Low activity of glutathione synthetase in cultured skin fibroblasts and/or red blood cells.
• Low levels of glutathione in red blood cells and/or cultured skin fibroblasts.
• Urinary 5-oxoproline (up to 1 g/kg/day)
• Mutation(s) in the glutathione synthetase (GSS) gene.
Heterozygous carriers have an enzyme activity of about 55% of the normal mean and normal levels of glutathione [18].
Epidemiology
More than seventy patients have been reported in more than 50 families worldwide.
Clinical description
According to the clinical symptoms, glutathione synthetase deficiency can be classified as mild, moderate or severe [19]:
Mild glutathione synthetase deficiency
These patients show mild hemolytic anemia as their only clinical symptom. In very rare cases, they may excrete excessive amounts of 5-oxoproline in their urine (reference range < 0.1 mol/mol creatinine) but they usually maintain sufficient cellular levels of glutathione to prevent accumulation of 5-oxoproline in body fluids.
Moderate glutathione synthetase deficiency
Patients with the moderate variant usually present in the neonatal period with severe and chronic metabolic acidosis, 5-oxoprolinuria, and mild/moderate hemolytic anemia.
Severe glutathione synthetase deficiency
Patients have symptoms as in moderate glutathione synthetase deficiency and, in addition, they develop progressive neurological symptoms, e.g. psychomotor retardation, mental retardation, seizures, spasticity, ataxia, and intention tremor. Some patients with severe glutathione synthetase deficiency also develop recurrent bacterial infections, probably due to defective granulocyte function. Retinal dystrophy has also been observed in adult patients[20].
Several patients died in early life.
Etiology
Glutathione synthetase catalyses the last step in the synthesis of glutathione and a deficiency results in low levels of glutathione (Figure 1). Acidosis is due to reduced feedback inhibition of γ-glutamyl cysteine synthetase in the gamma-glutamyl cycle, which ultimately leads to overproduction and accumulation of 5-oxoproline. The human glutathione synthetase enzyme is a homodimer with a subunit size of 52 kDa. The three-dimensional structure of the glutathione synthetase enzyme has been established [21]. The gene has been localized to chromosome 20q11.2 and its structure determined [22, 23]. Since the human genome contains only one glutathione synthetase gene, the various clinical forms of glutathione synthetase deficiency reflect different mutations or epigenetic modifications in the glutathione synthetase gene. Several mutations have been identified.
The mechanism of metabolic acidosis and 5-oxoprolinuria is the following: decreased levels of cellular glutathione lead to decreased feed-back inhibition of γ-glutamylcysteine synthetase. This results in excessive formation of the dipeptide γ-glutamylcysteine, which is converted by γ-glutamyl cyclotransferase into 5-oxoproline. The overproduction of 5-oxoproline exceeds the capacity of 5-oxoprolinase, and 5-oxoproline therefore accumulates in body fluids and is excreted in the urine.
Patients with glutathione synthetase deficiency accumulate the dipeptide γ-glutamylcysteine and cysteine in fibroblasts [24]. As γ-glutamylcysteine contains both reactive groups of glutathione (i.e. the sulphydryl and γ-glutamyl groups) it may to some extent compensate for glutathione in the cellular defence against oxidative stress.
Glutathione is participating in leukotriene C4 (LTC4) synthesis, the primary cysteinyl leukotriene. It has been shown that the synthesis of cysteinyl leukotriens is impaired in patients with glutathione synthetase deficiency [25].
Diagnostic methods
Urinary 5-oxoproline can be determined by gas chromatography-mass spectrometry [26]. Glutathione and sulphydryls in erythrocytes can be measured using 5,5'-dithiobis (2-nitrobenzoic acid), while glutathione in fibroblasts can be measured with the 5,5'-dithiobis (2-nitrobenzoic acid) glutathione recycling assay [16], or using HPLC [17].
Glutathione synthetase in erythrocytes and/or cultured fibroblasts can be measured as described elsewhere [1].
Mutation analysis of the glutathione synthetase gene (GSS) can be made as described by Shi et al. 1996 and Njålsson et al. 2003 [27, 28].
Differential diagnosis
For other causes of 5-oxoprolinuria, beside glutathione synthetase deficiency and 5-oxoprolinase deficiency [9, 29], please see Table 1.
Table 1 Causes of 5-oxoprolinuria beside glutathione synthetase deficiency and 5-oxoprolinase deficiency.
Genetic counseling
Families with glutathione synthetase deficiency can be referred for testing in order to analyze the enzyme activity as well as their mutations. It is essential to find out whether it is possible to correlate the genotype with the phenotype.
Antenatal diagnosis
Antenatal diagnosis is possible and can be made by:
• Mutation analysis of chorionic villi (the method of choice if the mutation in the family is known).
• Analysis of 5-oxoproline in amniotic fluid [30, 31].
• Analysis of glutathione synthetase activity in cultured amniocytes or chorionic villi [30].
Management including treatment
The clinical goals of management are correction of acidosis and early supplementation with vitamin C (ascorbic acid) and vitamin E (alpha-tocopherol) [19]. The acidosis is corrected with bicarbonate. The recommended dose of vitamin C is 100 mg/kg/day and of vitamin E 10 mg/kg/day. N-Acetylcysteine used to be recommended because it may protect cells from oxidative stress. However, cysteine has been shown to accumulate in tissues of patients with glutathione synthetase deficiency – at least in cultured fibroblasts [24]. Since cysteine is known to be neurotoxic in excessive amounts [32], treatment with N-acetylcysteine should not be recommended for patients with glutathione synthetase deficiency as this may increase the intracellular cysteine levels even more.
Patients with glutathione synthetase deficiency should avoid drugs known to precipitate hemolytic crises in patients with glucose-6-phosphate dehydrogenase deficiency.
Prognosis
A long-term follow up study of 28 patients with glutathione synthetase deficiency has showed that the factors most predictive of survival and long-term outcome are early diagnosis, correction of acidosis and early supplementation with vitamin C and vitamin E [19].
C. Gamma-glutamyl transpeptidase deficiency
Disease name
Gamma-glutamyl transpeptidase deficiency (OMIM 231950).
Definition and diagnostic criteria
Gamma-glutamyl transpeptidase deficiency is a very rare autosomal recessive disease characterized by increased glutathione concentration in plasma and urine. Central nervous system involvement may also be present. The diagnosis is established by:
• Low activity of γ-glutamyl transpeptidase in nucleated cells such as leukocytes or cultured skin fibroblasts. Erythrocytes also lack γ-glutamyl transpeptidase under normal conditions.
• High levels of glutathione in plasma and urine (up to 1 g/day in urine; controls < 10 mg). Cellular levels of glutathione are normal.
Epidemiology
Gamma-glutamyl transpeptidase deficiency is a very rare disease, which has been reported in seven patients in five families worldwide [33–37].
Clinical description
Five out of seven reported patients had central nervous system involvement [33–37]. Whether these symptoms are part of the clinical picture remains to be established. All patients have had glutathionuria (up to 1 g/day; controls < 10 mg). In addition, patients have increased urinary levels of gamma-glutamylcysteine and cysteine. Three patients with γ-glutamyl transpeptidase deficiency have been studied and found to have a complete deficiency of leukotriene D4 biosynthetsis [38].
Etiology
The human gamma-glutamyl transpeptidase gene family is composed of at least seven different gene loci and several of them are located on the long arm of chromosome 22 [9]. Gamma-glutamyl transpeptidase is a heterodimer with subunits of 21 kDa and 38 kDa. The enzyme is membrane-bound with its active site facing the external side of the cell. Erythrocytes lack gamma-glutamyl transpeptidase and this enzyme activity also varies in other tissues. Gamma-glutamyl transpeptidase catalyses the first step in the degradation of glutathione (Figure 1). No mutations have been identified in patients with gamma-glutamyl transpeptidase deficiency. A knock-out mouse for γ-glutamyl transpeptidase has been developed. Gamma-glutamyl transpeptidase deficiency leads to glutathionuria, glutathionemia, growth failure, cataracts, lethargy, shortened life span, and infertility. A closer study of the reproductive phenotype of γ-glutamyl transpeptidase deficient mice show that they are hypogonadal and infertile [39].
Diagnostic methods
Glutathione in plasma and urine can be determined by various chromatographic or calorimetric techniques.
Gamma-glutamyl transpeptidase in nucleated cells can be determined using the method described by Wright et al. [36].
Genetic counseling
The disease is transmitted as an autosomal recessive trait. Patients should be offered genetic counseling.
Antenatal diagnosis
Antenatal diagnosis has not been reported.
Management including treatment
No specific treatment has been proposed or tried. However, administration of N-acetylcysteine to γ-glutamyl transpeptidase deficient mutant mice for two weeks restored their fertility [39].
D. 5-Oxoprolinase deficiency
Disease name
5-Oxoprolinase deficiency (OMIM 260005)
Definition and diagnostic criteria
5-Oxoprolinase deficiency is a very rare autosomal recessive disease characterized by 5-oxoprolinuria and very heterogeneous clinical presentation (renal stone formation, enterocolitis, mental retardation, neonatal hypoglycemia, microcytic anemia and microcephaly).
The diagnosis of 5-oxoprolinase deficiency is established by:
• Low activity of 5-oxoprolinase in nucleated cells such as leukocytes or cultured skin fibroblasts (5-oxoprolinase is not present in erythrocytes).
• Elevated levels of 5-oxoproline in body fluids.
• Urinary 5-oxoproline.
Epidemiology
Eight patients have been reported worldwide.
Clinical description
All patients with 5-oxoprolinase deficiency have been identified because of 5-oxoprolinuria (4 to 10 g/day. Reference range < 0.1 mol/mol creatinine), but they lack a consistent clinical picture [9]. They have normal acid-base balance. Different clinical symptoms reported in individual patients with 5-oxoprolinase deficiency are renal stone formation, enterocolitis, mental retardation, neonatal hypoglycemia, microcytic anemia and microcephaly [40–45].
Etiology
5-Oxoprolinase catalyses a step in the gamma-glutamyl cycle (glutathione metabolism), the ring-opening of 5-oxoproline to yield glutamate. 5-Oxoprolinase is the enzyme in the gamma-glutamyl cycle with the lowest capacity (Figure 1).
The mammalian enzyme is not well studied, but it is apparently composed of two identical subunits. The mechanism leading to 5-oxoprolinuria is the following: decreased activity of 5-oxoprolinase leads to decreased conversion of 5-oxoproline to glutamate. Therefore, 5-oxoproline accumulates in body fluids and is excreted in the urine. The quantities are less than those found in patients with glutathione synthetase deficiency and therefore acid-base balance is usually normal.
Diagnostic methods
The level of 5-oxoproline in urine can be determined by gas chromatography-mass spectrometry [26]. Activity of 5-oxoprolinase in leukocytes and/or cultured fibroblasts can be measured according to the method described by Larsson et al. [46].
Differential diagnosis
For other causes of 5-oxoprolinuria, besides 5-oxoprolinase deficiency and glutathione synthetase deficiency [9, 29], please see Table 1.
Genetic counseling
Families with 5-oxoprolinase deficiency can be tested in order to analyze the enzyme activity. They should be referred for genetic counseling.
Antenatal diagnosis
Antenatal diagnosis of 5-oxoprolinase deficiency has not been reported to date.
Management including treatment
No specific treatment has been proposed or tried.
E. Dipeptidase deficiency
Disease name and synonyms
Dipeptidase deficiency
Cysteinylglycinuria
Cysteineglycinas deficiency
Definition and diagnostic criteria
Dipeptidase deficiency is an extremely rare diseases autosomal recessive manner, characterized by increased urinary excretion of cysteinylglycine and a pathological excretion pattern of leukotriens. The diagnosis is based on the finding of cysteinylglycinuria and decreased activity of dipeptidase, and is established by:
• Increased urinary excretion of cysteinyl glycine
• Normal concentration of cysteinyl glycine in plasma
• Low activity of dipeptidase in cultured skin fibroblasts and/or red blood cells.
Epidemiology
Dipeptidase deficiency has been suggested in only one patient worldwide. The diagnosis has not been confirmed by enzyme analysis in this patient.
Clinical description
One patient with a suspected membrane-bound dipeptidase deficiency has been described [47]. This was a 15-year-old boy who presented with mental retardation, mild motor impairment, and partial deafness. Biochemical investigations showed a normal level of cysteinyl glycine in plasma, an abnormal urinary profile with increased excretion of cysteinyl glycine (4972 mmol/mol of creatinine) and leukotrienes with increased leukotriene D4 and complete absence of LTE4. The concentration of leukotriene D(4) (LTD(4)), which is usually not detectable, was highly increased, whereas LTE(4), the major urinary metabolite in humans, was completely absent. These data suggest membrane-bound dipeptidase deficiency [47].
Etiology
Membrane-bound dipeptidase (E.C. 3.4.13.19) is the enzyme that hydrolyzes dipeptides, including cysteinylglycine compounds, such as the oxidized γ-glutamyltranspeptidase product cystinyl-bis-glycine and the conversion of leukotriene D4 to E4 [48]. Dipeptidase also hydrolyzes certain β-lactam antibiotics. The protein is 42 kDa unglycosylated and 63 kDa when glycosylated. The crystal structure of human membrane-bound dipeptidase has been reported [49]. Renal dipeptidase has been mapped to human chromosome 16 at q24 [50]. A deficiency of dipeptidase has been suspected in only one patient.
Diagnostic methods
The diagnosis is made by quantitative amino acid analysis of urine.
The activity of dipeptidase can be assayed with a HPLC method using glycyl-D-phenylalanine as described by Littlewood et al. 1989 [51]. To date, the corresponding enzyme deficiency has not been assayed in the patient with suspected dipeptidase deficiency.
Genetic counseling
Patients should be offered genetic counseling.
Antenatal diagnosis
Not relevant.
Management including treatment
No specific treatment has been proposed or tried.
Unresolved questions
• It remains to be established whether CNS symptoms reported in some patients with gamma-glutamylcysteine synthetase are related to the enzyme defect or not. Also, strategies for treatment need to be investigated.
• It remains to be established if free radicals are involved in the pathogenesis of glutathione synthetase deficiency and if patients with low levels of glutathione are more sensitive to oxidative stress.
• The relationship between genotype and phenotype in gamma-glutamyl transpeptidase deficiency remains to be established. Are the symptoms in the identified patients merely a coincidence?
• The relationship between genotype and phenotype in 5-oxoprolinase deficiency remains to be established. It is unknown whether symptoms in identified patients are merely a coincidence.
• In dipeptidase deficiency, it remains to be established if there is a relationship between the biochemical defect and the clinical symptoms.
• Prognosis of gamma-glutamylcysteine synthetase deficiency, gamma-glutamyl transpeptidase deficiency, 5-oxoprolinase deficiency and dipeptidase deficiency is unclear and difficult to predict as very few patients have been reported worldwide.
References
- Ristoff E, Augustson C, Geissler J, de Rijk T, Carlsson K, Luo JL, Andersson K, Weening RS, van Zwieten R, Larsson A, Roos D: A missense mutation in the heavy subunit of gamma-glutamylcysteine synthetase gene causes hemolytic anemia. Blood. 2000, 95 (7): 2193-2196.
CAS PubMed Google Scholar - Mañú Pereira M, Gelbart T, Ristoff E, Bergua JM, Kalko S, García Mateos E, Beutler E, J.L. VC: Chronic nonspherocytic haemolytic anaemia associated with severe neurological disease due to gamma-glutamylcysteine synthetase deficiency in a boy of Muslim origin. Manuscript. 2006
Google Scholar - Richards F, Cooper MR, Pearce LA, Cowan RJ, Spurr CL: Familial spinocerebellar degeneration, hemolytic anemia, and glutathione deficiency. Arch Intern Med. 1974, 134 (3): 534-537. 10.1001/archinte.134.3.534.
Article PubMed Google Scholar - Hamilton D, Wu JH, Alaoui-Jamali M, Batist G: A novel missense mutation in the gamma-glutamylcysteine synthetase catalytic subunit gene causes both decreased enzymatic activity and glutathione production. Blood. 2003, 102 (2): 725-730. 10.1182/blood-2002-11-3622.
Article CAS PubMed Google Scholar - Beutler E, Gelbart T, Kondo T, Matsunaga AT: The molecular basis of a case of gamma-glutamylcysteine synthetase deficiency. Blood. 1999, 94 (8): 2890-2894.
CAS PubMed Google Scholar - Hirono A, Iyori H, Sekine I, Ueyama J, Chiba H, Kanno H, Fujii H, Miwa S: Three cases of hereditary nonspherocytic hemolytic anemia associated with red blood cell glutathione deficiency. Blood. 1996, 87 (5): 2071-2074.
CAS PubMed Google Scholar - Beutler E, Moroose R, Kramer L, Gelbart T, Forman L: Gamma-glutamylcysteine synthetase deficiency and hemolytic anemia. Blood. 1990, 75 (1): 271-273.
CAS PubMed Google Scholar - Konrad PN, Richards F, Valentine WN, Paglia DE: Gamma-glutamyl-cysteine synthetase deficiency. A cause of hereditary hemolytic anemia. N Engl J Med. 1972, 286 (11): 557-561.
Article CAS PubMed Google Scholar - Larsson A, Ristoff E, Anderson ME: Glutathione synthetase deficiency and other disorders of the gamma-glutamyl cycle. The Metabolic and Molecular Bases of Inherited Disease. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, Childs B, Kinzler KW. New York , McGraw-Hill; 2005. Online. http://genetics.accessmedicine.com.
Google Scholar - Sierra-Rivera E, Summar ML, Dasouki M, Krishnamani MR, Phillips JA, Freeman ML: Assignment of the gene (GLCLC) that encodes the heavy subunit of gamma- glutamylcysteine synthetase to human chromosome 6. Cytogenet Cell Genet. 1995, 70 (3-4): 278-279.
Article CAS PubMed Google Scholar - Sierra-Rivera E, Dasouki M, Summar ML, Krishnamani MR, Meredith M, Rao PN, Phillips JA, Freeman ML: Assignment of the human gene (GLCLR) that encodes the regulatory subunit of gamma-glutamylcysteine synthetase to chromosome 1p21. Cytogenet Cell Genet. 1996, 72 (2-3): 252-254.
Article CAS PubMed Google Scholar - Huang CS, Chang LS, Anderson ME, Meister A: Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993, 268 (26): 19675-19680.
CAS PubMed Google Scholar - Shi ZZ, Osei-Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM, Lukin DJ, Danney CM, Matzuk MM, Lieberman MW: Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc Natl Acad Sci U S A. 2000, 97 (10): 5101-5106. 10.1073/pnas.97.10.5101.
Article PubMed Central CAS PubMed Google Scholar - Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW: Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000, 279 (2): 324-329. 10.1006/bbrc.2000.3930.
Article CAS PubMed Google Scholar - Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP: Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002, 277 (51): 49446-49452. 10.1074/jbc.M209372200.
Article CAS PubMed Google Scholar - Anderson ME: Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 1985, 113: 548-555.
Article CAS PubMed Google Scholar - Luo JL, Hammarqvist F, Andersson K, Wernerman J: Surgical trauma decreases glutathione synthetic capacity in human skeletal muscle tissue. Am J Physiol. 1998, 275 (2 Pt 1): E359-65.
CAS PubMed Google Scholar - Njalsson R, Ristoff E, Carlsson K, Winkler A, Larsson A, Norgren S: Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Hum Genet. 2005, 116 (5): 384-389. 10.1007/s00439-005-1255-6.
Article PubMed Google Scholar - Ristoff E, Mayatepek E, Larsson A: Long-term clinical outcome in patients with glutathione synthetase deficiency. J Pediatr. 2001, 139 (1): 79-84. 10.1067/mpd.2001.114480.
Article CAS PubMed Google Scholar - Ristoff E, Burstedt M, Larsson A, Wachtmeister L: Progressive retinal dystrophy in two sisters with glutathione synthetase (GS) deficiency. J Inherit Metab Dis. 2007, 30 (1): 102-10.1007/s10545-006-0412-y.
Article CAS PubMed Google Scholar - Polekhina G, Board PG, Gali RR, Rossjohn J, Parker MW: Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. Embo J. 1999, 18 (12): 3204-3213. 10.1093/emboj/18.12.3204.
Article PubMed Central CAS PubMed Google Scholar - Gali RR, Board PG: Sequencing and expression of a cDNA for human glutathione synthetase. Biochem J. 1995, 310 (Pt 1): 353-358.
Article PubMed Central CAS PubMed Google Scholar - Whitbread L, Gali RR, Board PG: The structure of the human glutathione synthetase gene. Chem Biol Interact. 1998, 111-112: 35-40. 10.1016/S0009-2797(97)00149-X.
Article CAS PubMed Google Scholar - Ristoff E, Hebert C, Njalsson R, Norgren S, Rooyackers O, Larsson A: Glutathione synthetase deficiency: is gamma-glutamylcysteine accumulation a way to cope with oxidative stress in cells with insufficient levels of glutathione?. J Inherit Metab Dis. 2002, 25 (7): 577-584. 10.1023/A:1022095324407.
Article CAS PubMed Google Scholar - Mayatepek E, Hoffmann GF, Carlsson B, Larsson A, Becker K: Impaired synthesis of lipoxygenase products in glutathione synthetase deficiency. Pediatr Res. 1994, 35 (3): 307-310.
Article CAS PubMed Google Scholar - Hoffmann G, Aramaki S, Blum-Hoffmann E, Nyhan WL, Sweetman L: Quantitative analysis for organic acids in biological samples: batch isolation followed by gas chromatographic-mass spectrometric analysis. Clin Chem. 1989, 35 (4): 587-595.
CAS PubMed Google Scholar - Njalsson R, Carlsson K, Winkler A, Larsson A, Norgren S: Diagnostics in patients with glutathione synthetase deficiency but without mutations in the exons of the GSS gene. Hum Mutat. 2003, 22 (6): 497-10.1002/humu.9199.
Article PubMed Google Scholar - Shi ZZ, Habib GM, Rhead WJ, Gahl WA, He X, Sazer S, Lieberman MW: Mutations in the glutathione synthetase gene cause 5-oxoprolinuria. Nat Genet. 1996, 14 (3): 361-365. 10.1038/ng1196-361.
Article CAS PubMed Google Scholar - Mayatepek E: 5-Oxoprolinuria in patients with and without defects in the gamma-glutamyl cycle. Eur J Pediatr. 1999, 158 (3): 221-225. 10.1007/s004310051054.
Article CAS PubMed Google Scholar - Erasmus E, Mienie LJ, de Vries WN, de Wet WJ, Carlsson B, Larsson A: Prenatal analysis in two suspected cases of glutathione synthetase deficiency. J Inherit Metab Dis. 1993, 16 (5): 837-843. 10.1007/BF00714275.
Article CAS PubMed Google Scholar - Manning NJ, Davies NP, Olpin SE, Carpenter KH, Smith MF, Pollitt RJ, Duncan SL, Larsson A, Carlsson B: Prenatal diagnosis of glutathione synthase deficiency. Prenat Diagn. 1994, 14 (6): 475-478. 10.1002/pd.1970140611.
Article CAS PubMed Google Scholar - Janaky R, Varga V, Hermann A, Saransaari P, Oja SS: Mechanisms of L-cysteine neurotoxicity. Neurochem Res. 2000, 25 (9-10): 1397-1405. 10.1023/A:1007616817499.
Article CAS PubMed Google Scholar - Goodman SI, Mace JW, Pollack S: Serum gamma-glutamyl transpeptidase deficiency. Lancet. 1971, 1 (7692): 234-235. 10.1016/S0140-6736(71)90970-6.
Article CAS PubMed Google Scholar - Hammond JW, Potter M, Wilcken B, Truscott R: Siblings with gamma-glutamyltransferase deficiency. J Inherit Metab Dis. 1995, 18 (1): 82-83. 10.1007/BF00711381.
Article CAS PubMed Google Scholar - O'Daley S: An abnormal sulphydryl compound in urine. Irish J Med Sci. 1968, 7: 578-579.
Google Scholar - Wright EC, Stern J, Ersser R, Patrick AD: Glutathionuria: gamma-glutamyl transpeptidase deficiency. J Inherit Metab Dis. 1980, 2 (1): 3-7. 10.1007/BF01805554.
Article CAS PubMed Google Scholar - Iida M, Yasuhara T, Mochizuki H, Takakura H, Yanagisawa T, Kubo H: Two Japanese brothers with hereditary gamma-glutamyl transpeptidase deficiency. J Inherit Metab Dis. 2005, 28 (1): 49-55. 10.1007/s10545-005-4417-8.
Article CAS PubMed Google Scholar - Mayatepek E, Okun JG, Meissner T, Assmann B, Hammond J, Zschocke J, Lehmann WD: Synthesis and metabolism of leukotrienes in gamma-glutamyl transpeptidase deficiency. J Lipid Res. 2004, 45 (5): 900-904. 10.1194/jlr.M300462-JLR200.
Article CAS PubMed Google Scholar - Kumar TR, Wiseman AL, Kala G, Kala SV, Matzuk MM, Lieberman MW: Reproductive defects in gamma-glutamyl transpeptidase-deficient mice. Endocrinology. 2000, 141 (11): 4270-4277. 10.1210/en.141.11.4270.
CAS PubMed Google Scholar - Schulman JD, Goodman SI, Mace JW, Patrick AD, Tietze F, Butler EJ: Glutathionuria: inborn error of metabolism due to tissue deficiency of gamma-glutamyl transpeptidase. Biochem Biophys Res Commun. 1975, 65 (1): 68-74. 10.1016/S0006-291X(75)80062-3.
Article CAS PubMed Google Scholar - Henderson MJ, Larsson A, Carlsson B, Dear PR: 5-Oxoprolinuria associated with 5-oxoprolinase deficiency; further evidence that this is a benign disorder. J Inherit Metab Dis. 1993, 16 (6): 1051-1052. 10.1007/BF00711529.
Article CAS PubMed Google Scholar - Roesel RA, Hommes FA, Samper L: Pyroglutamic aciduria (5-oxoprolinuria) without glutathione synthetase deficiency and with decreased pyroglutamate hydrolase activity. J Inherit Metab Dis. 1981, 4 (2): 89-90. 10.1007/BF02263605.
Article CAS PubMed Google Scholar - Bernier FP, Snyder FF, McLeod DR: Deficiency of 5-oxoprolinase in an 8-year-old with developmental delay. J Inherit Metab Dis. 1996, 19 (3): 367-368. 10.1007/BF01799269.
Article CAS PubMed Google Scholar - Cohen LH, Vamos E, Heinrichs C, Toppet M, Courtens W, Kumps A, Mardens Y, Carlsson B, Grillner L, Larsson A: Growth failure, encephalopathy, and endocrine dysfunctions in two siblings, one with 5-oxoprolinase deficiency. Eur J Pediatr. 1997, 156 (12): 935-938. 10.1007/s004310050746.
Article CAS PubMed Google Scholar - Mayatepek E, Hoffmann GF, Larsson A, Becker K, Bremer HJ: 5-Oxoprolinase deficiency associated with severe psychomotor developmental delay, failure to thrive, microcephaly and microcytic anaemia. J Inherit Metab Dis. 1995, 18 (1): 83-84. 10.1007/BF00711382.
Article CAS PubMed Google Scholar - Larsson A, Mattsson B, Wauters EA, van Gool JD, Duran M, Wadman SK: 5-oxoprolinuria due to hereditary 5-oxoprolinase deficiency in two brothers--a new inborn error of the gamma-glutamyl cycle. Acta Paediatr Scand. 1981, 70 (3): 301-308.
Article CAS PubMed Google Scholar - Mayatepek E, Badiou S, Bellet H, Lehmann WD: A patient with neurological symptoms and abnormal leukotriene metabolism: a new defect in leukotriene biosynthesis. Ann Neurol. 2005, 58 (6): 968-970. 10.1002/ana.20687.
Article CAS PubMed Google Scholar - Anderson ME, Allison RD, Meister A: Interconversion of leukotrienes catalyzed by purified gamma-glutamyl transpeptidase: concomitant formation of leukotriene D4 and gamma-glutamyl amino acids. Proc Natl Acad Sci U S A. 1982, 79 (4): 1088-1091. 10.1073/pnas.79.4.1088.
Article PubMed Central CAS PubMed Google Scholar - Nitanai Y, Satow Y, Adachi H, Tsujimoto M: Crystal structure of human renal dipeptidase involved in beta-lactam hydrolysis. J Mol Biol. 2002, 321 (2): 177-184. 10.1016/S0022-2836(02)00632-0.
Article CAS PubMed Google Scholar - Nakagawa H, Inazawa J, Inoue K, Misawa S, Kashima K, Adachi H, Nakazato H, Abe T: Assignment of the human renal dipeptidase gene (DPEP1) to band q24 of chromosome 16. Cytogenet Cell Genet. 1992, 59 (4): 258-260.
Article CAS PubMed Google Scholar - Littlewood GM, Hooper NM, Turner AJ: Ectoenzymes of the kidney microvillar membrane. Affinity purification, characterization and localization of the phospholipase C-solubilized form of renal dipeptidase. Biochem J. 1989, 257 (2): 361-367.
Article PubMed Central CAS PubMed Google Scholar - Oberholzer VG, Wood CB, Palmer T, Harrison BM: Increased pyroglutamic acid levels in patients on artificial diets. Clin Chim Acta. 1975, 62 (2): 299-304. 10.1016/0009-8981(75)90240-5.
Article CAS PubMed Google Scholar - Tham R, Nystrom L, Holmstedt B: Identification by mass spectrometry of pyroglutamic acid as a peak in the gas chromatography of human urine. Biochem Pharmacol. 1968, 17 (8): 1735-1738. 10.1016/0006-2952(68)90236-0.
Article CAS PubMed Google Scholar - Stokke O, Marstein S, Jellum E, Lie SO: Accumulation of pyroglutamic acid (5-oxoproline) in homocystinuria. Scand J Clin Lab Invest. 1982, 42 (4): 361-369.
Article CAS PubMed Google Scholar - Ghauri FY, McLean AE, Beales D, Wilson ID, Nicholson JK: Induction of 5-oxoprolinuria in the rat following chronic feeding with N-acetyl 4-aminophenol (paracetamol). Biochem Pharmacol. 1993, 46 (5): 953-957. 10.1016/0006-2952(93)90506-R.
Article CAS PubMed Google Scholar - Bonham JR, Rattenbury JM, Meeks A, Pollitt RJ: Pyroglutamicaciduria from vigabatrin. Lancet. 1989, 1 (8652): 1452-1453. 10.1016/S0140-6736(89)90158-X.
Article CAS PubMed Google Scholar - Croal BL, Glen AC, Kelly CJ, Logan RW: Transient 5-oxoprolinuria (pyroglutamic aciduria) with systemic acidosis in an adult receiving antibiotic therapy. Clin Chem. 1998, 44 (2): 336-340.
CAS PubMed Google Scholar - Jackson AA, Persaud C, Hall M, Smith S, Evans N, Rutter N: Urinary excretion of 5-L-oxoproline (pyroglutamic acid) during early life in term and preterm infants. Arch Dis Child Fetal Neonatal Ed. 1997, 76 (3): F152-7.
Article PubMed Central CAS PubMed Google Scholar - Goto A, Ishida A, Goto R, Hayasaka K, Nanao K, Yamashita A, Yamaguchi S, Takada G: Transient 5-oxoprolinuria in a very low-birthweight infant. J Inherit Metab Dis. 1992, 15 (2): 284-285. 10.1007/BF01799646.
Article CAS PubMed Google Scholar - Jackson AA, Badaloo AV, Forrester T, Hibbert JM, Persaud C: Urinary excretion of 5-oxoproline (pyroglutamic aciduria) as an index of glycine insufficiency in normal man. Br J Nutr. 1987, 58 (2): 207-214. 10.1079/BJN19870088.
Article CAS PubMed Google Scholar - Rizzo C, Ribes A, Pastore A, Dionisi-Vici C, Greco M, Rizzoni G, Federici G: Pyroglutamic aciduria and nephropathic cystinosis. J Inherit Metab Dis. 1999, 22 (3): 224-226. 10.1023/A:1005545012776.
Article CAS PubMed Google Scholar
Acknowledgements
These studies were supported by grants from the Swedish Research Council (4792), the Free Masons in Stockholm for Children's Welfare, the Memory Foundation of Golje, the HRH Crown Princess Lovisa Foundation, the Åke Wiberg Foundation, the Ronald McDonald Foundation, the Linnéa and Josef Carlsson Foundation, the Lennanders Foundation, the Swedish Society of Medicine, and the Samariten Foundation, which are gratefully acknowledged.
Author information
Authors and Affiliations
- Karolinska Institute, Department of Pediatrics, Karolinska University Hospital Huddinge, Stockholm, SE-141 86, Sweden
Ellinor Ristoff & Agne Larsson
Authors
- Ellinor Ristoff
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toEllinor Ristoff.
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ristoff, E., Larsson, A. Inborn errors in the metabolism of glutathione.Orphanet J Rare Dis 2, 16 (2007). https://doi.org/10.1186/1750-1172-2-16
- Received: 08 January 2007
- Accepted: 30 March 2007
- Published: 30 March 2007
- DOI: https://doi.org/10.1186/1750-1172-2-16