Deregulated β-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation (original) (raw)
Abstract
Aberrant activation of β-catenin contributes to the onset of a variety of tumors. We report that a tumor-derived β-catenin mutant induces accumulation and activation of the p53 tumor suppressor protein. Induction is mediated through ARF, an alternative reading frame product of the INK4A tumor suppressor locus, in a manner partially dependent on the transcription factor E2F1. In wild-type mouse embryo fibroblasts, mutant β-catenin inhibits cell proliferation and imposes a senescence-like phenotype. This does not occur in cells lacking either ARF or p53, where deregulated β-catenin actually overrides density-dependent growth inhibition and cooperates with activated Ras in transformation. Thus, the oncogenic activity of deregulated β-catenin is curtailed by concurrent activation of the p53 pathway, thereby providing a protective mechanism against cancer. When the p53 pathway is impaired, deregulated β-catenin is free to manifest its oncogenic features. This can occur not only by p53 mutations, but also by ablation of ARF expression, as observed frequently in early stages of colorectal carcinogenesis.
Keywords: ARF/β-catenin/p53/Ras/senescence
Introduction
β-catenin is a versatile protein. On the one hand, it is an important structural component of the cell adhesion machinery that mediates cell–cell interactions (Kemler, 1993; Ben-Ze’ev and Geiger, 1998). On the other hand, as a pivotal constituent of the Wnt–wingless pathway, β-catenin is also intimately involved in intracellular signal transduction (Cox and Peifer, 1998; Willert and Nusse, 1998).
The levels, subcellular localization and activity of β-catenin are tightly regulated within the cell (Aberle et al., 1997; Bienz, 1999; Clevers, 2000; Hecht et al., 2000; Rosin-Arbesfeld et al., 2000). The free cytosolic pool of β-catenin is unstable and subject to rapid proteolytic degradation, which requires its phosphorylation by the glycogen synthase kinase 3β (GSK-3β). Phosphorylation occurs on sites within the N-terminal domain of β-catenin, and is mediated through interaction of β-catenin with a complex including, in addition to GSK-3β, the APC (adenomatous polyposis coli) tumor suppressor protein and axin/conductin (Behrens et al., 1998; Ikeda et al., 1998). The N-terminally phosphorylated β-catenin then becomes a target for recognition by a specific SCF-type E3 ubiquitin ligase (Hart et al., 1999; Winston et al., 1999; Sadot et al., 2000), leading to its polyubiquitylation and subsequent proteasomal degradation.
Upon physiological activation of the Wnt–wingless pathway, β-catenin is stabilized and translocates into the nucleus. There, in complex with members of the TCF/LEF family of DNA binding proteins, it serves as a transcriptional regulator and induces the expression of cognate target genes (Behrens et al., 1996; Riese et al., 1997; van de Wetering et al., 1997). Aberrant stabilization of β-catenin, resulting in constitutive transcriptional activity, is encountered in a variety of cancers (Bienz, 2000; Peifer and Polakis, 2000; Polakis, 2000; Zhurinsky et al., 2000a). Stabilization can be achieved either through mutational inactivation of APC or axin, or through direct mutation of β-catenin. The latter mutations usually occur within the N-terminal domain of β-catenin, rendering it refractory to phosphorylation by GSK-3β (Polakis, 1999).
The c-myc and cyclin D1 genes, encoding important positive regulators of cell proliferation, have been identified as transcriptional targets of deregulated β-catenin (He et al., 1998; Shtutman et al., 1999; Tetsu and McCormick, 1999). Constitutive activation of those genes may account, at least in part, for the putative contribution of β-catenin deregulation to tumorigenesis. Additional β-catenin target genes such as WISP-1 and _PPAR_δ (He et al., 1998; Xu et al., 2000), may also play a positive role in cancer processes. The oncogenic potential of deregulated β-catenin has been convincingly demonstrated in vivo in mouse tumor models (Oshima et al., 1995; Gat et al., 1998; Chan et al., 1999).
The p53 tumor suppressor gene is also subject to frequent mutations in cancer (Levine, 1997). Such mutations primarily abolish the tumor inhibitory activities of the wild-type (wt) p53 protein. In non-stressed cells, p53 protein levels are usually very low, owing to rapid proteolytic degradation of wt p53. Numerous stress signals lead to activation and accumulation of p53 (Giaccia and Kastan, 1998; Prives, 1998; Lohrum and Vousden, 1999; Oren, 1999; Prives and Hall, 1999; Ashcroft et al., 2000). These signals include diverse types of DNA damage and chromosomal aberrations, as well as deregulation of various growth stimulatory signaling pathways. The latter type of deregulation may be achieved via aberrant activation of oncogenes such as c-myc and ras, or through the action of viral oncoproteins such as the products of the adenovirus E1A region. In several cases, including c-myc, ras, E2F1, and adenovirus E1A, induction of p53 relies to a great extent on another tumor suppressor protein, ARF (Bates et al., 1998; de Stanchina et al., 1998; Palmero et al., 1998; Stott et al., 1998; Zindy et al., 1998). ARF is the product of an alternative transcript of the INK4a tumor suppressor locus (Quelle et al., 1995; reviewed in Sharpless and DePinho, 1999; Sherr and Weber, 2000). ARF binds to the Mdm2 protein, which plays a key role in the degradation of p53 through its activity as a p53-specific E3 ubiquitin ligase (Honda and Yasuda, 1999). The binding of ARF to Mdm2 sequesters Mdm2 in the nucleolus, where it cannot target p53 for ubiquitylation; in addition, it directly inhibits the E3 activity of Mdm2 (Honda and Yasuda, 1999; Tao and Levine, 1999; Weber et al., 1999, 2000b; Lohrum et al., 2000). Hence, induction of ARF by deregulated oncogenes inhibits the ubiquitylation and subsequent proteasomal degradation of p53, resulting in accumulation of p53 and stimulation of p53-mediated cellular responses. This is believed to serve as a failsafe mechanism that protects against the tumorigenic consequences of oncogene activation (Sherr, 1998). In addition, ARF may also have p53-independent anti-proliferative functions (Carnero et al., 2000; Esteller et al., 2000; Weber et al., 2000a).
We reported previously that overexpression of β-catenin leads to accumulation of transcriptionally active p53 in transfected cells (Damalas et al., 1999). We now show that activated, tumor-derived mutant β-catenin triggers p53 accumulation when expressed at levels comparable with those present in cells derived from human colorectal cancer (CRC). The accumulation of p53 is due to induction of ARF expression. Maximal induction of ARF and p53 requires the presence of the E2F1 transcription factor. In normal primary mouse embryo fibroblasts (MEFs), deregulated β-catenin arrests cell proliferation and imposes a senescence-like phenotype that is dependent on both ARF and p53 function. Conversely, in ARF-deficient or p53-deficient MEFs, the same mutant β-catenin exerts a growth stimulatory effect, reflected in an ability to overcome density-dependent growth inhibition. Moreover, in the absence of either ARF or wt p53, deregulated β-catenin cooperates with oncogenic ras to enhance cell transformation.
Our findings support a model where the emergence of deregulated, constitutively active β-catenin is coupled with the transcriptional stimulation of ARF expression. This in turn triggers an anti-proliferative response, orchestrated largely through p53. Such anti-proliferative response is proposed to curb the potential oncogenic consequences of deregulated β-catenin, thereby providing a potent mechanism for tumor suppression. Elimination of this inhibitory arm, through inactivation of either p53 or ARF, might unveil the full oncogenic potential of deregulated β-catenin and allow it to play a more effective role in tumorigenesis. The observed silencing of ARF expression in a sizable fraction of colorectal adenomas, corresponding to early stages of cancer progression, is consistent with this model.
Results
Induction of p53 by oncogenic β-catenin is mediated through ARF
Experimental overexpression of β-catenin can induce p53 accumulation (Damalas et al., 1999). To investigate the molecular mechanism underlying this induction, primary MEFs were employed. wt MEFs were infected with a recombinant retrovirus encoding β-catenin S33Y, a constitutively active β-catenin mutant derived from a CRC patient (Morin et al., 1997). A retrovirus expressing only the puromycin resistance gene served as a negative control. Infection efficiencies were routinely close to 100%, as assessed by puromycin selection of representative infected cultures, and by infection of parallel cultures with a retrovirus encoding β-galactosidase (data not shown). The total levels of β-catenin in the infected MEFs, representing the combination of endogenous wt β-catenin and transduced mutant β-catenin, were only moderately elevated relative to the endogenous wt protein alone in control MEFs (Figure 1A). More importantly, these levels were comparable with those present in cell lines derived from human CRC, where β-catenin is deregulated owing to either direct β-catenin mutations (SW48 and HCT116) or inactivation of the APC tumor suppressor (SW480). As seen in Figure 1B, such levels of oncogenic β-catenin were already sufficient to elicit a marked increase in the steady state levels of p53.
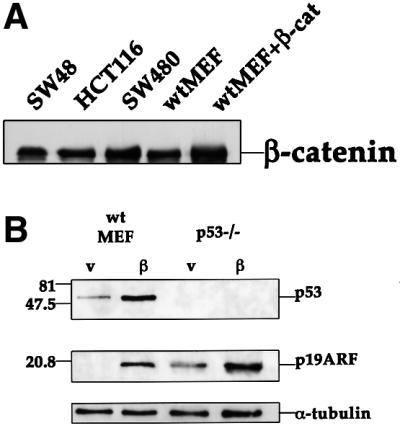

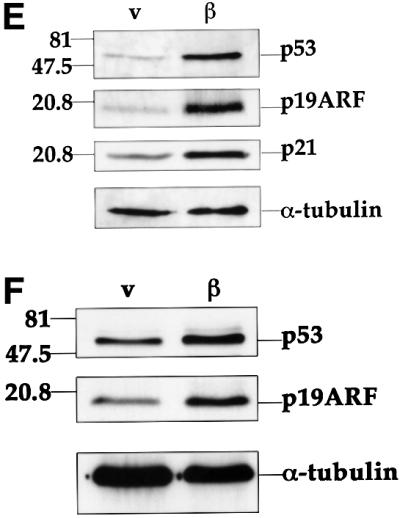
Fig. 1. Induction of p53 and ARF by deregulated β-catenin.(A) Comparison of β-catenin levels in different cell types. Cell extracts were prepared from CRC lines (SW48, HCT116, SW480) and from fibroblasts infected with recombinant retroviruses encoding either β-catenin S33Y (wtMEF + β-cat) or puromycin resistance only (wtMEF). Four micrograms of protein of each sample was subjected to western blot analysis with a monoclonal antibody directed against β-catenin (Sigma). (B) Low passage MEFs, derived from either wt or p53-null animals, were infected with recombinant retroviruses encoding either HA-tagged S33Y tumor-derived mutant β-catenin (β) or puromycin resistance only (v). Cells were trypsinized and replated 48 h later. Cells were re-fed the next day with medium containing 0.1% serum, and harvested after another 48 h. Twenty micrograms of protein of each sample was subjected to sequential western blot analysis with antibodies directed against p53, p19ARF and α-tubulin as a control for equal loading. (C) Low passage p53-null MEFs, infected with either β-catenin (β) or control retrovirus (v), were transiently transfected with a firefly luciferase reporter plasmid driven by the murine ARF promoter, either alone or together with a plasmid expressing dominant negative TCF4 (DN-TCF4). Transfections were done in triplicate. Luciferase activity was normalized for Renilla luciferase readings in the same extracts (see Materials and methods). (D) wt MEFs infected with a retrovirus encoding HA-tagged β-catenin S33Y (a–c) or with pBabe-puro control retrovirus (d–f) were fixed and stained with p19ARF-specific antiserum (a,d), B23 antiserum to visualize nucleoli (b,e) and DAPI to visualize nuclei (c,f). (E) wt MEFs were infected and processed essentially as in (B), except that blots were probed sequentially for p53, p21Waf1 and α-tubulin. Identical samples, run in parallel lanes of the same gel, were probed for p19ARF. (F) wt MEFs were infected and processed as in (B), except that the cells were maintained in medium containing 10% serum throughout the experiment.
Several oncoproteins trigger p53 accumulation through stimulation of ARF expression (Lowe, 1999; Sherr and Weber, 2000). We, therefore, investigated whether ARF plays a role in the induction of p53 by tumor-associated β-catenin. Analysis of infected wt MEFs revealed a dramatic increase in murine p19ARF protein levels in the presence of β-catenin S33Y (Figure 1B). Hence, deregulated β-catenin can effectively enhance ARF expression.
In p53-null MEFs, basal ARF expression is significantly elevated (Figure 1B), owing to relief of p53-dependent repression of ARF transcription (Robertson and Jones, 1998). Nevertheless, β-catenin elicited a further increase in ARF protein (2.5-fold after normalization for α-tubulin, Figure 1B). Remarkably, β-catenin augmented to a similar extent the transcriptional activity of the murine ARF promoter (Inoue et al., 1999) in these cells (Figure 1C). Moreover, activation of this promoter was effectively abrogated by overexpression of dominant negative TCF4 (DN-TCF4), known to block the transcriptional activity of β-catenin (Korinek et al., 1997). These findings imply that deregulated β-catenin can induce ARF expression at the transcriptional level.
The ARF protein induced by β-catenin resides predominantly in the nucleolus (Figure 1D, a), as confirmed by double staining for the nucleolar protein nucleophosmin/B23 (Figure 1D, b); this is consistent with earlier reports (Weber et al., 1999).
Upregulation of p53 by β-catenin was accompanied by elevated levels of p21, a p53 target gene product (Figure 1E), confirming that the upregulated p53 is functional. Moreover, induction of ARF and p53 by mutant β-catenin could be observed in the presence of either low (Figure 1E) or high (Figure 1F) serum concentrations, although its extent was milder in cells growing in high serum.
To further investigate whether physiological activation of β-catenin-mediated signaling can induce ARF expression, wt MEFs were treated with LiCl. LiCl is a selective inhibitor of GSK-3β; exposure of cells to LiCl results in β-catenin stabilization and augmentation of β-catenin-mediated signaling (Woodgett, 1994). As seen in Figure 2, high concentrations of LiCl elicited a significant increase in ARF protein levels. As expected, this was accompanied by a proportionate increase in p53 protein; induction of p53 by LiCl has recently been reported also by Mao et al. (2001).
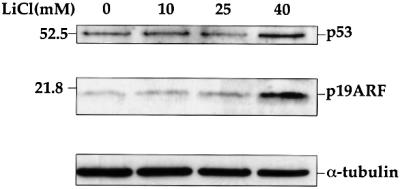
Fig. 2. Induction of ARF and p53 by LiCl. Early passage wt MEFs were plated at a density of 2 × 105/10-cm dish. Fourteen hours later, LiCl was added to the indicated final concentration, for an additional 48 h. Cells were then harvested and processed as in Figure 1.
The role of ARF in the induction of p53 by deregulated β-catenin was investigated with the aid of ARF-deficient MEFs (Kamijo et al., 1997). As seen in Figure 3, β-catenin S33Y was totally incapable of augmenting p53 in the absence of ARF, nor could it elevate p21 expression. It should be noted that very extensive β-catenin overexpression, for example by transient transfection with a CMV-driven expression plasmid, promotes p53 accumulation even in ARF-deficient immortalized NIH-3T3 cells (Damalas et al., 1999). Yet, the data in Figure 3 clearly demonstrates that ARF is essential for induction of active p53 by the more physiological levels of deregulated β-catenin achieved in the infected cells.
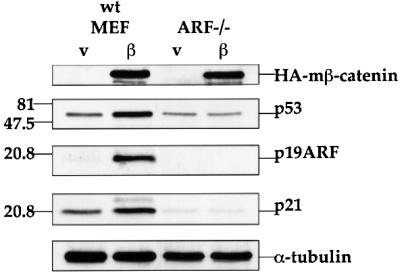
Fig. 3. ARF is required for accumulation and activation of p53 in response to oncogenic β-catenin. Early passage MEFs, derived from wt or ARF-deficient (ARF–/–) mice, were infected as in Figure 1B. Protein extracts (20 µg total protein/lane) were subjected to western blot analysis essentially as in Figure 1B, except that HA-tag specific antibodies were employed to visualize the mutant β-catenin (HA-mβ-catenin).
Together, these findings imply that deregulated β-catenin promotes ARF expression, most likely through enhanced transcription, thereby preventing p53 degradation and allowing accumulation of functional wt p53.
Role of E2F1 in the stimulation of the ARF–p53 pathway by deregulated β-catenin
Members of the E2F family of transcription factors play a critical role in cell-cycle progression (Nevins, 1998). E2F transcriptional activity is positively regulated by kinase complexes consisting of cyclin D and cdk4 or cdk6 (Sherr, 1996). Hence, increased expression of cyclin D proteins is expected to elevate E2F-mediated transcription. E2F1, a member of the E2F family, transactivates the ARF promoter (Bates et al., 1998) and was implicated in the induction of ARF expression by several oncoproteins (de Stanchina et al., 1998). The role of E2F1 in the upregulation of ARF and p53 by β-catenin was studied through the use of E2F1-null MEFs. As seen in Figure 4A, induction of both ARF and p53 by β-catenin S33Y was partially compromised in these cells; the fold induction, normalized for α-tubulin, is indicated below the respective lanes. Nevertheless, a residual increase in both proteins was still retained, indicating that while E2F1 is an important mediator of the effect of deregulated β-catenin on p53, its role is not exclusive.

Fig. 4. Involvement of E2F1 in the induction of the ARF–p53 pathway by β-catenin. (A) Induction of ARF and p53 in E2F1-null cells. MEFs from wt and E2F1-null mice were infected and analyzed as in Figure 3. Band intensities were determined by densitometry (NIH-Image). Numbers below lanes indicate the fold induction of the respective protein by mutant β-catenin, calculated relative to the parallel control virus-infected wt MEF sample, after normalizing for α-tubulin. (B) Western blot analysis of cyclin D1 protein in wt MEFs infected with either control (v) or mutant β-catenin (β) retrovirus. Infection and protein analysis were as in Figure 1B.
As expected (Shtutman et al., 1999; Tetsu and McCormick, 1999), infection of MEFs with the β-catenin S33Y retrovirus led to a substantial increase in cyclin D1 protein (Figure 4B). Deregulated β-catenin may thus stimulate ARF expression through induction of cyclin D1, leading to increased E2F1-mediated synthesis of ARF mRNA. The impact of cyclin D1 upregulation on ARF expression is, however, more complex. Specifically, ARF transcription is subject to positive regulation by another transcription factor, DMP1 (Inoue et al., 1999). DMP1 activity, in turn, is inhibited by cyclin D1 (Inoue and Sherr, 1998). Thus, cyclin D1 also possesses the potential to negatively regulate ARF expression through inhibition of DMP1. The factors that dictate the net effect of cyclin D1 induction on ARF expression still remain to be elucidated.
Sustained deregulation of β-catenin induces a senescence-like phenotype dependent on ARF and p53
To evaluate the phenotypic consequences of sustained β-catenin deregulation, MEFs of different genotypes were infected with retroviruses encoding either β-catenin S33Y or puromycin resistance only. Infected cultures were trypsinized, and an identical number of cells from each culture were re-plated at low density. Microscopic examination of the various cultures, several days later, revealed that wt MEFs infected with the control retrovirus reached a high cell density (Figure 5). In striking contrast, wt MEFs infected with β-catenin S33Y remained relatively sparse; moreover, many of the cells were flat and remarkably spread out, exhibiting a senescence-like morphology. The experiment shown in Figure 5 was performed in the presence of 10% serum. Essentially similar results were obtained in low (0.1%) serum, except that the morphological changes in β-catenin-infected wt MEFs were more dramatic, while all other cultures gradually ceased proliferation without displaying senescence-like features (data not shown).

Fig. 5. Overexpression of mutant β-catenin elicits a senescence-like phenotype dependent on ARF and p53. Early passage MEFs, derived from wt, ARF-null or p53-null mice, were infected with control retrovirus or retrovirus encoding HA-tagged β-catenin S33Y. Cells were trypsinized and replated 48 h later, and after an additional day fresh medium containing 10% serum was added. Cultures were maintained in the same medium for 6 days. Phase contrast photographs were taken at a magnification of 100×.
Growth rate analysis (Figure 6A) confirmed that the proliferation of MEFs overexpressing β-catenin S33Y was substantially retarded relative to their counterparts infected with the control virus. Unlike wt cells, MEFs lacking either ARF or p53 continued to proliferate after infection with the β-catenin S33Y retrovirus; in neither cell type was there any evidence for senescence-associated morphological alterations (Figure 5). In fact, overexpression of β-catenin S33Y in these cells exerted a measurable positive effect. After several days of cultivation, focal areas of high-cell density could be discerned in ARF-null cultures. While control ARF-null MEFs proliferated to saturation density and then stopped, cultures overexpressing β-catenin S33Y failed to stop at that point and continued to proliferate avidly (Figure 6B). Thus, in the absence of ARF, deregulated β-catenin can augment the ability of cells to overcome density-dependent growth inhibition, a feature that may facilitate tumor progression. The effect of β-catenin S33Y on p53-null MEFs was somewhat more complicated. Cells infected with the control virus grew to a high density, but then proliferation ceased and cell numbers actually went down (Figure 6C). That latter drop was abolished by overexpression of β-catenin S33Y, suggesting that under those conditions β-catenin may promote cell survival.

Fig. 6. Deregulated β-catenin inhibits cell proliferation in an ARF- and p53-dependent manner. Cultures of wt, ARF-null and p53-null MEFs were infected with control retrovirus (circles) or retrovirus encoding HA-tagged β-catenin S33Y (squares) and processed as in Figure 5. Infected cells were replated in 10-cm dishes, at a seeding density of 2 × 105 cells/dish. Triplicate cultures were counted at the indicated number of days after replating. The SE is indicated.
The dependence of the senescence-like phenotype on p53 was further confirmed by the use of a recombinant retrovirus encoding a temperature sensitive (ts) p53 mutant, p53Val135 (de Rozieres et al., 2000). p53–/– MEFs were first infected with the β-catenin S33Y retrovirus, and then re-infected with p53Val135. When the infected cultures were shifted to 32°C, resulting in activation of the ts p53 (Michalovitz et al., 1990), the cells assumed a typical senescence-like morphology (Figure 7A). This did not occur in the presence of activated p53 alone (control + p53Val135, 32°C). In contrast, avid proliferation with no morphological alterations, despite the presence of β-catenin, was seen at 37.5°C where the ts p53 assumes a mutant conformation devoid of wt p53 activity. It is noteworthy that under those conditions, allowing for relatively high levels of p53 even in the absence of deregulated β-catenin, p53 levels increased only mildly in response to excess β-catenin S33Y (Figure 7B). This raises the possibility that tumor-associated β-catenin can regulate not only the levels of p53, but also its specific biochemical activity.

Fig. 7. wt p53 activity is required for induction of a senescence-like phenotype by deregulated β-catenin. Early passage p53–/– MEFs were infected with control retrovirus or retrovirus encoding HA-tagged β-catenin S33Y. Forty-eight hours later cells were trypsinized, replated and subjected to a second round of infection with a retrovirus encoding the ts p53 mutant p53Val135. After 48 h fresh medium containing 10% serum was added, and some of the cultures were transferred to 32°C while the others were left at 37.5°C. (A) Phase contrast photographs taken 6 days after plating (magnification = 100×). (B) Western blot analysis of p53 protein levels in cultures maintained at 32°C and extracted 4 days after replating.
In conclusion, whereas deregulated β-catenin exerts potent anti-proliferative effects and elicits a senescence-like phenotype in the presence of wt p53 and ARF, inactivation of either ARF or p53 alleviates those anti-proliferative effects and reveals the potential growth-promoting features of deregulated β-catenin.
Tumor-associated β-catenin and activated Ras cooperate in transformation in the absence of ARF or p53
To further explore whether deregulated β-catenin can contribute to oncogenesis, we asked whether β-catenin S33Y cooperates with constitutively active mutant Ras in the transformation of cultured cells. This combination was chosen because deregulation of β-catenin is frequently followed by Ras mutations during colorectal carcinogenesis (Kinzler and Vogelstein, 1996). MEFs of various genotypes were sequentially infected with retroviruses expressing β-catenin S33Y and the tumor-associated mutant H-RasVal12. In wt MEFs, no transformed foci were detectable upon infection with either Ras alone or Ras plus β-catenin (data not shown). In contrast, RasVal12 elicited many transformed foci in ARF-null cells (Figure 8), as reported earlier (Kamijo et al., 1997). Importantly, while β-catenin alone did not give rise to a significant number of foci, the combination of β-catenin and activated Ras resulted in extensive transformation; foci were more numerous and substantially thicker than with Ras alone (Figure 8). Similarly, β-catenin S33Y also augmented Ras-mediated focus formation in p53-null MEFs, although overall numbers of foci were lower than in ARF-null MEFs (data not shown).

Fig. 8. Oncogenic β-catenin cooperates with Ras in transformation of ARF-null MEFs. Early passage ARF–/– MEFs (1.5 × 105 cells/10-cm dish) were infected with either control retrovirus or retrovirus encoding β-catenin S33Y. Two days later the cultures were trypsinized and replated again at 1.5 × 105 cells/10-cm dish, and subjected to a second round of infection with a retrovirus encoding the Val12 mutant H-Ras. Cultures were fixed 8–9 days later and stained with Giemsa stain. Triplicate dishes are shown. Where only a single oncogene is indicated, the cultures were actually also subjected to a second infection with control retrovirus to maintain the infection history of each culture equal.
Thus, constitutively activated Ras can cooperate with deregulated β-catenin to transform MEFs; this oncogenic activity of mutant β-catenin is, however, unleashed only upon inactivation of the p53 pathway.
Regulation of ARF by β-catenin in human CRC-derived cells
The oncogenic activity of β-catenin has been extensively studied in CRC cell lines, representing a type of cancer where deregulated β-catenin plays a pivotal role in tumorigenesis. It was, therefore, important to determine whether deregulated β-catenin can upregulate ARF expression in CRC cells. SW480 cells, in which β-catenin is deregulated owing to APC gene mutation, were transiently transfected with either DN-TCF4 or axin, both of which are potent negative regulators of β-catenin signaling (Behrens et al., 1998; Ikeda et al., 1998; Clevers, 2000). In a considerable minority of SW480 cells, ARF protein can easily be detected either in the nucleolus or in a mixed nucleolar/nucleoplasmic pattern (see Figure 9A). The overall fraction of ARF positive SW480 cells was found to be ∼15–20% (Figure 9B). It is noteworthy that constitutive ARF expression is not expected to exert marked anti-proliferative effects in SW480 cells, which carry mutant p53 (Rodrigues et al., 1990). In striking contrast, overexpression of either DN-TCF4 [Figure 9A, a–c; putative positive cells were identified by cotransfection with green fluorescent protein (GFP)] or axin (Figure 9A, d–f) reduced ARF staining to levels not significantly above background (see also Figure 9B). Hence, as in MEFs, constitutive β-catenin signaling drives ARF expression also in these CRC cells. Moreover, analysis of the transcriptional activity of the ARF promoter in SW480 cells revealed a substantial inhibition by either DN-TCF or axin (Figure 9C). Taken together, these results argue strongly that deregulated β-catenin can turn on ARF transcription in colorectal epithelial cells, thereby triggering the p53 pathway and placing a selective pressure for the subsequent inactivation of this protective pathway.
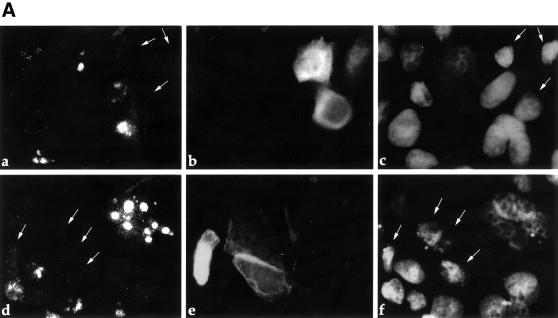
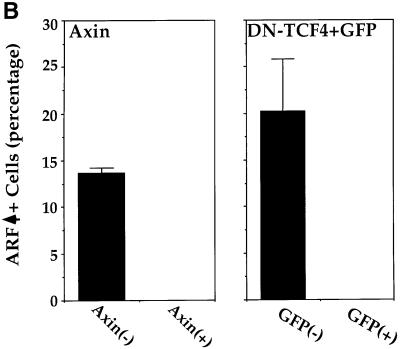
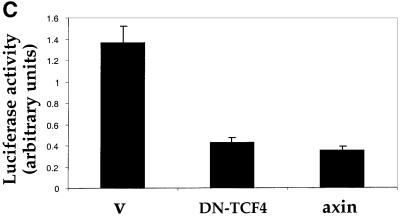
Fig. 9. ARF is induced by constitutive β-catenin signaling in SW480 CRC cells. (A) SW480 cells were plated at a density of 5 × 105 cells/well in a 6-well plate. Two days later, cultures were transiently transfected with either myc-tagged axin (5 µg) (a–c) or a combination of GFP (50 ng) plus DN-TCF4 (5 µg) (d–f). Twenty-four hours later cells were fixed. Endogenous p14ARF (a,c) was visualized by staining with polyclonal anti-p14ARF antibodies. Cells positive for transfected axin were visualized by staining with a monoclonal antibody directed against the myc tag (b). Putative DN-TCF4 transfectants were identified by GFP fluorescence (e). DAPI staining was employed to visualize nuclei (c,f). (B) Quantitative analysis of the experiment shown in (A). The left and right panels depict the data for SW480 cultures transfected with either axin or DN-TCF+GFP, respectively. (–) and (+) relate to the cell subpopulations staining negative or positive for the transfected protein (myc-tagged axin and GFP, respectively; GFP is expected to mark DN-TCF positive cells). In each case, the number relates to the percentage of cells within the given subpopulation where prominent nucleolar ARF staining was easily discernable. (C) SW480 cells were transiently transfected with a luciferase reporter driven by the mouse p19ARF promoter plus either control vector (v) or plasmids expressing DN-TCF4 or axin. Transfections were done in triplicate. Luciferase activity was normalized for Renilla luciferase readings in the same extracts (see Materials and methods).
Discussion
Accumulation of deregulated β-catenin is observed in a variety of human cancers. This occurs either as a consequence of mutations in the β-catenin gene itself, or owing to lesions in genes that control the ubiquitylation and degradation of cytosolic β-catenin, such as the APC tumor suppressor (Kinzler and Vogelstein, 1996) and axin (Satoh et al., 2000). In many cases, best exemplified by CRC, accumulation of transcriptionally active β-catenin is later followed by p53 mutations that inactivate the tumor suppressor functions of p53 (Kinzler and Vogelstein, 1996). We show here that deregulated β-catenin can activate the p53 pathway and trigger a p53-mediated anti-proliferative response, which in primary fibroblasts takes the form of a growth inhibition associated with senescence-like morphological features. Moreover, inactivation of the p53 pathway uncovers the oncogenic properties of deregulated β-catenin, measured by its ability to cooperate with mutant Ras in the transformation of primary MEFs. These observations may explain, at least in part, the selective pressure to inactivate the p53 pathway in tumors involving excessive β-catenin-dependent signaling.
Induction of p53 by deregulated β-catenin is strictly dependent on the ARF tumor suppressor. Deregulated β-catenin stimulates ARF expression and augments transcription from the ARF promoter in CRC-derived cells as well as in fibroblasts. This is completely abrogated by negative dominant TCF, strongly implicating the transcriptional activity of β-catenin in the induction of ARF and hence of p53. Of note, the β-catenin S33Y mutant is derived from a human tumor (Morin et al., 1997), and possesses enhanced transcriptional activity (Zhurinsky et al., 2000b).
The ability of β-catenin to activate p53 through upregulation of ARF adds it to a growing list of oncoproteins and growth regulatory proteins that can exert a similar effect (Bates et al., 1998; de Stanchina et al., 1998; Palmero et al., 1998; Radfar et al., 1998; Zindy et al., 1998; Eischen et al., 1999; Ries et al., 2000). Moreover, it explains the ability of overexpressed β-catenin to protect p53 against Mdm2-mediated proteasomal degradation (Damalas et al., 1999). Conceivably, the induced ARF protein binds to Mdm2, sequesters it in the nucleolus, and blocks the p53-specific E3 ubiquitin ligase activity of Mdm2 (Honda and Yasuda, 1999). Indeed, the ARF protein accumulated in response to β-catenin deregulation is largely nucleolar (Figures 1D and 9A).
Our findings predict that the oncogenic action of β-catenin will be facilitated in vivo not only by loss of wt p53 expression, but also by ablation of ARF expression. This is indeed the case, at least in CRC. While homozygous deletions of the INK4a/ARF locus are not associated with CRC (Cairns et al., 1995), many primary CRC tumors and cell lines exhibit complete or partial loss of ARF expression attributable to hypermethylation of the ARF promoter (Esteller et al., 2000). Importantly, silencing of the ARF gene occurs early in colorectal carcinogenesis, and is seen at equally high frequency already in adenomas (Esteller et al., 2000). We propose that ARF silencing enables emerging colorectal tumors to attenuate the activation of the p53 pathway by deregulated β-catenin, thereby averting the consequent restrictions on cell proliferation (Figure 10). The later emergence of mutations in the p53 gene itself may partly serve to enable progression of tumors that have maintained ARF expression. Alternatively, p53 mutations may provide additional features that are not enabled by the mere loss of ARF expression. Such mutations may eliminate the ability of tumor cells to undergo p53 activation through ARF-independent pathways, in response to conditions such as DNA damage, chromosomal aberrations or hypoxia (Oren, 1999; Ashcroft et al., 2000; Sherr and Weber, 2000). Moreover, p53 mutations may also contribute directly to tumorigenesis through dominant gain of function activities (Michalovitz et al., 1991; Dittmer et al., 1993). Indeed, while ARF promoter hypermethylation may be more frequent in CRC tumors retaining wt p53, it also occurs in a significant number of tumors carrying p53 mutations (Esteller et al., 2000).

Fig. 10. Schematic model depicting the relationship between deregulated β-catenin and the ARF–p53 pathway in MEFs and during carcinogenesis. In MEFs, ablation of ARF prevents the activation of p53 by deregulated β-catenin and spares the cells from p53-mediated growth inhibition (this study). In carcinogenesis, it is proposed that ARF silencing by promoter hypermethylation or by other mechanisms enables emerging tumor cells to benefit from the oncogenic activities of deregulated β-catenin while avoiding the inhibitory consequences of p53 activation. Additional genetic alterations, occurring at later stages of carcinogenesis, eventually generate a selective pressure for mutation of the p53 gene and lead to full malignancy. See Discussion for further details.
In many human tumors, including those driven by constitutive β-catenin signaling, activating Ras mutations are also observed; CRC is a good example (Kinzler and Vogelstein, 1996). As shown here, Ras can indeed cooperate with tumor-associated mutant β-catenin in the transformation of cultured cells. Of note, this oncogenic activity of β-catenin is not revealed as long as the p53 pathway is intact.
Recent findings suggest that the impact of Ras activation on p53 is largely dependent on the ARF status (Ries et al., 2000). When ARF is non-functional, Ras leads primarily to p53-independent induction of Mdm2 expression, which in turn enhances p53 degradation and incapacitates p53-mediated anti-proliferative signaling. Conversely, when ARF function is retained, deregulated Ras has the opposite outcome: ARF is induced and Mdm2 function is blocked, resulting in accumulation of active p53. One might, therefore, predict that the strongest selective pressure for elimination of wt p53 would be exerted in those tumors that experience sequential constitutive activation of β-catenin and Ras on a background of sustained ARF expression. This prediction remains to be proven.
Deregulation of β-catenin is primarily associated with epithelial tumors. While we show that CRC-derived epithelial cells induce ARF expression in response to the transcriptional activity of deregulated β-catenin (Figure 9), most of the work in the present study was performed with fibroblasts. It is, therefore, important to point out that deregulation of β-catenin is also implicated in the development of desmoid fibroma, a tumor resulting from excessive proliferation of fibroblasts, which arises frequently in individuals with germline mutations in the APC gene. A recent study employing APC-deficient Min mice revealed that loss of p53 is critical for the development of such tumors (Halberg et al., 2000). The ability of deregulated β-catenin to induce a vigorous p53-dependent anti-proliferative response offers a particularly appealing explanation for the need to dispose of p53 function in order for such fibroblastic tumors to arise.
In conclusion, our findings imply that the oncogenic activity of deregulated β-catenin places a selective pressure for the inactivation of the p53 pathway in emerging tumors. In some cases, the pressure is effectively alleviated early on by ARF silencing. In other cases, the tumor cells can apparently cope with this problem without turning off ARF expression, either because the extent of p53 activation is relatively mild and tolerable, or because other genetic alterations render the cells more refractory to some of the inhibitory effects of p53. In both types of cases, however, further progression down the road to malignancy often seems to entail the buildup of additional pressure on the p53 pathway, eventually resulting in direct mutational inactivation of the p53 gene itself.
Materials and methods
Cells
p53-null mice (Jacks et al., 1994) were obtained from Jackson Laboratories. Mouse MEFs were prepared from day 13.5 embryos. MEFs derived from E2F1 knockout mice and ARF knockout mice were obtained from L.Yamasaki and C.Sherr, respectively. HCT116, SW48 and SW480 cells were obtained from ATCC. MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal calf serum (FCS; Sigma), supplemented with non-essential amino acids and β-mercaptoethanol. HCT116 and SW48 cells were maintained in McCoy’s 5A medium plus 10% FCS. SW480 cells were maintained in DMEM plus 10% FCS.
Retroviral infection
High titer retroviral stocks were produced by transfecting retroviral constructs (pBabe-puro, pBabe-HA-β-catenin S33Y, pBabe-Ras Val12, or pBabep53Val135) into 293T cells (2 × 106/10-cm dish), by the calcium phosphate co-precipitation method, together with the ecotropic packaging vector pSV-ψ–E-MLV, providing ecotropic packaging helper function. Virus-containing culture supernatants were collected 24–72 h post-transfection, at 6 h intervals, and pooled together. Frozen low passage MEFs were thawed and plated at a density of 2 × 105/10-cm dish. Cells were infected 14 h later with filtered supernatants in the presence of polybrene (8 µg/ml; Sigma). Fresh supernatants were added three times, at 4 h intervals. Forty-eight hours post-infection, cells were trypsinized and replated; 24 h later, fresh medium containing 10% FCS was added to the cells. Whenever indicated, cells were replenished with medium containing 0.1% FCS. Cells were analyzed at the times indicated in the respective figure legends.
Immunoblot and immunostaining analysis
Cells were washed with ice-cold phosphate-buffered saline (PBS) and lyzed in NP-40 buffer (50 mM Tris pH 8, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40) containing protease inhibitors (phenylmethylsulfonyl fluoride and aprotinin). After 15 min on ice, with intermittent vigorous vortexing, lysates were cleared by centrifugation at 4°C. Samples corresponding to 20 µg total protein (Bio-Rad protein assay) were resolved on 12.5 or 15% SDS–polyacrylamide gels and transferred to nitrocellulose membranes (BA83, Schleicher & Schuell). Membranes were probed with polyclonal antibodies specific for mouse p53 (CM5, Novocastra), p19 ARF (gift of R.DePinho), mouse p21Waf1 (gift of C.Schneider), cyclin D1 (gift of T.Hunter) and Ha-tag (Y11; Santa Cruz). For the analysis of β-catenin levels in various cell lines (Figure 1A), only 4 µg total protein was loaded on each lane, and blots were probed with a monoclonal antibody specific for β-catenin (Sigma). Western blots were developed with the ECL kit (Amersham). Immunofluorescence microscopy was performed as described (Simcha et al., 1998). The myc tag was detected with the aid of the 9E10 monoclonal antibody (gift of G.Evan).
Luciferase assays
p53-null MEFs (20 000 cells/well, in a 6-well dish) were transfected with a combination of plasmids encoding firefly luciferase under control of the mouse ARF promoter (2.6 kb genomic DNA fragment, gift of C.Sherr) and Renilla luciferase under the cytomegalovirus (CMV) promoter (40 ng of each plasmid), with or without a dominant negative DN-TCF4 (Korinek et al., 1997) expression plasmid (200 ng). p53-null MEFs, rather than wt MEFs, were used in order to avoid activation of the endogenous p53 protein by the transfection procedure, potentially leading to p53-mediated repression of ARF promoter activity. Cells were harvested 48 h later.
For luciferase assays in SW480 cells, cells were plated at 5 × 105 cells/well in a 6-well dish. Two days later, the cells were transfected with a combination of plasmids encoding firefly luciferase under control of the mouse ARF promoter (1 µg) and Renilla luciferase under the CMV promoter (50 ng), with or without either axin (Shtutman et al., 1999; 4 µg) or dominant negative DN-TCF4 (4 µg). Cells were harvested 24 h later. Luciferase assays were performed with a commercial double luciferase kit (Promega), employing a TD-20e luminometer (Turner Design) as described (Damalas et al., 1999). Values of luciferase activity driven by the ARF promoter were normalized for Renilla luciferase readings in the same extracts.
Transformation assays
Transformation assays were performed essentially as described (Michalovitz et al., 1990), employing low passage MEFs in conjunction with retroviral infection. Cultures were maintained in culture for about 2 weeks and then washed twice with PBS and fixed with methanol for 5 min. Fixed cultures were stained with Giemsa stain.
Acknowledgments
Acknowledgements
We are indebted to B.Geiger for many inspiring discussions and continued support. We thank C.Sherr and M.Roussel for ARF-null MEFs, L.Yamasaki for E2F1-null MEFs, R.DePinho for p19ARF antiserum, K.Vousden for p14ARF antiserum, C.Schneider for p21 antiserum, T.Hunter for cyclin D1 antiserum, and G.Evan for the 9E10 antibody. This work was supported in part by grant RO1 CA 40099 from the National Cancer Institute (USA), the Center for Excellence Program of the Israel Science Foundation, the German–Israel Project Cooperation (DIP), the Cooperation Program in Cancer Research of the DKFZ and Israel’s Ministry of Science (MOS) and the Yad Abraham Center for Cancer Diagnosis and Therapy (to M.O.) and by grants from CaP CURE and the Israel Science Foundation (to A.B.-Z.).
References
- Aberle H., Bauer,A., Stappert,J., Kispert,A. and Kemler,R. (1997) β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J., 16, 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft M., Taya,Y. and Vousden,K.H. (2000) Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol., 20, 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates S., Phillips,A.C., Clark,P.A., Stott,F., Peters,G., Ludwig,R.L. and Vousden,K.H. (1998) p14ARF links the tumour suppressors RB and p53. Nature, 395, 124–125. [DOI] [PubMed] [Google Scholar]
- Behrens J., von Kries,J.P., Kuhl,M., Bruhn,L., Wedlich,D., Grosschedl, R. and Birchmeier,W. (1996) Functional interaction of β-catenin with the transcription factor LEF-1. Nature, 382, 638–642. [DOI] [PubMed] [Google Scholar]
- Behrens J., Jerchow,B.A., Wurtele,M., Grimm,J., Asbrand,C., Wirtz,R., Kuhl,M., Wedlich,D. and Birchmeier,W. (1998) Functional interaction of an axin homolog, conductin, with β-catenin, APC and GSK3β. Science, 280, 596–599. [DOI] [PubMed] [Google Scholar]
- Ben-Ze’ev A. and Geiger,B. (1998) Differential molecular interactions of β-catenin and plakoglobin in adhesion, signaling and cancer. Curr. Opin. Cell Biol., 10, 629–639 [DOI] [PubMed] [Google Scholar]
- Bienz M. (1999) APC: the plot thickens. Curr. Opin. Genet. Dev., 9, 595–603. [DOI] [PubMed] [Google Scholar]
- Bienz M. and Clevers,H. (2000) Linking colorectal cancer to Wnt signaling. Cell, 103, 311–320. [DOI] [PubMed] [Google Scholar]
- Cairns P. et al. (1995) Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nature Genet., 11, 210–212. [DOI] [PubMed] [Google Scholar]
- Carnero A., Hudson,J.D., Price,C.M. and Beach,D.H. (2000) p16INK4A and p19ARF act in overlapping pathways in cellular immortalization. Nature Cell Biol., 2, 148–155. [DOI] [PubMed] [Google Scholar]
- Chan E.F., Gat,U., McNiff,J.M. and Fuchs,E. (1999) A common human skin tumour is caused by activating mutations in β-catenin. Nature Genet., 21, 410–413. [DOI] [PubMed] [Google Scholar]
- Clevers H. (2000) Armadillo takes the APC shuttle. Nature Cell Biol., 2, E177–E178. [DOI] [PubMed] [Google Scholar]
- Cox R.T. and Peifer,M. (1998). Wingless signaling: the inconvenient complexities of life. Curr. Biol., 8, R140–R144. [DOI] [PubMed] [Google Scholar]
- Damalas A., Ben-Ze’ev,A., Simcha,I., Shtutman,M., Leal,J.F., Zhurinsky,J., Geiger,B. and Oren,M. (1999) Excess β-catenin promotes accumulation of transcriptionally active p53. EMBO J., 18, 3054–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rozieres S., Maya,R., Oren,M. and Lozano,G. (2000) The loss of mdm2 induces p53-mediated apoptosis. Oncogene, 19, 1691–1697. [DOI] [PubMed] [Google Scholar]
- de Stanchina E. et al. (1998) E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev., 12, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D., Pati,S., Zambetti,G., Chu,S., Teresky,A.K., Moore,M., Finlay,C. and Levine,A.J. (1993) Gain of function mutations in p53. Nature Genet., 4, 42–46. [DOI] [PubMed] [Google Scholar]
- Eischen C.M., Weber,J.D., Roussel,M.F., Sherr,C.J. and Cleveland,J.L. (1999) Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev., 13: 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M., Tortola,S., Toyota,M., Capella,G., Peinado,M.A., Baylin, S.B. and Herman,J.G. (2000) Hypermethylation-associated inacti vation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res., 60, 129–133. [PubMed] [Google Scholar]
- Gat U., DasGupta,R., Degenstein,L. and Fuchs,E. (1998) De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell, 95, 605–614. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Halberg R.B., Katzung,D.S., Hoff,P.D., Moser,A.R., Cole,C.E., Lubet,R.A., Donehower,L.A., Jacoby,R.F. and Dove,W.F. (2000) Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc. Natl Acad. Sci. USA, 97, 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart M. et al. (1999) The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol., 9, 207–210. [DOI] [PubMed] [Google Scholar]
- He T.C., Sparks,A.B., Rago,C., Hermeking,H., Zawel,L., da Costa,L.T., Morin,P.J., Vogelstein,B. and Kinzler,K.W. (1998) Identification of c-myc as a target of the APC pathway. Science, 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- Hecht A., Vleminckx,K., Stemmler,M.P., van Roy,F. and Kemler,R. (2000) The p300/CBP acetyltransferases function as transcriptional coactivators of β-catenin in vertebrates. EMBO J., 18, 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S., Kishida,S., Yamamoto,H., Murai,H., Koyama,S. and Kikuchi,A. (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J., 17, 1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K. and Sherr,C.J. (1998) Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol. Cell. Biol., 18, 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K., Roussel,M.F. and Sherr,C.J. (1999) Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc. Natl Acad. Sci. USA, 96, 3993–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T., Remington,L., Williams,B.O., Schmitt,E.M., Halachmi,S., Bronson,R.T. and Weinberg,R.A. (1994) Tumor spectrum analysis in p53-Mutant mice. Curr. Biol., 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Kamijo T., Zindy,F., Roussel,M.F., Quelle,D.E., Downing,J.R., Ashmun,R.A., Grosveld,G. and Sherr,C.J. (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell, 91, 649–659. [DOI] [PubMed] [Google Scholar]
- Kemler R. (1993) From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet., 9, 317–321. [DOI] [PubMed] [Google Scholar]
- Kinzler K.W. and Vogelstein,B. (1996) Lessons from hereditary colorectal cancer. Cell, 87, 159–170. [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker,N., Morin,P.J., van Wichen,D., de Weger,R., Kinzler,K.W., Vogelstein,B. and Clevers,H. (1997) Constitutive transcriptional activation by a β-catenin-Tcf complex in APC–/– colon carcinoma [see comments]. Science, 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lohrum M.A. and Vousden,K.H. (1999) Regulation and activation of p53 and its family members. Cell Death Differ., 6, 1162–1168. [DOI] [PubMed] [Google Scholar]
- Lohrum M.A., Ashcroft,M., Kubbutat,M.H. and Vousden,K.H. (2000) Identification of a cryptic nucleolar-localization signal in MDM2. Nature Cell Biol., 2, 179–181. [DOI] [PubMed] [Google Scholar]
- Lowe S.W. (1999) Activation of p53 by oncogenes. Endocr. Relat. Cancer, 6, 45–48. [DOI] [PubMed] [Google Scholar]
- Mao C.D., Hoang,P. and DiCorelto,P.E. (2001) Lithium inhibits cell cycle progression and induces stabilization of p53 in bovine aortic endothelial cells. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Michalovitz D., Halevy,O. and Oren,M. (1990) Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell, 62, 671–680. [DOI] [PubMed] [Google Scholar]
- Michalovitz D., Halevy,O. and Oren,M. (1991) p53 mutations: gains or losses? J. Cell. Biochem., 45, 22–29. [DOI] [PubMed] [Google Scholar]
- Morin P.J., Sparks,A.B., Korinek,V., Barker,N., Clevers,H., Vogelstein,B. and Kinzler,K.W. (1997) Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- Nevins J.R. (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ., 9, 585–593. [PubMed] [Google Scholar]
- Oren M. (1999) Regulation of the p53 tumor suppressor protein. J. Biol. Chem., 274, 36031–36034. [DOI] [PubMed] [Google Scholar]
- Oshima M., Oshima,H., Kitagawa,K., Kobayashi,M., Itakura,C. and Taketo,M. (1995) Loss of APC heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated APC gene. Proc. Natl Acad. Sci. USA, 92, 4482–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero I., Pantoja,C. and Serrano,M. (1998) p19ARF links the tumour suppressor p53 to Ras. Nature, 395, 125–126. [DOI] [PubMed] [Google Scholar]
- Peifer M. and Polakis,P. (2000) Wnt signaling in oncogenesis and embryogenesis a look outside the nucleus. Science, 287, 1606–1609. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2000) Wnt signaling and cancer. Genes Dev., 14, 1837–1851. [PubMed] [Google Scholar]
- Prives C. (1998) Signaling to p53: breaking the MDM2-p53 circuit. Cell, 95, 5–8. [DOI] [PubMed] [Google Scholar]
- Prives C. and Hall,P.A. (1999) The p53 pathway. J. Pathol., 187, 112–126. [DOI] [PubMed] [Google Scholar]
- Quelle D.E., Zindy,F., Ashmun,R.A. and Sherr,C.J. (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell, 83, 993–1000. [DOI] [PubMed] [Google Scholar]
- Radfar A., Unnikrishnan,I., Lee,H.W., DePinho,R.A. and Rosenberg,N. (1998) p19(Arf) induces p53-dependent apoptosis during abelson virus-mediated pre-B cell transformation. Proc. Natl Acad. Sci. USA, 95, 13194–13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries S., Biederer,C., Woods,D., Shifman,O., Shirasawa,S., Sasazuki,T., McMahon,M., Oren,M. and McCormic,F. (2000) Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19 ARF. Cell, 103, 321–330. [DOI] [PubMed] [Google Scholar]
- Riese J., Yu,X., Munnerlyn,A., Eresh,S., Hsu,S.C., Grosschedl,R. and Bienz,M. (1997) LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentaplegic. Cell, 88, 777–787. [DOI] [PubMed] [Google Scholar]
- Robertson K.D. and Jones,P.A. (1998) The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell. Biol., 18, 6457–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues N.R., Rowan,A., Smith,M.E.F., Kerr,I.B., Bodmer,W.F., Gannon,J.V. and Lane,D.P. (1990) p53 mutations in colorectal cancer. Proc. Natl Acad. Sci. USA, 87, 7555–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R., Townsley,F. and Bienz,M. (2000) The APC tumour suppressor has a nuclear export function. Nature, 406, 1009–1012. [DOI] [PubMed] [Google Scholar]
- Sadot E., Simcha,I., Iwai,K., Ciechanover,A., Geiger,B. and Ben-Ze’ev,A. (2000) Differential interaction of plakoglobin and β-catenin with the ubiquitin-proteasome system. Oncogene, 19, 1992–2001. [DOI] [PubMed] [Google Scholar]
- Satoh S. et al. (2000) AXIN1 mutations in hepatocellular carcinomas and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nature Genet., 24, 245–250. [DOI] [PubMed] [Google Scholar]
- Sharpless N.E. and DePinho,R.A. (1999) The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev., 9, 22–30. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. (1996) Cancer cell cycles. Science. 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. (1998) Tumor surveillance via the ARF-p53 pathway. Genes Dev., 12, 2984–2991. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Weber,J.D. (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev., 10, 94–99. [DOI] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky,J., Simcha,I., Albanese,C., D’Amico,M., Pestell,R. and Ben-Ze’ev,A. (1999) The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl Acad. Sci. USA, 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcha I., Shtutman,M., Salomon,D., Zhurinsky,J., Sadot,E., Geiger,B. and Ben-Ze’ev,A. (1998) Differential nuclear translocation and transactivation potential of β-catenin and plakoglobin. J. Cell Biol., 141, 1433–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott F.J. et al. (1998) The alternative product from the human CDKN2A locus, p14 (ARF), participates in a regulatory feedback loop with p53 and mdm2. EMBO J., 17, 5001–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W. and Levine,A.J. (1999) P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl Acad. Sci. USA, 96, 6937–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O. and McCormick,F. (1999) β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- van de Wetering M. et al. (1997) Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell, 88, 789–799. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Taylor,L.J., Roussel,M.F., Sherr,C.J. and Bar-Sagi,D. (1999) Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biol., 1, 20–26. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Jeffers,J.R., Rehg,J.E., Randle,D.H., Lozano,G., Roussel,M.F., Sherr,C.J. and Zambetti,G.P. (2000) p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev., 14, 2358–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J.D., Kuo,M.L., Bothner,B., DiGiammarino,E.L., Kriwacki,R.W., Roussel,M.F. and Sherr,C.J. (2000) Cooperative signals governing ARF-mdm2 interaction and nucleolar localization of the complex. Mol. Cell. Biol., 20, 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead I., Kirk,H. and Kay,R. (1995) Expression cloning of oncogenes by retroviral transfer of cDNA libraries. Mol. Cell. Biol., 15, 704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K. and Nusse,R. (1998) β-catenin: a key mediator of Wnt signaling. Curr. Opin. Genet. Dev., 8, 95–102. [DOI] [PubMed] [Google Scholar]
- Winston J.T., Strack,P., Beer-Romero,P., Chu,C.Y., Elledge,S.J. and Harper,J.W. (1999) The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev., 13, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett J.R. (1994) Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin. Cancer Biol., 5, 269–275. [PubMed] [Google Scholar]
- Xu L., Corcoran,R.B., Welsh,J.W., Pennica,D. and Levine,A.J. (2000) WISP-1 is a Wnt-1- and β-catenin-responsive oncogene. Genes Dev., 14, 585–595. [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. and Xiong,Y. (1999) Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell., 3, 579–591. [DOI] [PubMed] [Google Scholar]
- Zhurinsky J., Shtutman,M. and Ben-Ze’ev,A. (2000a) Plakoglobin and β-catenin: protein interactions, regulation and biological roles. J. Cell Sci., 113, 3127–3139. [DOI] [PubMed] [Google Scholar]
- Zhurinsky J., Shtutman,M. and Ben-Ze’ev,A. (2000b) Differential mechanisms of LEF/TCF family-dependent transcriptional activation by β-catenin and plakoglobin. Mol. Cell. Biol., 20, 4238–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F., Eischen,C.M., Randle,D.H., Kamijo,T., Cleveland,J.L., Sherr,C.J. and Roussel,M.F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev., 12, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]