Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain (original) (raw)
Abstract
The key enzyme in coronavirus polyprotein processing is the viral main proteinase, Mpro, a protein with extremely low sequence similarity to other viral and cellular proteinases. Here, the crystal structure of the 33.1 kDa transmissible gastroenteritis (corona)virus Mpro is reported. The structure was refined to 1.96 Å resolution and revealed three dimers in the asymmetric unit. The mutual arrangement of the protomers in each of the dimers suggests that Mpro self-processing occurs in trans. The active site, comprised of Cys144 and His41, is part of a chymotrypsin-like fold that is connected by a 16 residue loop to an extra domain featuring a novel α-helical fold. Molecular modelling and mutagenesis data implicate the loop in substrate binding and elucidate S1 and S2 subsites suitable to accommodate the side chains of the P1 glutamine and P2 leucine residues of Mpro substrates. Interactions involving the N-terminus and the α-helical domain stabilize the loop in the orientation required for _trans_-cleavage activity. The study illustrates that RNA viruses have evolved unprecedented variations of the classical chymotrypsin fold.
Keywords: 3C-like/catalytic dyad/coronavirus/proteinase/X-ray crystallography
Introduction
Transmissible gastroenteritis virus (TGEV) belongs to the Coronaviridae, a family of positive-strand RNA viruses. Coronaviruses have the largest RNA viral genomes known to date (28 500 nucleotides in the case of TGEV) and share a similar genome organization and common transcriptional and translational strategies with the Arteriviridae (den Boon et al., 1991; Cavanagh, 1997). TGEV infection is associated with severe and often fatal diarrhoea in young pigs (for reviews see Enjuanes and van der Zeijst, 1995; Saif and Wesley, 1999).
The viral proteins required for TGEV genome replication and transcription are encoded by the replicase gene (Eleouet et al., 1995; Penzes et al., 2001). This gene encodes two replicative polyproteins, pp1a (447 kDa) and pp1ab (754 kDa) that are processed by virus-encoded proteinases to produce the functional subunits of the replication complex (reviewed in Ziebuhr et al., 2000). The central and C-proximal regions of pp1a and pp1ab are processed by a 33.1 kDa viral cysteine proteinase which is called the ‘main proteinase’ (Mpro) or, alternatively, the ‘3C-like proteinase’ (3CLpro). The name ‘3C-like proteinase’ was introduced originally because of similar substrate specificities of the coronavirus Mpro and picornavirus 3C proteinases (3Cpro) and the identification of cysteine as the principal catalytic residue in the context of a predicted two-β-barrel fold (Gorbalenya et al., 1989a,b). Meanwhile however, several studies have revealed significant differences in both the active sites and domain structures between the coronavirus and picornavirus enzymes (Liu and Brown, 1995; Lu and Denison, 1997; Ziebuhr et al., 1997, 2000; Hegyi et al., 2002). Also, the crystal structures reported for a number of picornavirus 3C proteinases (Allaire et al., 1994; Matthews et al., 1994; Bergmann et al., 1997; Mosimann et al., 1997) have not been useful in predicting the three-dimensional structures of coronavirus main proteinases. Because of the large phylogenetic distance between the two groups of enzymes, we will use the term coronavirus Mpro throughout this article.
Sequence comparisons (Figure 1) and experimental data obtained for other coronavirus homologues allow us to predict that the mature form of the TGEV Mpro is released from pp1a and pp1ab by autoproteolytic cleavage at flanking Gln↓(Ser,Ala) sites (Eleouet et al., 1995; Hegyi and Ziebuhr, 2002). Accordingly, the TGEV Mpro has 302 amino acid residues that correspond to the pp1a/pp1ab residues 2879–3180. In vivo and in vitro analyses of avian infectious bronchitis virus (IBV), mouse hepatitis virus (MHV) and human coronavirus 229E (HCoV 229E) Mpro activities have shown consistently that the proteinase cleaves the replicase polyproteins at 11 conserved sites and, therefore, it seems reasonable to conclude that the Mpro-mediated processing pathways are conserved in all coronaviruses, including TGEV.

Fig. 1. Sequence comparison of coronavirus main proteinases. The alignment was produced using CLUSTAL X, version 1.81 (Thompson et al., 1997), and corrected manually on the basis of the three-dimensional structure of TGEV Mpro. The corresponding sequences of FIPV (strain 79–1146), HCoV (strain 229E), bovine coronavirus (BCoV, isolate LUN), MHV (strain JHM) and IBV (strain Beaudette) were derived from the replicative polyproteins of the respective viruses whose sequences are deposited at the DDBJ/EMBL/GenBank database (accession Nos: FIPV, AF326575; HCoV, X69721; BCoV, AF391542; MHV, M55148; IBV, M95169; TGEV, AJ271965). The β-strands and α-helices as revealed in the TGEV Mpro crystal structure (this study) are shown above the sequence alignment (see also Figures 4 and 5). Black background colour indicates the catalytic cysteine and histidine residues. Grey background colour indicates the key residue of the S1 subsite (TGEV Mpro His162) and its equivalents in other coronavirus main proteinases. Also shown in grey are the phenylalanine and tyrosine residues (TGEV Mpro Phe139 and Tyr160) that are proposed to stabilize the neutral state of His162 (see text for details).
Previous theoretical studies and experimental data have led to the following conclusions (Bazan and Fletterick, 1988; Gorbalenya et al., 1989a,b; Liu and Brown, 1995; Lu et al., 1995; Ziebuhr et al., 1995, 1997, 2000; Lu and Denison, 1997; Seybert et al., 1997; Ziebuhr and Siddell, 1999; Ng and Liu, 2000; Hegyi et al., 2002): (i) Corona virus main proteinases employ conserved cysteine and histidine residues in the catalytic site. In TGEV Mpro, these are Cys144 and His41. There has been some debate on the existence of a third residue in the catalytic centre. In common with picornavirus 3C proteinases, the catalytic centre of the coronavirus Mpro is predicted to be embedded in a chymotrypsin-like, two-β-barrel structure in which cysteine (rather than serine) serves as the principal nucleophile. (ii) Coronavirus main proteinases have well-defined substrate specificities. All known cleavage sites contain bulky hydrophobic residues (mainly leucine) at the P2 position, glutamine at the P1 position, and small aliphatic residues at the P1′ position. (iii) Coronavirus main proteinases possess a large C-terminal domain of ∼110 amino acid residues that is not found in other RNA virus 3C-like proteinases. The characterization of recombinant proteins, in which 33, 28 and 34 C-terminal amino acid residues were deleted from the IBV, MHV and HCoV main proteinases, respectively, resulted consistently in dramatic losses of proteolytic activity, suggesting that the C-terminal domain of Mpro contributes to proteolytic activity through undefined mechanisms.
The 1.96 Å TGEV Mpro crystal structure reported herein reveals the structural details of a unique catalytic system and facilitates the interpretation of previously published mutagenesis studies that have, at least in part, remained speculative due to the complete lack of structural information on ‘3C-like’ enzymes.
Results and discussion
Structure determination by MAD phasing
The presence of 10 methionine residues in the TGEV Mpro molecule suggested that selenomethionine-based multiwavelength anomalous dispersion (MAD; Hendrickson et al., 1990) could be used to solve the phase problem. The unit cell dimensions of the crystals (a = 72.8 Å, b = 160.1 Å, c = 88.9 Å, β = 94.3°, space group _P_21) and self-rotation calculations indicated the presence of as many as six TGEV Mpro molecules per asymmetric unit. In the MAD phasing process, we finally succeeded in locating 48 (out of 60) crystallographically independent selenium sites by the ‘Shake & Bake’ approach to direct methods (Weeks and Miller, 1999), without recourse to heavy atom derivatives or other methods of phasing (see Materials and methods). The phases obtained resulted in a readily interpretable electron density map.
Quality of the model
All six copies (designated A–F) of the TGEV Mpro in the asymmetric unit of the crystal could be built into well-defined electron density (Figure 2), which covered almost all of the 302 amino acid residues of each monomer. The only exceptions were the two C-terminal residues which were not visible in five of the six chains. Monomers A, E and F also lacked electron density for residue 300.

Fig. 2. Stereo view of a representative part of the electron density map. The 2|_F_o| – |_F_c| electron density map (1.96 Å resolution, contoured at 1σ above the mean) corresponds to Mpro residues 160–162 (Tyr–Met–His), a conserved motif in coronavirus main proteinases. The strong hydrogen bonding interaction between the Tyr160 hydroxyl group and His162 Nδ1 is indicated.
The final model comprises 1798 amino acid residues and 1006 water molecules, as well as 27 sulfate ions, nine dioxane molecules and six 2-methyl-2,4-pentanediol (MPD) molecules from the crystallization medium. The refinement converged to a final _R_-factor of 0.210 and an _R_free (Brünger, 1992) of 0.256, with good stereochemistry. Altogether, 88.4% of the amino acid residues were found in the most favoured regions of the Ramachandran plot, and 10.8% were in additionally allowed regions. Residues Asn70, Asn71 and Ser279 were in regions only generously allowed, but had clear electron density.
Domain structure
The six TGEV Mpro monomers present in the asymmetric unit are arranged in three dimers (Figure 3). Each monomer is folded into three domains, the first two of which are antiparallel β-barrels reminiscent of those found in serine proteinases of the chymotrypsin family (Figure 4). Residues 8–100 form domain I, and residues 101–183 make up domain II. The connection to the C-terminal domain III is formed by a long loop comprising residues 184–199. Domain III (residues 200–302) contains a novel arrangement of five α-helices. A deep cleft between domains I and II, lined by hydrophobic residues, constitutes the substrate-binding site. The catalytic site is situated at the centre of the cleft.

Fig. 3. Stereo depiction of the six molecules (three dimers) of TGEV Mpro in the asymmetric unit. The monomers A–F are shown in different colours; A = red, B = black, C = green, D = orange-red, E = yellow and F = cyan. Note the 2-fold symmetry axes between the monomers in each of the dimers, and between the two lower dimers in the figure (AB and EF). Each of the monomers measures ∼70 Å × 22 Å × 40 Å.

Fig. 4. A MOLSCRIPT diagram (Kraulis, 1991) showing the overall fold of TGEV Mpro (A) with the two β-barrel domains and the α-helical C-terminal domain. β-strands and helices are represented as arrows and cylinders, respectively. The β-barrels of each domain I and II are composed of six-stranded β-sheets (green). Domain III is composed mainly of α-helices (red). The structures of HAV 3Cpro (PDB code: 1HAV) (B) and α-chymotrypsin (4CHA, residues 12–15 and 147–148 are excised) (C) are shown for comparison.
The interior of the β-barrel of domain I consists entirely of hydrophobic residues. A short α-helix (helix A; Tyr53–Ser58) closes the barrel like a lid. Domain II is smaller than domain I and also smaller than the homologous domain II of chymotrypsin and hepatitis A virus (HAV) 3Cpro (Tsukada and Blow, 1985; Allaire et al., 1994; Bergmann et al., 1997). Several secondary structure elements of HAV 3Cpro (strands b2II and cII and the intervening loop) are missing in the TGEV Mpro. Also, the domain II barrel of the TGEV Mpro is far from perfect (Figure 4). The segment from Gly135 to Ser146 forms a part of the barrel, even though it consists mostly of consecutive loops and turns. In fact, in contrast to domain I, a structural alignment of domain II has proven difficult. The superposition of domains I and II of the TGEV Mpro onto those of the HAV 3Cpro yields an r.m.s.d. of 1.85 ± 0.05 Å for 114 equivalent (out of 184 compared) Cα pairs, while domain II alone displays an r.m.s.d. of 3.25 ± 0.28 Å for 57 (out of 85) Cα pairs.
Domain III is composed of five, mostly antiparallel, α-helices and the loops connecting them. The crossover angles are ∼90° between helices B and E, ∼30° between B and D, ∼20° between C and E, and ∼80° between E and F, whereas C–B and B–F are parallel to each other (see Figure 5). Interhelical contacts are mediated by hydrophobic side chains. The loops between the helices are quite long and fill up most of the interstitial space of domain III. Database searches (Holm and Sander, 1993; Gilbert et al., 1999) did not reveal other proteins or protein domains with the same topology as domain III. The N-terminal segment (residues 1–5) of the polypeptide chain folds onto domain III, placing the N-terminus of the protein within 17.0 (±2.7) Å of the C-terminus (Figure 4).
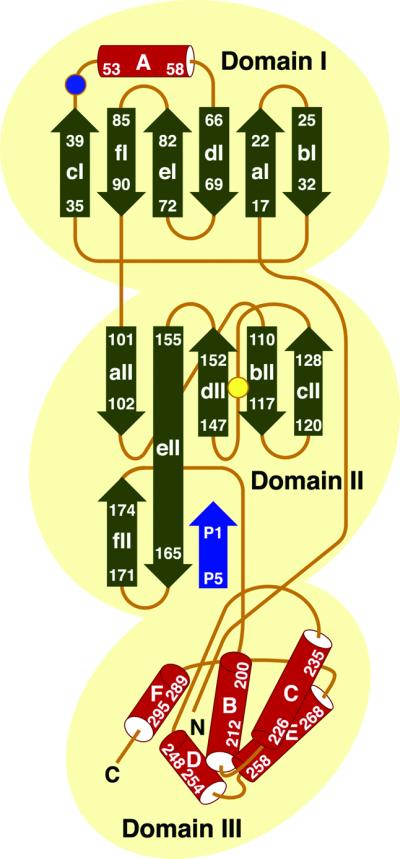
Fig. 5. Topological representation of the secondary structure elements of a TGEV Mpro monomer. α-helices and β-strands are represented as cylinders and arrows, respectively. Numbers indicate the N- and C-terminal residues of the secondary structure elements. Strands bI and cI are adjacent. Cys144 (yellow) and His41 (blue) are shown by circles. The positions of the N- and C-termini are indicated. Also, the presumed localization of the P5–P1 region of a model substrate is shown (blue) (for details, see text and Figure 7).
The six copies of the TGEV Mpro in the asymmetric unit of the crystal are highly similar. The core regions of domains I and II display an r.m.s.d. of 0.29 (±0.09) Å for 130 equivalent Cα atoms (monomer A as a reference; herein, geometrical values given are the r.m.s. over the six monomers, with the corresponding standard deviation). If all 299 well-determined Cα positions are included, the average r.m.s.d. for all monomers is 0.57 (±0.18) Å. The largest deviations of the main chain trace are in: (i) the N-terminal segment from residues 1 to 4 (average r.m.s.d. 1.69 ± 0.91 Å); (ii) the flexible surface loop from residues 216 to 225 (average r.m.s.d. 0.99 ± 0.51 Å); (iii) the C-terminus of helix E and the loop region between residues 267 and 276 (average r.m.s.d. 0.99 ± 0.42 Å); and (iv) the segment 294–300 following the C-terminal F helix (average r.m.s.d. 1.55 ± 0.44 Å). In addition to being flexible and at the surface of the molecules, segments (ii) and (iii) are involved in interdimer crystal contacts in some but not all of the six protomers. Surprisingly, the regions with the highest r.m.s.d. are not the regions with the highest temperature factors, except for the C-terminal domain of monomer F which does have high temperature factors (∼70 Å2; whole model 47 Å2, including all 1006 water molecules).
Active site
The active site of the coronavirus Mpro is similar to those of the picornavirus 3C proteinases, as had been predicted earlier (Gorbalenya et al., 1989b). The mutual arrangement of the nucleophilic Cys144 and the general acid–base catalyst His41 of TGEV Mpro is identical to that of the HAV 3Cpro Cys172 and His44 residues and the Ser195 and His57 residues of chymotrypsin. The distance between the sulfur atom of Cys144 and the Nε2 of His41 is 4.05 (±0.04) Å, i.e. longer than the corresponding cysteine– histidine distances in HAV 3Cpro (3.92 Å; Bergmann et al., 1997), poliovirus (PV) 3Cpro (3.4 Å; Mosimann et al., 1997) and papain (3.65 Å; Kamphuis et al., 1984) (Figure 6B and C). In contrast to papain, but in agreement with the picornavirus 3C proteinases, the sulfur atom is in the plane of the histidine imidazole. There are clear indications from the difference Fourier synthesis (Figure 6A) that Cys144 is oxidized, at least to the stage of the sulfinic acid, -SO2–, and probably to the sulfonic acid, -SO3–, in all six copies of TGEV Mpro in the crystal. Such oxidation could occur during the time required for crystallization or during X-ray data collection, and would lead to inactivation of the enzyme. Refinement of the corresponding derivatives was, however, not successful.

Fig. 6. Active site of the TGEV Mpro. (A) Difference electron density (|_F_o| – |_F_c| at 3.0σ above the mean; red) for the oxidized active site Cys144, indicating three oxygen atoms bound to the sulfur. (B) The catalytic Cys144 and His41 residues are shown. The region forming the oxyanion hole (main chain amides of Gly142, Thr143 and Cys144) is highlighted in pink. The water molecule, which occupies a position equivalent to that of the catalytic aspartate of serine proteinases, is shown together with its hydrogen-bonding partners, His41, His163 and Asp186. (C) Superposition of the active site residues of chymotrypsin (shown in red) with the spatially equivalent residues of TGEV Mpro (blue) and HAV 3Cpro (green). The equivalent to the third catalytic residue (Asp102) of chymotrypsin is Asp84 in HAV 3Cpro (side chain oriented differently) and Val84 in TGEV Mpro.
It is generally assumed that the native state of the active site of papain-like cysteine proteinases is a thiolate– imidazolium ion pair formed by cysteine and histidine residues (Polgár, 1974). In proteinases of the papain family, an asparagine is the third member of the catalytic triad. Chymotrypsin and other members of this serine proteinase family have a catalytic triad consisting of Ser195...His57...Asp102. In HAV 3Cpro, Asp84 is present at the required position, although its side chain points away from His44, making its role disputable (Malcolm, 1995; Bergmann et al., 1997). PV 3Cpro, human rhinovirus (HRV) 3Cpro and HRV 2Apro have a glutamate or aspartate in the proper orientation to accept a hydrogen bond from the active site histidine (Matthews et al., 1994; Mosimann et al., 1997; Petersen et al., 1999). In contrast, TGEV Mpro has Val84 in the corresponding position, with its side chain pointing away from the catalytic site (Figure 6B and C). A buried water molecule is found in the place that normally would be occupied by the side chain of the third member of the catalytic triad. This water molecule makes hydrogen bonds to His41 Nδ1, His163 Nδ1 and Asp186 Oδ1 (Figure 6B). His163 is not conserved among coronavirus main proteinases and its substitution by leucine (Mpro-H163L) had no significant effect on the proteolytic activity in the standard peptide assay (see Materials and methods), as compared with the activity of the wild-type Mpro (Table I). Asp186 makes a salt bridge to Arg40 that appears to be required to maintain the active site geometry, since both Asp186 and Arg40 are absolutely conserved among coronaviruses. Through this (and other) interaction(s), the polypeptide segment 184–199, which connects domains II and III and is probably involved in substrate binding (see below), is held in the proper position. Taken together, the data contradict a direct involvement of His163 or Asp186 in catalysis, making the TGEV Mpro a clear case of a viral cysteine proteinase employing only a catalytic dyad.
Table I. Enzymatic activities of TGEV Mpro mutants.
| Plasmid | Oligonucleotides used for cloning or mutagenesis (5′→3′) | Protein | Mpro amino acids | Activity (%)a |
|---|---|---|---|---|
| pMal-Mpro | TCAGGTTTGCGGAAAATGGCAC, | Mpro | Ser1–Gln302 | 100 |
| AAAAGGATCCTTACTGAAGATTTACACCATACATTTG | ||||
| pMal-MproΔ184–302 | TCAGGTTTGCGGAAAATGGCAC, | MproΔ184–302 | Ser1–Gly183 | <0.02 |
| AAAGGATCCTTAACCACCGTACATTTCTCCTTCAAAATT | ||||
| pMal-MproΔ200–302 | TCAGGTTTGCGGAAAATGGCAC, | MproΔ200–302 | Ser1–Ser199 | 0.4 |
| AAAGGATCCTTATGACATGACATTAGTACCTTCCAATTG | ||||
| pMal-MproΔ1–5/Δ200–302 | ATGGCACAGCCTAGTGGTCTTGTA, | MproΔ1–5/Δ200–302 | Met6–Ser199 | 0.6 |
| AAAGGATCCTTATGACATGACATTAGTACCTTCCAATTG | ||||
| pMal-MproΔ1–5 | ATGGCACAGCCTAGTGGTCTTGTA, | MproΔ1–5 | Met6–Gln302 | 0.3 |
| AAAAGGATCCTTACTGAAGATTTACACCATACATTTG | ||||
| pMal-Mpro-H163L | GTATACATGCATCTCTTAGAACTTGGAAATGGCTCGCAT, | Mpro-H163L | Ser1–Gln302 | 98 |
| TCCAAGTTCTAAGAGATGCATGTATACAAAATAGAGAAT | (His163→Leu) | |||
| pMal-Mpro-C144A | AGCTGGTACTGCTGGATCAGTAGGTTATGTGTTAGAA, | Mpro-C144A | Ser1–Gln302 | <0.02 |
| CTACTGATCCAGCAGTACCAGCTATAAAAGATCCTTT | (Cys144→Ala) |
Substrate hydrolysis by cysteine and serine proteinases occurs through a covalent tetrahedral intermediate resulting from attack of the active site nucleophile on the carbonyl carbon of the scissile bond. The developing oxyanion is stabilized by strong hydrogen bonds donated by amide groups of the enzyme. This so-called ‘oxyanion hole’ is also found in TGEV Mpro. It is made up by the main chain amides of Gly142, Thr143 and Cys144 (Figure 6B).
Substrate-binding site
The specificity of Mpro for a very limited range of amino acids at the P1, P2 and P4 positions resembles the substrate specificity of picornavirus 3C proteinases (Palmenberg, 1990; Ziebuhr et al., 2000). This leads us to believe that, similarly to 3Cpro (Matthews et al., 1994; Bergmann et al., 1997; Mosimann et al., 1997), specific substrate binding by Mpro is ensured by well-defined S4, S2 and S1 specificity pockets. In order to visualize potential interactions with the substrate, we have modelled a pentapeptide representing the P5–P1 residues of a TGEV Mpro cleavage site (Asn–Ser–Thr–Leu–Gln, pp1a amino acids 2874–2878; Hegyi and Ziebuhr, 2002) into the substrate-binding cleft of Mpro (Figure 7). The model is based on the assumption that Mpro binds substrates in a manner analogous to that found in complexes of chymotrypsin-like proteinases with peptide inhibitors. X-ray structures have shown that the P4–P1 residues of peptide inhibitors assume a common main chain conformation when bound to these proteinases, with the P4 and P3 residues adopting a β conformation and the P2 and P1 residues assuming a specific main-chain conformation suitable to place their side chains in the pre-formed S1 and S2 specificity pockets (James et al., 1980; Fujinaga et al., 1985, 1987, Matthews et al., 1999). These studies lead us to suggest that the residues P5 to P3 of Mpro substrates may form an antiparallel β-sheet with segment 164–167 of the long strand eII on one side, and with the segment 186–191 (which links domains II and III) on the other. Hydrogen bonding interactions are likely between the main chain amide and carbonyl oxygen atoms of substrate residues Thr(P3), Ser(P4) and Asn(P5) and the main chain atoms of TGEV Mpro residues Glu165, Ser189 and Gly167 (see Figure 7).

Fig. 7. Stereo diagram of a P5–P1 substrate (Asn–Ser–Thr–Leu–Gln, red; corresponding to the TGEV Mpro N-terminal autoprocessing site) modelled into the active site cleft of the TGEV Mpro. Hydrogen bonds are depicted by dotted lines.
S1 subsite
It has been shown for the HAV, HRV and PV 3Cpro enzymes that the imidazole side chain of a conserved histidine, which is located in the centre of a hydrophobic pocket, interacts with the P1 carboxamide side chain of the substrate. This interaction is generally accepted to determine the picornavirus 3Cpro specificity for glutamine at P1 (Matthews et al., 1994, 1999; Bergmann et al., 1997; Mosimann et al., 1997). Mutational analyses revealed that any replacement of His162 completely abolished the proteolytic activities of the HCoV and feline infectious peritonitis virus (FIPV) Mpro enzymes (Ziebuhr et al., 1997; Hegyi et al., 2002). The structure shows that the imidazole side chain of His162 is positioned suitably to interact with a P1 glutamine side chain. His162 is located at the very bottom of a hydrophobic pocket which is formed by residues Phe139 and the main-chain atoms of Ile140, Leu164, Glu165 and His171. The side chain of Glu165 forms an ion pair (2.96 ± 0.14 Å) with His171. This salt bridge is itself on the periphery of the molecule, forming part of the ‘outer wall’ of the S1 subsite. Accordingly, mutants of the HCoV 229E Mpro, in which the residue equivalent to His171 had been replaced by alanine, serine or threonine, retained significant proteolytic activities (Ziebuhr et al., 1997). In order to interact with the P1 glutamine side chain of the substrate, His162 has to maintain a neutral state over a wide pH range. Most probably, this is achieved by two important interactions: (i) stacking onto the phenyl ring of Phe139, at a distance of 3.53 ± 0.18 Å; and (ii) accepting a hydrogen bond from the buried Tyr160 hydroxyl group which has no other hydrogen-bonding partner. The role proposed for the hydroxyl group of Tyr160 is strongly supported by FIPV Mpro mutagenesis studies in which the proteolytic activities of Y160F, Y160G, Y160A and Y160T mutants were shown to be dramatically reduced (Hegyi et al., 2002). Tyr160 is part of the absolutely conserved coronavirus Mpro sequence signature, 160Tyr-X-His162 (Figures 1 and 2), whereas Gly(Ala)-X-His is found at the equivalent sequence position in most 3C and 3C-like proteinases (Gorbalenya et al., 1989a). Accordingly, in the 3C and 3C-like proteinases, stabilization of histidine in the neutral tautomeric state has to be ensured by other residues. Notably, in the case of PV 3Cpro, this involves a tyrosine residue (Tyr138) which, however, is provided by a different part of the structure (β-strand cII; Mosimann et al., 1997). For HAV 3Cpro, other mechanisms are proposed (Bergmann et al., 1997).
Halfway down the S1 subsite of TGEV Mpro, there is dumbbell-shaped electron density which we have assigned to two water molecules, although theoretically they are too close to one another (2.10 ± 0.16 Å). One of them makes a hydrogen bond with Nε2 of His162, while the second one, unusually for water, makes no additional contacts. In our model of the substrate complex, these two water molecules mark the position of the carboxamide group of the P1 glutamine side chain.
S2 subsite
Coronavirus main proteinases have a strong preference for leucine at the P2 position (Ziebuhr et al., 2000). The putative S2 subsite identified in the structure is a hydrophobic pocket that is suitably positioned and large enough to accommodate a leucine side chain easily. The S2 pocket is lined by the side chains of Leu164 (the main chain of which forms part of the S1 subsite, see above), Pro188, Ile51, His41 and Thr47 (Figure 7). In our electron density maps, part of the S2 subsite (of all six copies of the monomer) harbours extra electron density that we interpreted as an MPD molecule from the crystallization medium. In the HAV 3Cpro, the corresponding subsite is formed by different parts of the polypeptide chain. It is also smaller and can accommodate the side chains of serine and threonine (Bergmann et al., 1997).
Quaternary structure
The quaternary arrangement of the proteinase is a homodimer, with three copies in the asymmetric unit (monomers A and B, C and D, and E and F). All dimers have approximate C2 symmetry (Figure 3) and ∼1580 (±199) Å2 of each monomer, i.e. 11–12% of its solvent-accessible surface, are buried upon dimerization. The dimer formation is driven mainly by intermolecular interactions between domains II and III of one monomer and the N-terminal residues of the other (see below for further details). In contrast, the domain III–domain III interface appears to be the consequence rather than the cause of other intermolecular interactions. It involves a relatively small area of 337 ± 45 Å2 and comprises only two hydrogen bonds, between the amide group of Gly281 (molecule A) and the main-chain oxygen of Ser279 (molecule B), as well as its symmetry mate, Gly281B...Ser279A (3.22 ± 0.37 Å, averaged over all six monomers).
Interestingly, the N-terminal residues of each monomer are relatively close to the substrate-binding site of the other monomer in the dimer. The following observations for monomer A hold true for all other monomers. The NH3+ group of Ser1A, which is the P1′ residue of the autocleavage reaction of TGEV Mpro, is 11.9 ± 1.6 Å from the active site Cys144B Sγ of the second molecule in the dimer but as much as 34.2 ± 0.9 Å away from its own active site cysteine. Ser1A is in contact with residues participating in the substrate-binding site of monomer B. Its NH3+ group makes a salt bridge (4.99 ± 1.04 Å) to the carboxylate of Glu165B (Figure 8). This glutamate, which is absolutely conserved among coronaviruses, is part of the S1 subsite (see above), where it also interacts with His171. Although these two side chains form the ‘wall’ of the specificity site, they have their polar groups oriented towards the surface of the proteinase molecule and away from the substrate’s P1 glutamine. An intermolecular ionic interaction between Arg4A and Glu286B (6.0 ± 0.7 Å) appears to play a role in positioning the N-terminal residues. Because of the 2-fold non-crystallographic symmetry (NCS), the same interaction occurs between Arg4B and Glu286A. Residues 6A–8A form a short β-strand interacting with strand cII of monomer B (at Val124B). Most of the interactions between the N-terminus of molecule A and the region next to the S1 subsite of molecule B constitute a perfect fit. Given the fact that the P′ residues in serine and cysteine proteinases constitute the leaving group of the cleavage reaction and, in coronavirus main proteinases, are not subject to stringent specificity requirements, it is quite conceivable that, after autoproteolysis, the N-terminus of one monomer slides over the active site of the partner monomer and adopts the position seen in our crystal structure, i.e. with Ser1A interacting with Glu165B at the ‘outer wall’ of the S1 subsite. This, in turn, would suggest that the dimer we are seeing corresponds to the product of the autolysis reaction and that this occurs in trans. Molecular modelling revealed that binding of the Mpro N-terminus in the active site cleft of the same molecule would require remodelling of the entire N-terminal segment and beyond (residues 1–13; data not shown), making cleavage in cis less likely. There is additional experimental evidence supporting these conclusions. First, dilution experiments with MHV Mpro translated in vitro contradict _cis_-cleavage activity (Lu et al., 1996). Secondly, the fact that, early in infection, Mpro remains part of a relatively stable 150 kDa precursor protein in which it is flanked by hydrophobic domains (Schiller et al., 1998) argues against rapid autoprocessing in cis. The proposed model of intermolecular self-processing would imply that components of the replication complex could first be anchored to membranes (i.e. the site of RNA replication) in an uncleaved form, and only later, when the precursor proteins accumulate to high local concentrations, will Mpro release itself by intermolecular cleavage, thereby triggering the complete spectrum of _trans_-processing reactions.

Fig. 8. Intra- and intermolecular contacts of the TGEV Mpro N-terminus. (A) MOLSCRIPT stereo representation of a TGEV Mpro dimer. Molecule A is coloured from blue at the N-terminus, via green (domain II), to red (C-terminus), while molecule B is shown in grey. The catalytic Cys144 and His41 residues are labelled in both monomers. (B) Detailed view of the interactions made by the N-terminal segment (blue) and domains II/III of monomer A as well as domains II/III of monomer B. Residues critically involved in these interactions are designated by the single-letter code and shown in ball-and-stick representation (see text for details). The N- and C-termini of molecule A are indicated.
Intramolecular interactions of the N-terminus
A specific conformation of the N-terminal segment allows it to ‘squeeze’ residues 1–8 in between domains II and III of the same monomer and domains II and III of monomer B (see above and Figure 8). In this context, the N-terminus also interacts with domains II and III of its own protomer. For example, the side-chain amino group of Lys5A makes strong intramolecular hydrogen bonds with Ser110A Oγ of domain II (2.83 ± 0.15 Å), and with the Glu286A main chain oxygen (2.80 ± 0.07 Å), as well as with Glu291A Oε1 (2.74 ± 0.13 Å) of domain III. Furthermore, the side chain of Leu3A completes a hydrophobic patch on domain III which includes Phe206A, Ala209A, Phe287A, Val292A, the Cβ atom of Gln295A and Met296A; these residues belong to helices B and F. All sequenced members of the coronavirus proteinase family have a hydrophobic residue in position 3, while glycine is absolutely conserved in position 2 (see Figure 1). The latter residue adopts the αL conformation which is easily accessible only to glycine. To investigate the functional significance of these interactions, a recombinant protein, MproΔ1–5, in which the N-terminal residues Ser1–Lys5 were removed from the Mpro sequence, was expressed and tested for proteolytic activity in a _trans_-cleavage assay using a 15mer peptide representing the N-terminal TGEV Mpro autoprocessing site. As shown in Table I, the activity of MproΔ1–5 was decreased to only 0.3% of the Mpro activity. We conclude from these data that, indeed, residues 1–5 may be critically involved in stabilizing the mutual orientation of domains II and III and thus, indirectly, in maintaining the proper orientation of the intervening loop region (residues 184–199). If this hypothesis is correct, then the deletion of domain III should have similarly detrimental effects on the proteolytic activity and, in fact, the published data (see Introduction) seem to support this conclusion. To corroborate this hypothesis further, an additional set of Mpro mutants was characterized in which we used the structural information to remove domain III completely. In this approach, the probability of domain III misfolding, which might have been the cause of Mpro inactivation in previous studies using randomly ‘truncated’ coronavirus main proteinases (Lu and Denison, 1997; Ziebuhr et al., 1997; Ng and Liu, 2000), should be significantly reduced. The TGEV Mpro deletion mutants tested for activity comprised (i) domains I and II (MproΔ184–302); (ii) domains I and II together with the entire loop region (MproΔ200–302); or (iii) domains I and II combined with the loop region but lacking the five N-terminal residues (MproΔ1–5/Δ200– 302). As Table I shows, MproΔ200–302 had clearly detectable (albeit significantly reduced) activity (0.4% of Mpro). Similarly, the mutant MproΔ1–5/Δ200–302 had significantly reduced activity (0.6% of Mpro). In sharp contrast, no activities were detectable for MproΔ184–302 and the active site mutant, Mpro-C144A (the latter being used as a negative control). The fact that residues 184–199 proved to be indispensable for proteolytic activity supports our model of substrate binding (Figure 7) in which residues of the loop are predicted to be critically involved in the formation of a β-sheet-type structure with the substrate (see above). The data also show that an intact N-terminus and the C-terminal domain are required for full activity. The structure suggests that the additional α-helical domain III as well as the N-terminal residues help fix domains II and the loop 184–199 in a catalytically competent orientation. It will be interesting to investigate whether similar mechanisms are also operating in other 3C-like proteinases with (smaller) C-terminal domains (e.g. arteriviruses and potyviruses; Ziebuhr et al., 2000; Hegyi et al., 2002).
Beyond its presumed role in proteolytic activity, domain III may have other functions, which remain to be determined. In contrast to picornavirus 3C proteinases for which RNA-binding activities are well established (Andino et al., 1993; Leong et al., 1993; Xiang et al., 1995), the Mpro structure does not support such an activity for the coronavirus main proteinase. Thus, calculation of the electrostatic potential (Nicholls et al., 1991) does not reveal an overall basic character of domain III, nor are there distinct patches of basic or aromatic residues (data not shown). The same applies to domains I and II. Also, the conserved picornavirus sequence motif, KFRDI, located between domains I and II, as well as the small helices and reverse turns that together form the RNA-binding site of HAV 3Cpro (Bergmann et al., 1997) are missing in the TGEV Mpro structure.
Conclusion
The crystal structure of TGEV Mpro shows that coronaviruses have evolved proteinases in which a thiolate– imidazolium catalytic dyad has been combined with a two-β-barrel fold. This framework is extended further by a novel α-helical domain that, together with the N-terminal residues 1–5, appears to be involved in proteolytic activity by maintaining the proper positioning of the presumed substrate-binding loop, 184–199. We are confident that the first crystal structure of a non-picornaviral chymotrypsin-like cysteine proteinase will facilitate further molecular modelling of other members of the huge family of RNA viral ‘3C-like’ enzymes for which structural information is still lacking.
Materials and methods
Protein purification and crystallization
Recombinant TGEV Mpro was expressed and purified as previously described for the HCoV and FIPV main proteinases (Ziebuhr et al., 1997; Hegyi et al., 2002). Briefly, the coding sequence of the TGEV Mpro was inserted into the _Xmn_I and _Bam_HI sites of pMal-c2 plasmid DNA (New England Biolabs). The resulting plasmid, pMal-Mpro, was used to transform Escherichia coli TB1 cells. The maltose-binding protein (MBP)–TGEV Mpro fusion protein was purified by amylose–agarose chromatography, cleaved with factor Xa, and the recombinant Mpro (residues Ser1–Gln302) was purified by hydrophobic interaction, anion exchange and size exclusion chromatography (Hegyi et al., 2002). The purified and concentrated TGEV Mpro (12.5 mg/ml) was stored in 12 mM Tris–HCl pH 7.5, 120 mM NaCl, 1 mM dithiothreitol (DTT), 0.1 mM EDTA. This protein solution was used to crystallize Mpro by the hanging drop vapour diffusion method at 4°C. The best crystals, which were of triangular shape and had dimensions of ∼0.3 × 0.25 × 0.3 mm, were obtained by using 100 mM HEPES pH 8.8, 1.8 M ammonium sulfate, 6% MPD, 5 mM DTT and 4% dioxane as the reservoir and grew in ∼10 days.
Incorporation of selenomethionine
The Mpro structure could not be solved using conventional molecular replacement techniques. Therefore, selenomethionine (SeMet)-substituted TGEV Mpro was produced. The coding sequence of the MBP–TGEV Mpro fusion protein was inserted into pET-11d (Novagen), and the resulting plasmid, pET-TGEV-Mpro, was used to transform the methionine-auxotrophic 834(DE3) E.coli strain (Novagen), which was propagated in minimal medium containing 40 µg/ml seleno-l-methionine. The SeMet-substituted TGEV Mpro was purified as described above and concentrated to 9.5 mg/ml. Crystals of the SeMet-substituted Mpro were grown as decribed for the native protein but using 2 M ammonium sulfate and 8% MPD.
Diffraction data collection
Crystals used for data collection were rinsed with mustard oil and cryo-cooled in liquid nitrogen. Diffraction data up to 1.95 Å resolution were collected from native crystals at 100 K on the X-ray diffraction beamline at ELETTRA (Sincrotrone Trieste, Trieste, Italy), using a Mar165 CCD detector (Table II). MAD data sets were collected to 2.8 Å resolution at four wavelengths using a Mar165 CCD detector on beamline BW7A of the EMBL Outstation at DESY (Hamburg, Germany). SeMet data sets were collected for the _f_” maximum and _f_′ minimum wavelengths. Additional data were collected at remote wavelengths below and above the Se K-edge (Table II). Data integration and scaling were performed using DENZO and SCALEPACK (Otwinowski and Minor, 1997).
Table II. Summary of X-ray diffraction data from crystals of native and SeMet-substituted Mpro.
| | | Peak | Edge | High | Low | | | | | | | ---------------------------------------- | ------------------- | ------- | ------- | ------- | ------- | ------- | ------- | ------ | ------ | | Beamline | XRDa | BW7Ab | | | | | | | | | Data sete | Native | P1 | P2 | P3 | E1 | E2 | H1 | H2 | L1 | | Wavelength (Å)d | 0.99983 | 0.97487 | 0.97845 | 0.97848 | 0.97864 | 0.97874 | 0.95583 | 0.9080 | 1.0022 | | Resolution (Å) (highest resolution bin)c | 50–1.95 (1.98–1.95) | 30–2.8 | 30–2.8 | 30–2.8 | 30–2.8 | 30–2.8 | 30–2.8 | 30–2.8 | 30–2.8 | | Completeness (%)c | 98.9 (97.0) | 99.9 | 98.1 | 99.7 | 99.9 | 99.7 | 99.7 | 98.8 | 97.3 | | Mosaicity (°) | 0.62 | 0.4 | 0.6 | 0.7 | 0.4 | 0.6 | 0.4 | 0.6 | 0.4 | | _R_merge (%)c,f | 4.2 (22.1) | 10.5 | 11.4 | 10.6 | 8.1 | 8.2 | 8.6 | 7.2 | 8.0 | | _R_rim (%)c,g | 4.6 (27.1) | 12.1 | 13.0 | 12.3 | 9.2 | 8.9 | 10.2 | 7.5 | 10.3 | | _R_pim (%)c,h | 1.8 (15.2) | 6.1 | 6.6 | 6.4 | 4.7 | 4.5 | 5.2 | 3.2 | 5.4 | | Redundancyc | 5.4 (2.9) | 3.8 | 3.8 | 3.9 | 3.8 | 3.9 | 3.7 | 3.6 | 2.9 | | I/σ(I)c | 13.5 (4.0) | 5.4 | 4.7 | 4.8 | 6.1 | 4.1 | 4.1 | 4.9 | 2.5 |
Structure determination
The unit cell dimensions, as well as the self-rotation function (ALMN; CCP4, 1994), implied that several monomers were present in the asymmetric unit. A Matthews coefficient (Matthews, 1968) of 2.3 Å3/Da and a solvent content of 51% were obtained assuming six molecules in the asymmetric unit. The bottleneck of the structure determination was the identification of the 60 selenium positions (six monomers with 10 Se each). Solving the problem by SnB v2.0 (Weeks and Miller, 1999) required data of increased precision, which were obtained by averaging of several data sets and monitoring the process by _R_pim (Weiss and Hilgenfeld, 1997). Only after we had combined three merged peak-wavelength data sets with two merged edge-wavelength data sets (redundancy = 18) were we able to obtain 105 solutions (from 5000 trials) with significantly reduced minimal function values (_R_min = 0.49, CC = 0.51; Hauptman, 1991) (details to be published elsewhere). The positions of the best 60 atom solutions from SnB were examined for NCS. In total, 37 positions were found to obey a 2-fold NCS. This symmetry predicted a further 11 positions. All 48 positions were used in MLPHARE (CCP4, 1994) for phasing, followed by solvent flattening and NCS averaging in DM (Cowtan and Main, 1996). The resulting electron density maps were of sufficient quality for chain tracing. The first monomer was built manually into the experimental electron density map, using the program ‘O’ (Jones et al., 1991). All other monomers were generated by NCS. NCS restraints were applied during the initial stages of refinement at low resolution and later gradually released as the resolution limit was extended to 1.96 Å.
Cycles of adjustments to the model with O and subsequent refinement using the program CNS (Brünger et al., 1998) converged to an _R_free of 0.256 and a crystallographic _R_-factor of 0.210. Data quality and refinement statistics are given in Table III. The quality of the structural model and its agreement with the structure factors were checked with programs PROCHECK (Laskowski et al., 1993), WHATCHECK (Vriend, 1990) and SFCHECK (Vaguine et al., 1999). Solvent accessibility was calculated using the algorithm of Lee and Richards (1971; program NACCESS), using a solvent probe of radius 1.4 Å. The molecular diagrams were drawn using MOLSCRIPT (Kraulis, 1991) and rendered with RASTER 3D (Bacon and Anderson, 1988). Atomic coordinates and structure factors have been submitted to the RCSB Protein Data Bank under accession code 1LVO.
Table III. Phasing statistics, refinement statistics and model quality.
| Phasing | |
|---|---|
| FOMa before solvent flattening | 0.48 |
| FOMa after solvent flattening (no averaging) | 0.72 |
| FOMa after solvent flattening (with averaging) | 0.79 |
| Refinement | |
| Resolution (Å) | 50–1.96 |
| _R_-factorb | 0.210 |
| _R_free | 0.256 |
| No. of non-hydrogen atoms [average _B_-value (Å2)] | |
| Protein (main chain) | 7198 (46.1) |
| Protein (side chain) | 6613 (47.2) |
| Water | 1006 (50.3) |
| MPD | 48 (67.6) |
| Sulfate | 135 (57.1) |
| Dioxane | 54 (71.7) |
| R.m.s. deviation from ideal geometry | |
| Bonds (Å) | 0.017 |
| Angles (°) | 1.9 |
| Improper dihedral angles (°) | 1.16 |
Proteolytic activities of TGEV Mpro mutants
For the expression of Mpro proteins with N- and C-terminal deletions (MproΔ184–302, MproΔ200–302, MproΔ1–5 and MproΔ1–5/Δ200–302), the corresponding Mpro coding sequences were amplified by PCR and inserted into _Xmn_I–_Bam_HI-digested pMal-c2 plasmid DNA. To substitute the Mpro residues Cys144 (by Ala) and His163 (by Leu), the corresponding codons were replaced in pMal-Mpro by site-directed mutagenesis using a recombination-PCR method (Yao et al., 1992). The details of the primers used for cloning and mutagenesis and the amino acid sequences of the recombinant proteins expressed and tested for proteolytic activity are given in Table I. The plasmid DNAs were transformed into E.coli TB1 cells and the recombinant proteins were synthesized, affinity purified and cleaved with factor Xa as described previously (Hegyi et al., 2002). The purity and structural integrity of the mutant proteins were analysed by SDS–PAGE. The control protein for this experiment, wild-type TGEV Mpropro, was purified in an identical manner. Enzymatic activities of the mutant proteins were measured by using a peptide cleavage assay (Ziebuhr et al., 1997) with a peptide substrate representing the N-terminal TGEV Mpro autoprocessing site (H2N-VSVNSTLQSGLRKMA-COOH; letters in bold indicate the scissile bond that is cleaved by Mpro).
Acknowledgments
Acknowledgements
We thank the staff of ELETTRA (Trieste, Italy) and the EMBL Outstation at DESY (Hamburg, Germany) for help with data collection. Access to these research infrastructures was supported by the European Commission (contract numbers HPRI-CT-1999-00033 and HPRI-CT-1999-00017, respectively). We thank M.S.Weiss and D.Pal for their advice and helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft awarded to J.Z. (Zi 618/2), S.G.S. (Si 357/2) and R.H. (Hi 611/2). R.H. thanks the Fonds der Chemischen Industrie.
References
- Allaire M., Chernaia,M.M., Malcolm,B.A. and James,M.N. (1994) Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature, 369, 72–76. [DOI] [PubMed] [Google Scholar]
- Andino R., Rieckhof,G.E., Achacoso,P.L. and Baltimore,D. (1993) Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J., 12, 3587–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon D.J. and Anderson,W.F. (1988) A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graphics, 6, 219–220. [Google Scholar]
- Bazan J.F. and Fletterick,R.J. (1988) Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl Acad. Sci. USA, 85, 7872–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann E.M., Mosimann,S.C., Chernaia,M.M., Malcolm,B.A. and James,M.N. (1997) The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol., 71, 2436–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. (1997) Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol., 142, 629–633. [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan K.D. and Main,P. (1996) Phase combination and cross validation in iterated density-modification calculations. Acta Crystallogr. D, 52, 43–48. [DOI] [PubMed] [Google Scholar]
- den Boon J.A., Snijder,E.J., Chirnside,E.D., de Vries,A.A., Horzinek,M.C. and Spaan,W.J. (1991) Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol., 65, 2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleouet J.F., Rasschaert,D., Lambert,P., Levy,L., Vende,P. and Laude,H. (1995) Complete sequence (20 kilobases) of the polyprotein-encoding gene 1 of transmissible gastroenteritis virus. Virology, 206, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L. and van der Zeijst,B.A.M. (1995) Molecular basis of transmissible gastroenteritis virus epidemiology. In Siddell,S.G. (ed.), The Coronaviridae. Plenum Press, New York, NY, pp. 337–376.
- Fujinaga M., Delbaere,L.T.J., Brayer,G.D. and James,M.N.G. (1985) Refined structure of α-lytic protease at 1.7 Å resolution. J. Mol. Biol., 184, 479–502. [DOI] [PubMed] [Google Scholar]
- Fujinaga M., Sielecki,A.R., Read,R., Ardelt,W., Laskowski,M.,Jr and James,M.N.G. (1987) Crystal and molecular structures of the complex of α-chymotrypsin with its inhibitor turkey ovomucoid third domain at 1.8 Å resolution. J. Mol. Biol., 195, 397–418. [DOI] [PubMed] [Google Scholar]
- Gilbert D., Westhead,D., Nagano,N. and Thornton,J. (1999) Motif-based searching in TOPS protein topology databases. Bioinformatics, 15, 317–326. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Donchenko,A.P., Blinov,V.M. and Koonin,E.V. (1989a) Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett., 243, 103–114. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin,E.V., Donchenko,A.P. and Blinov,V.M. (1989b) Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res., 17, 4847–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptman H.A. (1991) A minimal principle in the phase problem. In Moras,D., Pojamy,A.D. and Thierry,J.C. (eds), Crystallographic Computing 5, From Chemistry to Biology. IUCr and Oxford University Press, Oxford, pp. 324–332.
- Hegyi A. and Ziebuhr,J. (2002) Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol., 83, 595–599. [DOI] [PubMed] [Google Scholar]
- Hegyi A., Friebe,A., Gorbalenya,A.E. and Ziebuhr,J. (2002) Mutational analysis of the active centre of coronavirus 3C-like proteases. J. Gen. Virol., 83, 581–593. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A., Horton,J.R. and LeMaster,D.M. (1990) Seleno methionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J., 9, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- James M.N.G, Sielecki,A.R., Brayer,G.D., Delbaere,L.T.J. and Bauer,C.A. (1980) Structure of product and inhibitor complexes of Streptomyces griseus protease A at 1.8 Å resolution: a model for serine protease catalysis. J. Mol. Biol., 144, 43–88. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kamphuis I.G., Kalk,K.H., Swarte,M.B. and Drenth,J. (1984) Structure of papain refined at 1.65 Å resolution. J. Mol. Biol., 179, 233–256. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT—a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lee B. and Richards,F.M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol., 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Leong L.E., Walker,P.A. and Porter,A.G. (1993) Human rhinovirus-14 protease 3C (3Cpro) binds specifically to the 5′-noncoding region of the viral RNA. J. Biol. Chem., 268, 25735–25739. [PubMed] [Google Scholar]
- Liu D.X. and Brown,T.D. (1995) Characterisation and mutational analysis of an ORF 1a-encoding proteinase domain responsible for proteolytic processing of the infectious bronchitis virus 1a/1b polyprotein. Virology, 209, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Lu,Y. and Denison,M.R. (1996) Intracellular and _in vitro_-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology, 222, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. and Denison,M.R. (1997) Determinants of mouse hepatitis virus 3C-like proteinase activity. Virology, 230, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Lu,X. and Denison,M.R. (1995) Identification and characteriz ation of a serine-like proteinase of the murine coronavirus MHV-A59. J. Virol., 69, 3554–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm B.A. (1995) The picornaviral 3C proteinases: cysteine nucleophiles in serine proteinase folds. Protein Sci., 4, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B.W. (1968) Solvent content of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Matthews D.A. et al. (1994) Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell, 77, 761–771. [DOI] [PubMed] [Google Scholar]
- Matthews D.A. et al. (1999) Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl Acad. Sci. USA, 96, 11000–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann S.C., Cherney,M.M., Sia,S., Plotch,S. and James,M.N. (1997) Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol., 273, 1032–1047. [DOI] [PubMed] [Google Scholar]
- Ng L.F. and Liu,D.X. (2000) Further characterization of the coronavirus infectious bronchitis virus 3C-like proteinase and determination of a new cleavage site. Virology, 272, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–325. [DOI] [PubMed] [Google Scholar]
- Palmenberg A.C. (1990). Proteolytic processing of picornaviral polyprotein. Annu. Rev. Microbiol., 44, 603–623. [DOI] [PubMed] [Google Scholar]
- Penzes Z. et al. (2001) Complete genome sequence of transmissible gastroenteritis coronavirus PUR46-MAD clone and evolution of the purdue virus cluster. Virus Genes, 23, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J.F., Cherney,M.M., Liebig,H.D., Skern,T., Kuechler,E. and James,M.N. (1999) The structure of the 2A proteinase from a common cold virus: a proteinase responsible for the shut-off of host-cell protein synthesis. EMBO J., 18, 5463–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgár L. (1974) Mercaptide–imidazolium ion-pair: the reactive nucleophile in papain catalysis. FEBS Lett., 47, 15–18. [DOI] [PubMed] [Google Scholar]
- Saif L.J., and Wesley,R. (1999) Transmissible gastroenteritis virus. In Straw,B.E.S., Allaire,W.L. Mengeling,W.L. and Taylor,D.J. (eds), Diseases of Swine, 8th edn. Iowa State University Press, Ames, Iowa, pp. 295–325.
- Schiller J.J., Kanjanahaluethai,A. and Baker,S.C. (1998) Processing of the coronavirus MHV-JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology, 242, 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., Ziebuhr,J. and Siddell,S.G. (1997) Expression and characterization of a recombinant murine coronavirus 3C-like proteinase. J. Gen. Virol., 78, 71–75. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada H. and Blow,D.M. (1985) Structure of α-chymotrypsin refined at 1.68 Å resolution. J. Mol. Biol., 184, 703–711. [DOI] [PubMed] [Google Scholar]
- Vaguine A.A., Richelle,J. and Wodak,S.J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D, 55, 191–205. [DOI] [PubMed] [Google Scholar]
- Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graphics, 8, 52–56. [DOI] [PubMed] [Google Scholar]
- Weeks C.M. and Miller,R. (1999) The design and implementation of SnB version 2.0. J. Appl. Crystallogr., 32, 120–124. [Google Scholar]
- Weiss M.S. and Hilgenfeld,R. (1997) On the use of merging _R_-factor as a quality indicator for X-ray data. J. Appl. Crystallogr., 30, 203–205. [Google Scholar]
- Xiang W., Harris,K.S., Alexander,L. and Wimmer,E. (1995) Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol., 69, 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z., Jones,D.H. and Grose,C. (1992) Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl., 1, 205–207. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J. and Siddell,S.G. (1999) Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol., 73, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Herold,J. and Siddell,S.G. (1995) Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J. Virol., 69, 4331–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Heusipp,G. and Siddell,S.G. (1997) Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol., 71, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder,E.J. and Gorbalenya,A.E. (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol., 81, 853–879. [DOI] [PubMed] [Google Scholar]