Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction (original) (raw)
Abstract
Polyglutamine (polyGln) aggregation is implicated in the disease progression of Huntington's disease and other expanded CAG repeat diseases. PolyGln aggregation in vitro follows a simple nucleated growth polymerization pathway without apparent complications such as populated intermediates, alternative assembly pathways, or secondary nucleation phenomena. Previous analysis of the aggregation of simple polyGln peptides revealed that the critical nucleus (the number of monomeric units involved in the formation of an energetically unfavorable aggregation nucleus) is equal to one, suggesting that polyGln nucleation can be viewed as an unfavorable protein folding reaction. We provide here a method for experimentally determining the number of elongation growth sites per unit weight for any polyGln aggregate preparation, a key parameter required for completion of the nucleation kinetics analysis and determination of the thermodynamics of nucleation. We find that, for the polyGln peptide Q47, the second-order rate constant for fibril elongation is 11,400 liters/mol per s, whereas Kn*, the equilibrium constant for nucleation of aggregation, is remarkably small, equal to 2.6 × 10-9. The latter value corresponds to a free energy of nucleus formation of +12.2 kcal/mol, a value consistent with a highly unfavorable folding reaction. The methods introduced here should allow further analysis of the energetics of polyGln nucleus formation and accurate comparisons of the seeding capabilities of different fibril preparations, a task of increasing importance in the amyloid field.
Keywords: amyloid, Huntington's disease, nucleated growth polymerization, nucleation free energy
Among the family of coagulation mechanisms by which proteins can noncovalently aggregate (1), a particularly important limiting case is nucleated growth polymerization, which is implicated in crystallization and some cases of amyloid fibril formation. Consistent with its association with the formation of more ordered aggregates, this mechanism requires the energetically unfavorable formation of a nucleus from bulk phase monomers, followed by growth of the nucleus by monomer addition (2). Although many amyloid assembly reactions exhibit the kinetic lag phase (3-7) that is a signature feature of nucleated growth polymerization, other important criteria for a nucleated growth mechanism are sometimes more technically difficult to satisfy (2), leading to some uncertainty about the aggregation mechanism. In fact, some amyloidogenic proteins under certain solution conditions (8, 9) appear to undergo amyloid growth via pathways that are more similar to classical colloidal coagulation (1, 10) and do not appear to be nucleation-dependent.
In contrast to many amyloid systems, polyglutamine (polyGln) aggregation, which is associated with Huntington's disease (11, 12), behaves in an manner more consistent with theory, allowing a clear observation and analysis of nucleated growth polymerization kinetics. Thus, simple polyGln peptides appear not to condense into oligomeric and protofibrillar forms, observed in other amyloid assembly reactions (13), that can complicate kinetic analysis. Furthermore, amyloid-like (14) polyGln aggregates do not significantly fracture under normal growth conditions, which in other systems can complicate kinetics analysis because of secondary nucleation (2, 7, 15). These features of polyGln behavior allow a detailed analysis of the nucleation kinetics of aggregation, leading to the surprising observation that the nucleus is an alternatively folded, high-energy state of the monomer, and indicating that the repeat length dependence of polyGln aggregation (16, 17) derives from an increase in the nucleation equilibrium constant, Kn*, with increasing repeat length (18) (Fig. 1).
Fig. 1.

Nucleated growth polymerization with monomer addition. (a) Previously described mechanism in which nucleation is an unfavorable folding event within monomeric polyGln; adapted from ref. 18. (b) Addition of tagged monomer to the growing end of a polyGln aggregate is unstable and prone to dissociate unless followed by polymer growth through subsequent rounds of monomer addition.
A continuing challenge to our ability to fully characterize the nucleation kinetics of polyGln aggregation has been our inability to determine the value of Kn*. This number cannot be determined directly through physical measurement of nuclei, because (i) nucleation is a very rare event and (ii) nuclei, once formed, quickly either collapse to bulk phase monomer or proceed down the productive aggregation pathway. At the same time, although a complex kinetic parameter 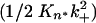 containing Kn* can be derived from nucleation kinetics analysis (2, 18), indirect determination from the kinetics analysis is hampered by our ignorance of the value of k+, the second-order rate constant for aggregate (and nucleus) elongation (Fig. 1). In turn, our inability to determine k+ experimentally stems from our ignorance of the effective molecular weight of polyGln aggregates, that is, the number of productive elongation sites per weight of aggregate. The experimentally determined rate constant is a pseudofirst-order rate constant containing an embedded fibril concentration value (19, 20).
containing Kn* can be derived from nucleation kinetics analysis (2, 18), indirect determination from the kinetics analysis is hampered by our ignorance of the value of k+, the second-order rate constant for aggregate (and nucleus) elongation (Fig. 1). In turn, our inability to determine k+ experimentally stems from our ignorance of the effective molecular weight of polyGln aggregates, that is, the number of productive elongation sites per weight of aggregate. The experimentally determined rate constant is a pseudofirst-order rate constant containing an embedded fibril concentration value (19, 20).
We describe here simple methods for determining the number of elongation sites in a preparation of polyGln aggregates. We use this method to determine the average functional molecular weight of a polyGln aggregate preparation and use this value to determine a second-order rate constant, k+, for elongation of the aggregate. The rate constant is used to complete the nucleation kinetics analysis of a Q47 peptide to provide an estimate of the nucleation equilibrium constant Kn*. As expected, Kn* is a very low number, illustrating the difficulty in directly observing the nucleus.
Materials and Methods
Materials and General Methods. All synthetic peptides were obtained from the Keck Biotechnology Center at Yale University, New Haven, CT. Most were purified by reverse-phase HPLC, and structures were confirmed by electrospray MS, as described (14, 17, 21); the Q30 peptide used in a sample work-up was not purified before use. Long polyGln peptides after purification typically contain two to four major repeat length constituents, whose weight average repeat length is reported (14, 17). The biotinylated polyGln peptides used in these experiments contained biotin added to the N terminus as a modified glutamine side chain of biotin with a short polyethylene glycol spacer (Nova Biochem 04-12-1250), which is the meaning of the prefix “biotin-PEG” in this article. Biotin-labeled Aβ(1-40) has been described (22). The HPLC sedimentation assay for estimation of polyGln monomer concentration (17, 18, 21) and the microtiter plate elongation assay (22, 23) have been described.
Preparation and Characterization of Q47 Aggregates. Six types of aggregates were prepared and analyzed, all from Q47 polyGln. The basic aggregation conditions (PBS, 37°C; PBS, frozen at -20°C; 50 mM Tris·HCl, pH 7.5, 37°C), which generate aggregates with ribbon, protofibril, and fibril morphologies, respectively, have been described (14). These aggregates were either used with no further treatment or exposed to six 30-s pulses of sonication, with a 1-min interval between each two pulses, on ice. The weight concentrations of the aggregates were determined by dissolving an aliquot of the aggregate suspension in formic acid, diluting into HPLC running buffer, and analyzing on an analytical HPLC system (21).
Solution Phase Determination of Aggregate Growing Ends. The basic protocol consists of incubation of a known weight concentration of aggregate (typically 190-260 ng/ml) with 0.5-1.0 μM biotin-PEG-polyGln at 25°C. To determine the fmol of biotin-PEG-Q29 bound, 500-μl aliquots of the aggregate suspension were collected by centrifugation, incubated with europium-complexed streptavidin [diluted 1:1,000 from the commercial preparation (PerkinElmer) into PBS] for 60 min at room temperature in the dark, and washed three times by centrifugation and resuspension in PBS; the pellet was resuspended in 100 μl of enhancement solution (PerkinElmer); and time-resolved fluorescence counts were determined in the PerkinElmer/EG&G Wallac Victor2 microtiter plate reader. All centrifuge steps were at 20,800 × g in an Eppendorf centrifuge at 4°C for 1 h. Counts were converted to moles of europium by using a standard curve and to moles of streptavidin by using the manufacturer's determination of seven molecules of europium per streptavidin molecule. It was assumed in our calculation that one molecule of tetrameric streptavidin corresponds to one molecule of biotin-PEG-Q29 on the aggregate. Nonspecific binding, determined by controls lacking unlabeled Q30 in the work-up (see below), was subtracted from the binding values. At a concentration of 0.5 μM biotin-PEG-Q29, the amount of stably bound biotin-PEG-Q29 in these experiments (10-40 fmol) represents <0.02% of the available biotin-PEG-Q29.
The above basic format was run in two different modes. In the first mode, time points were worked up by addition of 30 μMQ30 and incubation at 25°C for 15 min, collection of aggregate by centrifugation (as above), resuspension of aggregates in 30 μM Q30, incubation at 37°C for 60 min, and collection of aggregates, washing, and processing with streptavidin-europium as described above. As discussed in Results, this protocol gives approximate binding results and a good estimate of the time course of binding.
In the second, preferred, mode, the entire reaction mixture containing aggregates and biotin-PEG-Q29 monomer was quenched after 20 min of coincubation (the end of the binding reaction as determined by experiments done in the first mode; see Results) by adding Q30 to 30 μM and immediately worked up according to the general protocol described above. This method gives a more precise estimate of the total amount of biotin-polyGln bound. As described in Results, the precision of this method was determined in a study of the 37°C time dependence of biotin incorporation after quenching.
In these experiments, we used Q47 aggregates as an example of the type of aggregate of interest in determining the concentration of aggregation growth sites. We used biotin-labeled Q29 as a peptide that supports elongation on a variety of aggregates from different polyGln repeat lengths (17). We used unlabeled and unpurified Q30 as a relatively low-cost dilution peptide of a repeat length essentially identical to biotinyl-Q29.
Results
Previous analysis of polyGln aggregate growth (23) suggests a multiphase growth mechanism including an initial reversible binding step (“docking”) that can be distinguished from subsequent, rate-limiting, rearrangements (“locking”) to complete the elongation cycle (24). We reasoned that it might be possible to slow down the elongation reaction sufficiently, for example, by lowering the reaction temperature, so that a similar reversible binding step might be observable and quantifiable in solution/suspension phase, to effectively yield the number of functional growth sites in the aggregate preparation.
We incubated biotinyl-Q29 with a known weight of Q47 aggregates in PBS at 25°C and removed aliquots for analysis. Because we expected that loosely bound (docked) biotin-PEG-Q29 would dissociate from the aggregates unless trapped by subsequent rounds of elongation (Fig. 1_b_), we analyzed data points by adding unlabeled Q30, collecting the aggregates by centrifugation at low temperature, washing, and incubating with fresh unlabeled Q30, before quantifying the biotin (see Materials and Methods). The results (Fig. 2) showed a time-dependent binding of biotin-PEG-Q29 to a plateau value reached after 10 min of incubation. Further, conducting a parallel experiment on the same weight of a sonicated aggregate preparation increased the plateau value (Fig. 2), consistent with elongation kinetics data from other amyloid systems (3). Fig. 2 also shows that a polyGln aggregate prepared under different growth conditions yields a different plateau amplitude. These amplitude values can be interpreted as the number of fmol of growth sites per weight of aggregate. The fragile nature of the bound biotin-PEG-Q29 (Fig. 1_b_) is illustrated by the control curves in Fig. 2 a and b, generated by incubating and washing equivalent aliquots of the Q47 aggregate/biotin-PEG-Q29 mixture, but without adding unlabeled Q30. No signal is obtained in these controls, presumably because without multiple rounds of elongation the biotin-PEG-Q29 dissociates (Fig. 1_b_) during work-up; we interpret this finding as an indication of the specificity of biotin-PEG-Q29 binding. It remains possible, however, that binding to nonspecific sites might also be stabilized in the presence of unlabeled excess polyGln.
Fig. 2.

Time-dependent binding of biotinylated peptides to aggregates. (a) Binding of biotin-PEG-Q29 to Q47 aggregates grown in PBS at 37°C (squares). (b) Binding of biotin-PEG-Q29 to Q47 aggregates grown in Tris·HCl at 37°C (diamonds). In a and b,Q47 aggregates were sonicated (empty symbols) or not (filled symbols). Each time point contained 500 μl of either 190 ng/ml (squares) or 260 ng/ml (diamonds) aggregate and was developed by incubation 15 min at 25°C with 30 μM unlabeled Q30. Controls are Q47 aggregates with (○) or without (•) sonication, with no added unlabeled Q30 during workup (see Materials and Methods). (c) Binding of heterogeneous biotin-labeled peptides to Q47 (PBS, 37°C) aggregates: 0.5 μM biotin-PEG-PGQ9-P2,3, with (▵) or without (○)30 μM unlabeled Q30 added during work-up; 0.1 μM biotin-Aβ(1-40), with 5 μM unlabeled Aβ(1-40) added during work-up (▴). In c, all time points consisted of 500 μl of 228 ng/ml Q47 aggregates.
Additional controls, however, also suggest that the plateau values seen in Fig. 2 a and b are associated with binding to specific aggregate elongation sites. Fig. 2_c_ shows that when a parallel experiment is conducted with a biotin-labeled polyGln mutant, PGQ9-P2,3, that is incapable of undergoing elongation (25), only background binding is observed. In another control, biotinyl-Aβ(1-40) exhibits only background levels of binding to Q47 aggregates (Fig. 2_c_), consistent with previous observations (22) of poor cross-seeding between polyGln and Aβ. The low binding amplitudes obtained in the Fig. 2_c_ experiments suggest that the major portion of binding seen for elongation-competent forms of polyGln is associated with productive elongation sites.
We were concerned that some potential amplitude might be lost in the experimental format underpinning Fig. 2 during the 25°C incubation after quenching. To obtain a more accurate value for the number of growing ends, we conducted the experiment summarized in Fig. 3. Here, after incubating with biotin-PEG-Q29 for 20 min at 25°C, an excess of Q30 was added to the incubation mixture, and the temperature was shifted immediately to 37°C. The advantage of this approach is that the initially bound biotin-PEG-Q29 should be rapidly “capped” and immobilized by rounds of Q30 elongation, which is strongly favored at 37°C. The disadvantage is that the remaining biotinyl-Q29, although diluted by the unlabeled Q30, will still add to the aggregate during the incubation step required for locking in the biotin-PEG-Q29. Fig. 3 shows that this time-dependent incorporation of biotin-PEG-Q29 (as well as unlabeled Q30) at 37°C can be corrected for by extrapolation to zero time (Fig. 3_b_). Fig. 3_b_ shows that the extrapolated value matches the experimental value for an aliquot taken immediately after quenching with Q30 (and without 37°C incubation). In the example shown, the extrapolation yields a value of ≈22 fmol of biotin-PEG-Q29 bound to 190 ng of Q47 aggregates, corresponding to ≈20% better binding than what is observed when the 25°C postquenching incubation step is included. These results suggest that the short incubation at 25°C after addition of unlabeled Q30 is detrimental, and at the same time that substituting a 37°C incubation in its place, followed by extrapolation to zero time, is unnecessary. That is, it is both simple and accurate to quench with excess unlabeled Q30 and immediately centrifuge. [Nonetheless, all of the data described here were collected with the 37°C incubation and extrapolation to zero time (Fig. 3_b_).]
Fig. 3.

Time and concentration dependence of biotinyl-Q29 binding to Q47 aggregates. (a and b) Biotin-PEG-Q29 (0.5 μM) was incubated with 240 ng/ml Q47 aggregates at 25°C for 20 min, then 30 μM unlabeled Q30 monomer was added and the reaction shifted to 37°C. □, fmol biotin-PEG-Q29 bound, as stabilized by added unlabeled Q30; ○, fmol biotin-PEG-Q29 bound, in control lacking unlabeled Q30; ♦, percentage of unlabeled Q30 remaining monomeric in a centrifugation supernatant, as determined by analytical HPLC. (c) Amount of biotinyl-PEG-Q29 bound in 30 min at 25°C.
Fig. 3_c_ provides data on the concentration dependence of biotin-PEG-Q29 binding and shows that (i) the values obtained by using the above methods reflect the saturation of a limited number of polyGln binding sites, and (ii) the concentration of 0.5 μM used throughout this study is essentially saturating.
The above extrapolation analysis was conducted multiple times on a variety of polyGln aggregates. The average values for each aggregate are listed in Table 1. Table 1 shows that preparing Q47 aggregates by different methods produces aggregates that contain different numbers of binding sites for polyGln per weight of aggregate, and that the number of binding/elongation sites per weight of aggregate goes up when aggregates are sonicated. Furthermore, Fig. 4_b_ shows that the pseudofirst-order rate constants for aggregate elongation (example, Fig. 4_a_) increase in proportion with the number of biotin-PEG-Q29 binding sites per weight of aggregate. Fig. 4_b_ and Table 1 suggest that aggregates grown in Tris buffer in the absence of NaCl undergo elongation reactions slightly more efficiently than aggregates grown in PBS, consistent with different morphologies seen in electron micrographs (14).
Table 1. Properties of polyglutamine aggregates.
| Q47 aggregate* | Fmol biotin-Q30† | Fmol/ug aggregate‡ | [ends], nM, in elongated reaction§ | k+, M—1·s—1¶ | Monomers per end in aggregate∥ | Fmol/ug aggregate, microplate** |
|---|---|---|---|---|---|---|
| PBS/37°C nonsonicated | 21.1 ± 0.4 | 221 | 0.42 | 11,400 | 690 | 175 |
| PBS/37°C sonicated | 29.2 ± 0.1 | 305 | 0.58 | 9,880 | 500 | 236 |
| PBS/frozen nonsonicated | 40.0 ± 0.6 | 308 | 0.76 | 9,270 | 500 | ND |
| PBS/frozen sonicated | 46.0 ± 0.1 | 354 | 0.88 | 10,500 | 435 | ND |
| Tris/37°C nonsonicated | 28.0 ± 0.1 | 215 | 0.60 | 12,600 | 714 | 159 |
| Tris/37°C sonicated | 40.0 ± 0.6 | 308 | 0.86 | 12,800 | 500 | 224 |
Fig. 4.

Correlation of experimentally determined growing ends to pseudofirst-order rate constants for elongation. (a) Determination of k*, the pseudofirst-order rate constant for elongation of Q47 monomers by Q47 aggregates (grown in PBS, 37°C, and nonsonicated), with unpolymerized monomer determined by the HPLC sedimentation assay. (b) For six different aggregates (see Table 1), correlation of growing end concentration of an aggregate suspension and the pseudofirst-order elongation rate constant for that aggregate suspension mixed with monomeric polyGln (aggregates made in PBS, ♦; aggregates made in Tris·HCl, ▪).
Because microtiter plate elongation assays exhibit a relatively rapid binding (docking) step (23, 26), we investigated whether the microtiter plate assay format might prove a convenient alternative means for determining the concentration of aggregate growing ends. We conducted our standard microtiter plate elongation assay (22, 23) by using variants of the aggregate preparations described here. A typical result is shown in Fig. 5. As shown in Fig. 5_b_, extrapolation of the slow kinetic phase back to the y axis provides an estimate of the amplitude of the fast phase, in fmol. After subtraction of background binding, as indicated by a nonzero extrapolation of the fast phase (see Fig. 5_a_), this amplitude is taken as the number of elongation sites in the immobilized aggregate. The values obtained (Table 1, last column) agree well with those determined in solution phase, but exhibit systematically lower values that might be caused by masking of aggregate growth sites upon binding to the plastic surface. We think the suspension phase method is more rigorous and reliable than the microtiter plate assay.
Fig. 5.

Estimation of growing ends by using a microtiter plate elongation assay. Time-dependent addition of biotin-PEG-Q29 to various Q47 aggregates adhered to plastic microtiter plate wells: PBS, 37°C aggregates (squares); Tris·HCl, 37°C aggregates (diamonds); with (empty symbols) and without (filled symbols) sonication. (a) Overall time course. (b) Extrapolation of slow, second kinetic phase back to zero time to obtain the approximate amplitude of the fast phase.
The values listed in Table 1 are also consistent with an analysis of electron micrographs. The ribbon-like structures of polyGln aggregates grown in PBS at 37°C (17) consist of filaments with diameters of ≈4.5 nm and average length 200 nm. Assuming a cylindrical shape with these dimensions and a packing density of 1.35 g/cm3 typical of globular proteins (27), one obtains an average filament size corresponding to ≈400 Q47 molecules per filament, or, assuming filament growth from both ends, 200 molecules per growing end. This finding is in reasonably good agreement with the numbers generated by the growing end analysis described here (Table 1). There are several possible explanations for why our EM analysis might result in underestimates, including the fact that many filaments extended beyond the boundary of the EM grid, and the possibility that all visible ends in the grid may not be competent.
With reliable values of the molar concentration of Q47 aggregates, and therefore reliable values for the second-order elongation rate constants for these aggregates, it becomes possible to calculate an estimate of the nucleation equilibrium constant, Kn*, for Q47 aggregation. The calculation is grounded in the nucleation kinetics analysis of polyGln aggregation (18) by using the equation 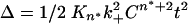 . Plots of the extent of reaction vs. time2 (Fig. 6_a_) yield slopes
. Plots of the extent of reaction vs. time2 (Fig. 6_a_) yield slopes 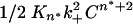 . A plot of the log of these slopes vs. the log of concentration C (Fig. 6_b_) yields a slope equal to n* + 2. The slope of the log-log plot in Fig. 6_b_ is 2.87, corresponding to a critical nucleus, n*, essentially equal to 1, confirming previous determinations on a variety of polyGln repeat lengths (18). The _x_-intercept of the log-log plot in Fig. 6_b_, -0.7668, is equal to log
. A plot of the log of these slopes vs. the log of concentration C (Fig. 6_b_) yields a slope equal to n* + 2. The slope of the log-log plot in Fig. 6_b_ is 2.87, corresponding to a critical nucleus, n*, essentially equal to 1, confirming previous determinations on a variety of polyGln repeat lengths (18). The _x_-intercept of the log-log plot in Fig. 6_b_, -0.7668, is equal to log 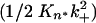 . Using for the second-order rate constant of aggregate elongation, k+, the value 11,400 liters/mol per s (Table 1) for Q47 aggregates grown in PBS at 37°C, we obtain _K_n* = 2.6 × 10-9. [In this calculation, we assume that the rate constants for elongation of the nucleus and elongation of the aggregate are identical; it is not possible to independently determine the rate constant for elongation of the nucleus (2).]
. Using for the second-order rate constant of aggregate elongation, k+, the value 11,400 liters/mol per s (Table 1) for Q47 aggregates grown in PBS at 37°C, we obtain _K_n* = 2.6 × 10-9. [In this calculation, we assume that the rate constants for elongation of the nucleus and elongation of the aggregate are identical; it is not possible to independently determine the rate constant for elongation of the nucleus (2).]
Fig. 6.

Analysis of nucleation kinetics. (a) Representative time2 plots of the early portion of Q47 (PBS, 37°C) at different concentrations (▴, 36 μM, left y axis; •, 105 μM, right y axis). (b) Log-log plot of the slopes of the time2 plots vs. Q47 concentration. The slope of the log-log plot is 2.87 (_R_2 = 0.977).
Discussion
The nucleation kinetics of protein aggregation can be mathematically modeled by analytic, numerical, or hybrid approaches (2). Although numerical approaches can mathematically describe the data, the resulting parameters may be difficult to interpret in physical terms (2). Our analytic approach is based on a nucleation mechanism in which (i) the nucleus is in a (pre)-thermodynamic equilibrium with monomer, (ii) the nucleus is less stable than monomer and is in fact the least stable species on the aggregation pathway, and (iii) rounds of elongation, described by elongation rate constants, stabilize the growing aggregate and strongly disfavor its dissociation (2). Application of this approach to polyGln aggregation gave the surprising result that the nucleus is an unstable form of the monomer (18). Because CD experiments suggest that monomeric polyGln is substantially disordered (17, 28), we attribute nucleation of polyGln aggregation to a highly unfavorable folding reaction within the monomer (18). Although the folding of monomeric proteins is first-order, so that the mole fraction of protein folded under particular conditions is independent of concentration, nonetheless the absolute concentration of folded protein is expected to increase as total protein concentration increases. Monomeric folding thus satisfies the (relatively weak) concentration dependence of nucleation kinetics observed in polyGln aggregation (18). It seems reasonable to extend this mechanistic model by attempting to elucidate the thermodynamics describing the nucleation step, i.e., the presumed folding reaction within the polyGln monomer.
The suspension phase titration of aggregate growth sites described here shows saturation and specificity for aggregation-competent polyGln sequences. Within a series of aggregates the number of growth sites varies with the elongation rate constants for each of the aggregates used as a seed. The data are also in good agreement with estimates from analysis of EMs of aggregates and an alternative, microtiter plate assay. This assay should be amenable to the study of at least some other amyloid systems.
The elongation rate constant of ≈104 M-1·s-1 is substantially below the diffusion limits for second-order reactions. This finding is probably because the slow step in elongation is not the initial productive encounter (docking) but rather a rearrangement within the docked monomer (locking) (24).
The very low value for the nucleation equilibrium constant Kn* obtained here (2.6 × 10-9) is not surprising for a nucleation reaction. The value suggests that the steady-state concentration of nuclei in a test tube polyGln aggregation experiment, in which monomer concentrations of 20 μM are typical, would be in the range of 5 × 10-14 M, or 50 fM. This estimate suggests that it will prove very difficult indeed to directly observe and characterize free aggregation nuclei experimentally. As described (18), the repeat length dependence of polyGln aggregation appears to be controlled by the dependence of Kn* on repeat length. Thus, the Kn* for polyGln peptides shorter than Q47 is expected to be even smaller than 2.6 × 10-9, whereas the value for longer repeats will be somewhat larger than this number.
The value of 2.6 × 10-9 for Kn* corresponds to a free energy of nucleation of 12.2 kcal/mol (using the expression Δ_G_ = -RTlnKeq), in contrast to negative free energies of folding for typical globular protein that fall in the range -5 to -15 kcal/mol (29). It should be possible to expand such nucleation kinetics studies to learn more about the nucleation reaction. Derived values and how they change with solution conditions should prove invaluable in providing experimental benchmarks for supporting computational modeling of polyGln solution structure and aggregation properties (30, 31).
There has been much recent interest in the degree to which fibril structure dictates elongation efficiencies in homogeneous and heterogeneous seeding reactions (22, 32-35). Although it is possible to highly accurately determine pseudofirst-order rate constants for cross-seeding reactions (22), these rate constants inevitably include an embedded, inscrutable fibril concentration term, making it difficult to extract and interpret relative rate constants in structure-function terms. The methods introduced here should remove this technical barrier.
Quantifying growth sites on polyGln aggregates immediately yields two physical parameters potentially relevant to polyGln aggregation and disease risk for expanded CAG repeat diseases. The repeat length dependence of Kn* is the only known biophysical parameter paralleling the polyGln repeat length correlation with disease risk and thus continues to provide support for a role for aggregation in the disease process. It will remain premature to categorically rule out a disease role for polyGln aggregation (36) until the detailed chronology of appearance in disease tissue and cell models is established for the rich variety of aggregate morphologies, functionalities, and sizes known to be formed by polyGln proteins (37-39).
Acknowledgments
This work was supported by National Institutes of Health Grant R01 AG19322 (to R.W.).
Author contributions: R.W. designed research; A.M.B. and A.K.T. performed research; A.M.B., A.K.T., and R.W. analyzed data; and R.W. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: polyGln, polyglutamine.
References
- 1.Anderson, V. J. & Lekkerkerker, H. N. (2002) Nature 416**,** 811-815. [DOI] [PubMed] [Google Scholar]
- 2.Ferrone, F. (1999) Methods Enzymol. 309**,** 256-274. [DOI] [PubMed] [Google Scholar]
- 3.Jarrett, J. T. & Lansbury, P. T., Jr. (1992) Biochemistry 31**,** 12345-12352. [DOI] [PubMed] [Google Scholar]
- 4.Harper, J. D. & Lansbury, P. T., Jr. (1997) Annu. Rev. Biochem. 66**,** 385-407. [DOI] [PubMed] [Google Scholar]
- 5.Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M. & Biere, A. L. (1999) J. Biol. Chem. 274**,** 19509-19512. [DOI] [PubMed] [Google Scholar]
- 6.Hamada, D. & Dobson, C. M. (2002) Protein Sci. 11**,** 2417-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins, S. R., Douglass, A., Vale, R. D. & Weissman, J. S. (2004) PLoS 2**,** 1582-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modler, A. J., Gast, K., Lutsch, G. & Damaschun, G. (2003) J. Mol. Biol. 325**,** 135-148. [DOI] [PubMed] [Google Scholar]
- 9.Hurshman, A. R., White, J. T., Powers, E. T. & Kelly, J. W. (2004) Biochemistry 43**,** 7365-7381. [DOI] [PubMed] [Google Scholar]
- 10.Myers, D. (1999) Surfaces, Interfaces, and Colloids (Wiley, New York).
- 11.Bates, G. (2003) Lancet 361**,** 1642-1644. [DOI] [PubMed] [Google Scholar]
- 12.Michalik, A. & Van Broeckhoven, C. (2003) Hum. Mol. Genet. 12**,** Suppl. 2, R173-R186. [DOI] [PubMed] [Google Scholar]
- 13.Goldsbury, C., Frey, P., Olivieri, V., Aebi, U. & Muller, S. A. (2005) J. Mol. Biol. 352**,** 282-298. [DOI] [PubMed] [Google Scholar]
- 14.Chen, S., Berthelier, V., Hamilton, J. B., O'Nuallain, B. & Wetzel, R. (2002) Biochemistry 41**,** 7391-7399. [DOI] [PubMed] [Google Scholar]
- 15.Padrick, S. B. & Miranker, A. D. (2002) Biochemistry 41**,** 4694-4703. [DOI] [PubMed] [Google Scholar]
- 16.Scherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R., Hasenbank, R., Bates, G. P., Lehrach, H. & Wanker, E. E. (1999) Proc. Natl. Acad. Sci. USA 96**,** 4604-4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, S., Berthelier, V., Yang, W. & Wetzel, R. (2001) J. Mol. Biol. 311**,** 173-182. [DOI] [PubMed] [Google Scholar]
- 18.Chen, S., Ferrone, F. & Wetzel, R. (2002) Proc. Natl. Acad. Sci. USA 99**,** 11884-11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naiki, H. & Gejyo, F. (1999) Methods Enzymol. 309**,** 305-318. [DOI] [PubMed] [Google Scholar]
- 20.O'Nuallain, B., Shivaprasad, S., Kheterpal, I. & Wetzel, R. (2005) Biochemistry 44**,** 12709-12718. [DOI] [PubMed] [Google Scholar]
- 21.Wetzel, R. (2005) in The Protein Folding Handbook: Part II, eds. Buchner, J. & Kiefhaber, T. (Wiley, Weinheim, Germany), pp. 1170-1214.
- 22.O'Nuallain, B., Williams, A. D., Westermark, P. & Wetzel, R. (2004) J. Biol. Chem. 279**,** 17490-17499. [DOI] [PubMed] [Google Scholar]
- 23.Berthelier, V., Hamilton, J. B., Chen, S. & Wetzel, R. (2001) Anal. Biochem. 295**,** 227-236. [DOI] [PubMed] [Google Scholar]
- 24.Esler, W. P., Stimson, E. R., Jennings, J. M., Vinters, H. V., Ghilardi, J. R., Lee, J. P., Mantyh, P. W. & Maggio, J. E. (2000) Biochemistry 39**,** 6288-6295. [DOI] [PubMed] [Google Scholar]
- 25.Thakur, A. & Wetzel, R. (2002) Proc. Natl. Acad. Sci. USA 99**,** 17014-17019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esler, W. P., Stimson, E. R., Ghilardi, J. R., Felix, A. M., Lu, Y. A., Vinters, H. V., Mantyh, P. W. & Maggio, J. E. (1997) Nat. Biotechnol. 15**,** 258-263. [DOI] [PubMed] [Google Scholar]
- 27.Fischer, H., Polikarpov, I. & Craievich, A. F. (2004) Protein Sci. 13**,** 2825-2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett, M. J., Huey-Tubman, K. E., Herr, A. B., West, A. P., Ross, S. A. & Bjorkman, P. J. (2002) Proc. Natl. Acad. Sci. USA 99**,** 11634-11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Creighton, T. E. (1984) Proteins: Structures and Molecular Properties (Freeman, New York).
- 30.Khare, S. D., Ding, F., Gwanmesia, K. N. & Dokholyan, N. V. (2005) PLoS Comput. Biol. 1**,** e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, X., Vitalis, A., Wyczalkowski, M. & Pappu, R. V. (2005) Proteins: Structure, Function, and Bioinformatics, in press.
- 32.Xing, Y., Nakamura, A., Korenaga, T., Guo, Z., Yao, J., Fu, X., Matsushita, T., Kogishi, K., Hosokawa, M., Kametani, F., Mori, M. & Higuchi, K. (2002) J. Biol. Chem. 277**,** 33164-33169. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka, M., Chien, P., Naber, N., Cooke, R. & Weissman, J. S. (2004) Nature 428**,** 323-328. [DOI] [PubMed] [Google Scholar]
- 34.Vanik, D. L., Surewicz, K. A. & Surewicz, W. K. (2004) Mol. Cell 14**,** 139-145. [DOI] [PubMed] [Google Scholar]
- 35.Derkatch, I. L., Uptain, S. M., Outeiro, T. F., Krishnan, R., Lindquist, S. L. & Liebman, S. W. (2004) Proc. Natl. Acad. Sci. USA 101**,** 12934-12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slow, E. J., Graham, R. K., Osmand, A. P., Devon, R. S., Lu, G., Deng, Y., Pearson, J., Vaid, K., Bissada, N., Wetzel, R., et al. (2005) Proc. Natl. Acad. Sci. USA 102**,** 11402-11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukai, H., Isagawa, T., Goyama, E., Tanaka, S., Bence, N. F., Tamura, A., Ono, Y. & Kopito, R. R. (2005) Proc. Natl. Acad. Sci. USA 102**,** 10887-10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wetzel, R. (2005) in Genetic Instabilities and Neurological Diseases, eds. Wells, R. & Ashizawa, T. (Elsevier, San Diego), in press.
- 39.Osmand, A. P., Berthelier, V. & Wetzel, R. (2006) Methods Enzymol., in press. [DOI] [PubMed]