Association Mapping in Structured Populations (original) (raw)
Abstract
The use, in association studies, of the forthcoming dense genomewide collection of single-nucleotide polymorphisms (SNPs) has been heralded as a potential breakthrough in the study of the genetic basis of common complex disorders. A serious problem with association mapping is that population structure can lead to spurious associations between a candidate marker and a phenotype. One common solution has been to abandon case-control studies in favor of family-based tests of association, such as the transmission/disequilibrium test (TDT), but this comes at a considerable cost in the need to collect DNA from close relatives of affected individuals. In this article we describe a novel, statistically valid, method for case-control association studies in structured populations. Our method uses a set of unlinked genetic markers to infer details of population structure, and to estimate the ancestry of sampled individuals, before using this information to test for associations within subpopulations. It provides power comparable with the TDT in many settings and may substantially outperform it if there are conflicting associations in different subpopulations.
Introduction
Association mapping has been advocated as the method of choice for identifying loci involved in the inheritance of complex traits (e.g., Risch and Merikangas 1996). In its simplest form, this method involves identifying markers with significant allele-frequency differences between individuals with the phenotype of interest (“cases”) and a set of unrelated control individuals. A statistical association between genotypes at a marker locus and the phenotype is usually considered to be evidence of close physical linkage between the marker and a disease locus.
However, it is well known that the presence of population structure can result in “spurious associations”—that is, associations between a phenotype and markers that are not linked to any causative loci (e.g., Lander and Schork 1994). Such associations can occur when the disease frequency varies across subpopulations, thereby increasing the probability that affected individuals will be sampled from particular subpopulations. Any marker allele that is in high frequency in the overrepresented subpopulations will then be associated with the phenotype (Ewens and Spielman 1995; Pritchard and Rosenberg 1999).
In response to this problem, Spielman et al. (1993) proposed the “transmission/disequilibrium test” (TDT), which uses the genotypes of parents of affected individuals to ensure that association between a marker allele and the phenotype is detected only if the marker is linked to a disease locus, even in the presence of population structure. A number of similar family-based methods have since been devised to treat a variety of genetic models and using different relatives (e.g., Boehnke and Langefeld 1998; Lazzeroni and Lange 1998; Spielman and Ewens 1998).
Despite the success and popularity of family-based methods such as the TDT, the problem of how to perform valid case-control association studies is of considerable importance. In many situations, the case-control study design has substantial practical advantages over family-based designs. Collecting DNA from relatives of affected individuals is often much harder than is collecting DNA from unrelated controls, particularly in the case of late-onset diseases. As a result, case-control studies tend to be cheaper than family-based studies of the same sample size. Further, the ability to use unrelated controls suggests the possibility of independent studies reusing databases of control genotype data, thus reducing genotyping costs.
One possible approach to eliminating spurious associations in case-control studies would be to avoid performing such studies in populations where structure is clearly present—for example, by identifying culturally defined ethnic groups. However, cultural groups may not accurately reflect underlying genetic population structure. Because of this, even in apparently culturally homogeneous populations, concerns over the possible presence of “cryptic” population structure make it difficult to assess the true significance of associations found using conventional methods. In response to this, Pritchard and Rosenberg (1999) suggested using background levels of association in multilocus genotype data from cases and controls to identify situations in which population structure may lead to spurious associations. This leaves open the question of how to proceed in situations where population structure does appear to be a problem.
In this study, we develop a test for association that is valid in the presence of population structure. Our test exploits the methods of Pritchard et al. (2000), which use the genotypes of a sample of individuals at a series of unlinked markers to identify the presence of population structure, and to assign the sampled individuals to putative “unstructured” subpopulations which do not exhibit association between unlinked markers. Our test is based on the idea that any association between a candidate allele and the phenotype within a subpopulation cannot be due to population structure. We envisage a two-stage procedure for identifying significant associations in studies with large numbers of candidate loci (e.g., genomewide screens). First, the method of Pritchard et al. (2000) would be applied to the SNP or microsatellite candidates to learn about population structure, and to assign the sampled individuals to putative subpopulations. Second, our test for association would be applied to identify candidates that exhibit a significant association with the phenotype within subpopulations.
In the next section, we provide a brief background of the model and methods used by Pritchard et al. (2000) for inferring population structure. We then develop a test that makes use of this inferred structure, and we use simulation studies to check whether it properly corrects for the problem of spurious associations. We also perform power comparisons with the TDT. Although, as we discuss later, direct comparison of the methods is difficult, the simulations suggest that the power of our test is comparable with that of the TDT in many settings and can be higher than the TDT if different marker alleles are associated with the phenotype in different subpopulations. We conclude with a discussion of the results and of how our work relates to other recent work on association mapping in structured populations.
Background on Inference of Population Structure from Genetic Markers
In this section we summarize the models and methods used by Pritchard et al. (2000), implemented in the program structure, available from http://www.stats.ox.ac.uk/~pritch/home.html. The method uses genotypic correlations among unlinked markers to learn about the structure of the population from which a sample has been taken and about the genetic backgrounds of the sampled individuals. Genic associations among unlinked markers are assumed to be the result of population structure, and, loosely speaking, individuals are assigned to subpopulations in such a way as to eliminate these associations.
Specifically, we model the sampled individuals as having inherited their genes from a pool of K “unstructured” subpopulations (where K may be unknown). The subpopulations are “unstructured” in that within each subpopulation all loci are in Hardy-Weinberg equilibrium, with no linkage disequilibrium between loci provided they are not tightly linked. The allele frequencies at each locus within each subpopulation are assumed to be unknown. Each individual’s genetic background is represented by a vector _q_=(_q_1,…,q K), where q k is the proportion of the individual’s genome which originated in subpopulation k. This provides a flexible way of capturing various patterns of admixture.
For example, in a sample of African Americans, a typical individual might have 5%–20% European admixture (Parra et al. 1998), whereas some individuals may have substantially more or less. Such a sample could be modeled using _K_=2 subpopulations (African and European), with typical individuals having _q_1 in the range (.05,.2) and _q_2 in the range (.8,.95), but with some individuals having more extreme values. The challenge is to infer this kind of pattern using genetic data. Pritchard et al. (2000) provide a method of performing such inference, even when little is known about either the number of subpopulations that have contributed to the sample or the allele frequencies in these putative subpopulations. They use a Markov chain Monte Carlo method to estimate the number of subpopulations, the allele frequencies in each subpopulation, and the value of q for each sampled individual. The method can be applied to most of the commonly used genetic markers, including microsatellites and SNPs, and can produce accurate results using modest numbers of loci, even when popular clustering algorithms such as Neighbor-Joining are relatively uninformative. The accuracy of the inference depends on the sample size, the number of loci used, and on the magnitude of allele-frequency differences between the subpopulations. Examples of applications of this method are given later. See Pritchard et al. (2000) for further details.
A Test for Association in Structured Populations
In order to test for association in the presence of population structure we replace the standard null hypothesis of no overall association between allele frequencies at the candidate locus and phenotype, with a null hypothesis of no such association within subpopulations. Since, by definition, the subpopulations are “unstructured,” any association between a candidate locus and phenotype within subpopulations cannot be due to structure, and so our test should not suffer from spurious associations.
Formally, we test a null hypothesis _H_0 that subpopulation allele frequencies at the candidate locus are independent of phenotype, against an alternative hypothesis _H_1 where the subpopulation allele frequencies at the candidate locus depend on phenotype. Let C denote the list of genotypes of all sampled individuals at the candidate locus, _P_0 and _P_1 denote subpopulation allele frequencies at the candidate locus under _H_0 and _H_1 respectively, and Q denote the collection of vectors q representing the genetic backgrounds of sampled individuals. We do not restrict the number of alleles at the candidate locus. Below, we describe specific models _Pr_0(C;_P_0,Q) and _Pr_1(C;_P_1,Q) for the distribution of C under _H_0 and _H_1, respectively. A natural measure of the relative support for _H_0 and _H_1 is the likelihood ratio
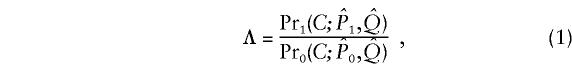
where 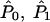 and
and 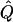 are estimates of _P_0, _P_1 and Q. Large values of Λ indicate that the alternative model (in which allele frequencies at the candidate locus depend on the phenotype) is substantially better than the null model. We approximate the significance of a particular value of Λ by simulation (see below).
are estimates of _P_0, _P_1 and Q. Large values of Λ indicate that the alternative model (in which allele frequencies at the candidate locus depend on the phenotype) is substantially better than the null model. We approximate the significance of a particular value of Λ by simulation (see below).
In testing for association in the presence of population structure, we proceed as follows. We begin with a sample of cases and controls, each of which is genotyped at a number of unlinked marker loci and apply the method of Pritchard et al. (2000) to infer the population structure and the ancestry, Q, of the sampled individuals. The unlinked marker loci might be a series of randomly chosen markers from across the genome or—in a study, such as a genome screen, that considered markers in many candidate genes—would include the candidate loci themselves. Having estimated Q, we then perform the modified test of association described above, for each candidate locus. In the Discussion section, we describe a validation procedure that can be used to check, for a given data set, whether this procedure has provided an adequate correction for stratification. We denote our approach using the acronym STRAT: STRuctured population Association Test.
Models for the Distribution of C
In order to compute the test statistic, Λ, we define explicit probability models _Pr_0(·) and _Pr_1(·) for the probability distribution of C, the collection of alleles at the candidate locus, under _H_0 and _H_1 respectively. Let p(φ)kj denote the frequency of allele j at the candidate locus in subpopulation k among individuals with phenotype φ. Under _H_0, the frequencies p(φ)kj are independent of φ, and we can write p(φ)_kj_=p kj. Let _P_0 denote the allele frequencies at the candidate locus under _H_0: _P_0={p kj:_k_=1,…,K;_j_=1,…,J}, where J is the number of alleles. Let _P_1 be the allele frequencies at the candidate locus under _H_1: _P_1={p(φ)kj:_k_=1,…,K;_j_=1,…,J,φ=0,1}. Then we assume that, under _H_0, the distribution of C is given by
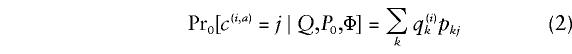
independently for each (i,a), and under _H_1 the distribution of C is given by
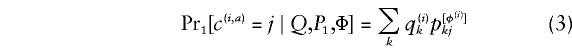
independently for each (i,a). The assumption of independence of the observations under _H_1 corresponds to certain implicit assumptions about the mode of transmission of the disease. The power of our test will depend on this assumption, but the size of our test (significance under _H_0) will not. In calculating the test statistic Λ we used the program structure (Pritchard et al. 2000) to estimate 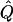 , and the EM algorithm (Dempster et al. 1977) to estimate
, and the EM algorithm (Dempster et al. 1977) to estimate 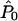 and
and 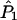 .
.
Assessing Significance Probabilities
We approximate the significance probability of observed data C by simulation, as follows. We simulate C(1),C(2),…,C(M) as independent random draws from 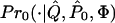 , and approximate the significance probability by
, and approximate the significance probability by
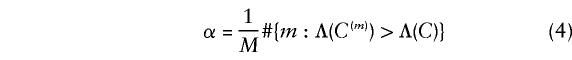
where #A denotes the number of members of the set A. This approximation will be reasonable, provided that (under _H_0) 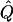 and
and 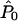 are accurate estimates of the “true” underlying values of Q and _P_0, as then C is also approximately a random draw from
are accurate estimates of the “true” underlying values of Q and _P_0, as then C is also approximately a random draw from 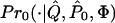 .
.
Details of Simulation Studies
In order to validate the statistical methods described in this paper, we present applications to a range of models. There are, of course, many conceivable models, including a variety of possible models of population structure, of population allele frequencies at the candidate locus, and of the genetic basis (if any) of the disease. Here, we have chosen three rather different models of population structure and a variety of candidate allele frequencies and disease models (under both the null and alternative hypotheses). Our population models are as follows:
- A.
Two discrete subpopulations; two-fold difference in disease frequency between the subpopulations. The divergence of the subpopulations is chosen to be typical of moderately divergent human populations from the same continent. - B.
Two subpopulations of equal size which recently merged to form an admixed population; eight-fold difference in disease frequency between the original subpopulations. Population separation modeled as being typical of human populations from different continents. - C.
An admixed population in which most individuals have a large portion of their ancestry from one subpopulation, and some small portion of ancestry from a second, quite divergent subpopulation; eight-fold difference in disease frequency between the original subpopulations. This is intended to model sampling from an African-American population with some European admixture. Subpopulation allele frequencies were taken from real microsatellite data from Europeans and Africans, in order to capture the appropriate degree of population divergence.
For each population model, we simulated genotype data for samples of unrelated affected and control individuals (see below). These data sets are designated as A, B, and C, to correspond with models A, B, and C, respectively.
Details of Data Sets
The data sets used in this paper were generated as follows.
- A.
We simulated genotypes of 150 affected and 150 control individuals at 100 unlinked microsatellite loci, using standard coalescent techniques (Hudson 1990). We assumed that the individuals were sampled from two subpopulations (each of constant effective size 2_N_ chromosomes) that had split from a single ancestral population (also of size 2_N_) at a time 0.05_N_ generations in the past, with no subsequent migration. Microsatellite mutation was modeled by a simple stepwise mutation process, with the mutation parameter θ=4_N_μ set at 16.0 per locus.
These parameters correspond to an expected value of (δμ)2 of 0.8 (Goldstein et al. 1995), which is less than the average observed at dinucleotide loci between continental groups (Feldman et al. 1999) but is typical of fairly divergent populations within continents; for example, recent comparisons among African populations have produced estimated values of (δμ)2 in the range of 0.5–1.4 (Cooper et al. 1999; N. Rosenberg, unpublished data).
We assumed that half of the controls came from each of the two subpopulations, but 100 of the affected individuals were from subpopulation 1, and just 50 from subpopulation 2. This implies that the risk of disease in subpopulation 1 is about two-fold higher than in population 2 (see Pritchard and Rosenberg [1999]). - B.
We simulated genotypes of 500 affected and 500 control individuals at 150 unlinked microsatellite loci, with stepwise mutation and θ=16 per locus, sampled from an admixed population formed in the following way. Two discrete subpopulations of equal size—formed as in A above, but with divergence time between the subpopulations set to 0.15_N_ generations—are fused to produce a single population, which undergoes two generations of random mating. The divergence time between the two discrete subpopulations corresponds to an expected (δμ)2 of 2.4, which is within the range observed at dinucleotide loci between non-African groups from different continents and is less than that observed between Africans and non-Africans (e.g., see Cooper et al. 1999; Feldman et al. 1999). We made the simplifying assumption of independence among loci.
We modeled the ascertainment of affected and control individuals as follows. Each control individual is a random draw from the population, so that the probability that a control individual has i grandparents from subpopulation 1, and 4-i grandparents from subpopulation 2, is
where _i_∈{0,...,4}. The risk of disease for individuals with i grandparents from subpopulation 1 was assumed proportional to 1+(_R_-1)i/4 where we took _R_=8. The probability that an affected individual has i grandparents in subpopulation 1 is then proportional to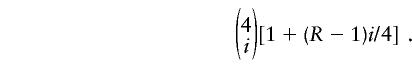
(Note that there are two types of pedigrees for individuals with _i_=2; our simulations incorporated this feature.) - C.
We simulated four data sets of genotypes of 400 affected and 400 control individuals at 120 microsatellite loci, under the null hypothesis of no true associations. These data are intended to approximate a population of African Americans, with an average of 20% European admixture (this is consistent with estimates given by Parra et al. [1998]). We assume that the disease of interest has an eight-fold higher prevalence among Europeans, and, hence, affected individuals tend to have a larger-than-usual degree of European ancestry. Thus, alleles that are common in Europeans are overrepresented among cases, and we might expect that a naive test of association that ignores population structure would be liable to a high rate of false positives. We look to see how pronounced this effect is and also whether we can correct for it appropriately.
In order to make these data more realistic, the simulations were based on estimated microsatellite allele frequencies in Africans and Europeans at sixty microsatellites reported in Jorde et al. (1995, 1997). The allele frequency estimates were from samples of 72 individuals of African origin (Pygmy, Nguni, San, Sotho, or Tswana), and 120 individuals of European origin (British, Finnish, French, or Polish). For the purposes of the simulation, the estimated allele frequencies in Africans and Europeans were considered to be true population frequencies. In order to simulate 120 loci (when the original data set contained 60 loci), we used each set of estimated allele frequencies twice.
For each individual, we first simulated q (as described below), where q is the fraction of European ancestry and 1-q the fraction of African ancestry. Then, at each locus, two alleles were drawn independently with probability q from the European, and probability 1-q from the African allele-frequency distributions.
While there are data about the average amount of European admixture in African Americans, there seems to be little information about the distribution across individuals in the amount of admixture. For the purpose of these simulations, we assumed that q is distributed as a beta distribution with parameters (1,4). The mean of this distribution is 0.2, and the central 80% is between about 0.03 and 0.45. Control individuals were sampled from this distribution.
In order to simulate cases, we assumed a disease with eight-fold higher risk in Europeans (_q_=1), than in Africans (q_=0), with the risk changing linearly with q. We used rejection sampling to simulate q of affected individuals from this distribution: that is, we drew q from the beta prior, and accepted it with probability (1+7_q)/8. If it was rejected, we drew a new q from the prior. We repeated this process until a q was accepted.
The data were analyzed as follows. First, we performed χ2 tests of association at each locus, ignoring the presence of population structure (pooling rare alleles as was done by Pritchard and Rosenberg [1999]). We then used the test described by Pritchard and Rosenberg (1999) to check whether there was evidence for mismatching of the case and control samples. Finally, we inferred q for the sampled individuals, assuming two populations, and used STRAT to estimate P values for each locus. Our results focus on the empirical distribution of P values under the χ2 and STRAT approaches.
Simulation of Candidate Loci
In order to study the performance of the proposed test of association, we simulated a large number of independent biallelic candidate loci for each of the individuals in data sets A and B. Each locus was assumed to have two alleles, A and a, where allele A is at frequencies _p_1 and _p_2 in subpopulations 1 and 2, respectively. (When simulating data set A, but not data set B, we reassigned phenotypes to individuals in each simulation, according to the probabilities given above.) We performed tests of association (TDT, χ2, and STRAT) between the genotypes at each candidate and the assigned phenotypes.
For simulating under the null model (candidates not associated with the phenotype), genotypes at the candidate loci were assigned to individuals at random, conditional on _p_1 and _p_2, and the individuals’ (true) ancestries. For simulating under the alternative model (candidate associated with the phenotype), we assume a model in which the relative risk for individual i depends on his/her genetic background q(i), and genotype C(i) at the candidate locus. (In considering C(i) we label each allele copy according to both its type and subpopulation of origin. For example, we write A_1 for a copy of the A allele which was transmitted from an ancestor in subpopulation 1.) Specifically, we assume that Pr_[i affected|q(i),C(i)]=α_q(i)R C(i), where α_q(i) is a constant that depends on q(i), and R C(i) is a risk factor for genotype C(i). These risk factors are specified by assigning relative risks R _A_1,R _A_2,R _a_1,R _a_2 (table 2) to alleles _A_1,_A_2,_a_1,_a_2 and assuming that risks combine multiplicatively; for example, the relative risk for an individual with genotype _A_1_a_2 is R A_1_R _a_2. We then simulate the genotypes of affected individuals by evaluating
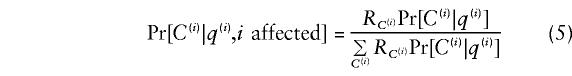
for all possible genotypes. In the discrete population case, we can simplify the notation by defining R_1≡_R _A_1/R _a_1, R_2≡_R _A_2/R _a_2.
Table 2.
Comparison of the Power of the STRAT and the TDT[Note]
| 2A. Discrete Populations | |||||
|---|---|---|---|---|---|
| Estimated Probability of Significant Results at _P_= | |||||
| .05 | .01 | ||||
| R _A_1, R _A_2 and _p_1, _p_2 | STRAT | TDT | STRAT | TDT | Equiv n |
| 1.5, 1.5: | |||||
| .1, .1 | .27 | .36 | .11 | .16 | 110 |
| .1, .5 | .41 | .49 | .20 | .25 | 120 |
| .1, .9 | .25 | .34 | .09 | .14 | 110 |
| 1.0, 2.0: | |||||
| .1, .1 | .35 | .19 | .16 | .06 | 335 |
| .1, .5 | .63 | .44 | .39 | .22 | 245 |
| .1, .9 | .21 | .10 | .07 | .03 | 450 |
| 2.0, .5: | |||||
| .1, .1 | .62 | .31 | .36 | .13 | 345 |
| .1, .5 | .87 | .06 | .71 | .01 | >105 |
| .1, .9 | .72 | .16 | .49 | .05 | 1,100 |
| 2B. Admixed Populations | |||||
| Estimated Probability of Significant Results at _P_= | |||||
| .05 | .01 | ||||
| R _A_1, R _a_1, R _A_2, R _a_2 and _p_1, _p_2 | STRAT | TDT | STRAT | TDT | Equiv n |
| 1.3, 1.0, 1.3, 1.0: | |||||
| .1, .1 | .34 | .45 | .14 | .23 | 350 |
| .1, .5 | .52 | .75 | .27 | .52 | 290 |
| .1, .9 | .28 | .80 | .11 | .58 | 120 |
| 1.0, 1.0, 2.0, 1.0: | |||||
| .1, .1 | .71 | .65 | .47 | .41 | 570 |
| .1, .5 | 1.00 | 1.00 | .98 | 1.00 | 370 |
| .1, .9 | .92 | 1.00 | .76 | 1.00 | 140 |
| 2.0, 1.0, 1.0, 2.0: | |||||
| .1, .1 | .58 | .35 | .33 | .16 | 930 |
| .1, .5 | .78 | .05 | .57 | .01 | >105 |
| .1, .9 | .39 | .12 | .19 | .04 | 2,600 |
Note that, in this alternative model, the candidate marker can be interpreted in two ways. Either it is the functional site itself, or it is a marker which is in linkage disequilibrium with a functional site. In the latter case, the relative risk incurred by the alleles at the candidate marker will normally be less than the risk of having the functional allele itself, unless the two loci are in perfect association.
TDT Comparisons
We also performed a separate series of simulations of the TDT (Spielman et al. 1993), generating samples of 150 and 500 parent-offspring trios under population models A and B, respectively, for each of the parameter combinations in tables 1 and 2. For each candidate locus, we resimulated the q(i) independently for each affected individual and then simulated both their genotypes and those of their parents accordingly. For population model A, the two parents were sampled from the same population as the offspring. For population model B, the parents were from generation 1 in the admixed population, and the actual ancestry of each parent could be determined from the simulated value of q(i) for the affected offspring. When simulating the alternative model, we assumed that there was no recombination between the candidate and a nearby disease gene, in which case the parents’ untransmitted alleles are random draws from the appropriate subpopulation frequencies (Spielman et al. 1993). We produced the data marked “Equiv _n_” (table 2) by running TDT simulations under each model with different numbers of trios and finding the number of trios for which the power of the TDT, at the 0.05 and 0.01 levels, was approximately the same as the power estimated for STRAT at the fixed sample size (150 or 500 cases).
Table 1.
Estimated Rejection Rates by Test[Note]
| 1A. Discrete Populations | |||
|---|---|---|---|
| _p_1, _p_2 | STRAT | TDT | χ2 |
| _P_=.05 | |||
| .5, .5 | .050 | .050 | .049 |
| .1, .1 | .048 | .049 | .049 |
| .5, .1 | .050 | .050 | .437 |
| .9, .1 | .049 | .050 | .769 |
| _P_=.01 | |||
| .5, .5 | .011 | .010 | .011 |
| .1, .1 | .009 | .010 | .009 |
| .5, .1 | .009 | .010 | .260 |
| .9, .1 | .010 | .009 | .649 |
| _P_=10-3 | |||
| .5, .1 | 0.83×10-3 | 0.98×10-3 | .112 |
| .9, .1 | 0.83×10-3 | 0.90×10-3 | .506 |
| _P_=10-4 | |||
| .5, .1 | 0.97×10-4 | 0.95×10-4 | .046 |
| .9, .1 | 0.80×10-4 | 0.76×10-4 | .370 |
| 1B. Admixed Populations | |||
| _p_1, _p_2 | STRAT | TDT | χ2 |
| _P_=.05 | |||
| .5, .5 | .048 | .049 | .054 |
| .1, .1 | .050 | .049 | .051 |
| .5, .1 | .044 | .050 | .623 |
| .9, .1 | .033 | .050 | .998 |
| _P_=.01 | |||
| .5, .5 | .010 | .010 | .010 |
| .1, .1 | .010 | .009 | .010 |
| .5, .1 | .008 | .010 | .370 |
| .9, .1 | .005 | .010 | .979 |
| _P_=10-3 | |||
| .5, .1 | 0.83×10-3 | 0.86×10-3 | .156 |
| .9, .1 | 0.45×10-3 | 1.06×10-3 | .874 |
| _P_=10-4 | |||
| .5, .1 | 0.85×10-4 | 0.38×10-4 | .050 |
| .9, .1 | 0.61×10-4 | 0.92×10-4 | .662 |
Results of Simulation Studies
Accuracy of the Inference of Population Structure
We begin by presenting results from using the method of Pritchard et al. (2000) to estimate the ancestry of the individuals in data sets A, B, and C. With data sets A and B, the method correctly infers that the underlying population structure consists of two subpopulations. (In both cases, essentially all of the posterior weight is on the model of two subpopulations; the choice of prior has little influence. See Pritchard et al. [2000] for further discussion—and caveats—regarding the estimation of the number of subpopulations.)
It is of particular interest to examine the assignment of individuals to subpopulations. For data set A (samples from two discrete subpopulations), the assignment is essentially perfect (fig. 1). In the case of data set B, where most individuals are admixed, the problem is more difficult, but the algorithm still performs well (fig. 2), accurately estimating the ancestry of most individuals.
Figure 1.

Summary of estimates of the ancestry of individuals in data set A (discrete populations). The dashed and solid lines show histograms of estimated values of q(i)1 (the proportion of ancestry of individual i in subpopulation 1) for individuals from subpopulations 1 and 2, respectively. Using this data set, the classification of individuals to subpopulations is essentially perfect.
Figure 2.

Summary of estimates of the ancestry of individuals in the population with recent admixture (data set B). The peaks, from left to right, represent histograms of estimated values of q(i)1 (the proportion of ancestry of individual i in subpopulation 1) for individuals with 0, 1, 2, 3, and 4 grandparents, respectively, in subpopulation 1. The solid lines show the results for the entire sample, while the dotted lines show the results for those individuals who are affected. These data were simulated under a model in which an individual’s risk of disease increases with their amount of ancestry in subpopulation 1, with the result that affected individuals tend to have a greater proportion of ancestry in subpopulation 1 than do controls. This effect can be seen here, as the proportion of affected individuals in each peak increases from left to right. Our test for association uses the estimated q(i) to control for the presence of biased sampling.
Figure 2 illustrates the biased sampling of affected individuals that occurs as a result of the difference in disease frequency among subpopulations. In the model used to generate this data set, the risk of disease increases with the amount of ancestry that an individual has in subpopulation 1, which means that affected individuals tend to have a greater proportion of ancestry in subpopulation 1 than do controls. This effect can be seen in the figure, in which the proportion of affected individuals in each peak increases from left to right. By inferring that this is the situation, our method can control for this ascertainment bias, whereas a standard association test that ignored the presence of population structure would be liable to an excessive rate of false positives.
Inference of population structure is most difficult in the case of data set C, because virtually everybody in the sample is admixed, and there is little information about the allele frequencies in Europeans. The result is that, although the estimated q increases roughly linearly with the amount of European ancestry, it does not provide an accurate estimate of the “true” q (one realization is shown in fig. 3). In a sense, the data do not fit the assumed model of population structure particularly well, and, in fact, the model with three subpopulations has a higher posterior probability than does the model with two. It is probably the case that adding even a small number of European individuals to the data set would greatly improve the inference.
Figure 3.

Plot of estimated q against actual ancestry for one realization of data set C. This data set is intended to be a rough model of an African American population, with an average of 20% European admixture. Controls are marked by a plus sign (+) and cases by an unblackened circle (○). It is assumed that the disease of interest is more common among individuals with substantial European ancestry, and, hence, the distribution of cases is shifted toward the right, relative to controls.
It is also interesting to consider the estimated value of the parameter α, which was used by Pritchard et al. (2000) to parametrize the extent of admixture in the population. For data set A, this is close to 0 (∼0.03); for data sets B and C, it is ∼1.25 and ∼2.5, respectively, indicating that most individuals are of mixed ancestry.
Validity
As described above, we simulated a large number of independent biallelic candidate loci for each of the individuals in data sets A and B, under a range of models. We used the estimated ancestry of individuals (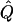 ), obtained above, in testing for association at each locus.
), obtained above, in testing for association at each locus.
Table 1 summarizes the results of some of these simulations under the null hypothesis (genotype independent of phenotype). These results show that STRAT rejects the null hypothesis at the appropriate rate (at P values of 0.05, 10-2, 10-3, and 10-4) or is slightly conservative. This holds even when a naive test of association (using the χ2) is wildly inaccurate. Our estimated P values perform well even for data set B, where the inferred ancestry (Q) is not perfect.
We simulated four data sets from model C. The degree of sampling bias generated by the parameters chosen was not extreme, and for one realization there was a fairly similar (but not identical) distribution of q in cases and controls. Using the test of Pritchard and Rosenberg (1999) to check for mismatching of the case and control samples yielded a nonsignificant result for this case (_P_=.21), and significant results for the other three (P<10-4). We focus on these latter three. Figure 4 shows a plot of the cumulative distribution of P values for the three data sets. While the χ2 test produced an excess of small P values for these data sets, the distribution of P values obtained using STRAT is close to the desired (uniform) distribution. It seems that although our estimation of q was rather imprecise, it enabled an adequate correction for these data.
Figure 4.

Cumulative plot of estimated P values across the 120 loci for three realizations of data set C. The three solid lines show the distribution of P values obtained using a χ2 test which ignores population structure. These indicate an excess of small P values. The three dashed lines show the distributions of P values obtained using STRAT. These fall close to the diagonal (which is the ideal distribution) or (in one case) appear slightly conservative.
Power
We have also examined the power of our test under a number of alternative models (typical results shown in table 2). For each model, we compared the power of the STRAT to that of the TDT, assuming the same number of affected individuals for each test. It is not entirely clear that this is the appropriate comparison since, for a given number of affecteds, the TDT requires 50% more genotyping at the candidate locus while the STRAT may require additional genotyping of unlinked markers. Nonetheless, these comparisons allow some qualitative predictions about the relative power of these tests:
- 1.
When the same allele is associated in both subpopulations, the TDT tends to be slightly, but not dramatically, more powerful (top three lines for both models, table 2). Note, however, that if we compare the two tests on the basis of the number of individuals genotyped, the STRAT is slightly more powerful than the TDT in several of these cases (table 2, last column). - 2.
When there is an association in one subpopulation but not the other, the STRAT is often more powerful than the TDT (middle three lines for both models, table 2). An exception arises under the admixture model, when the candidate allele frequencies are quite different in the two subpopulations (admixture model, lines 5 and 6). In this case the TDT has very good power, for reasons that are discussed in detail by McKeigue (1997) and are exploited further by McKeigue (1998). - 3.
If different alleles are associated in each subpopulation, the STRAT retains power, whereas the TDT may have little or no power (bottom two lines for both models, table 2). Such a situation might arise when, through random evolutionary sampling, a particular allele at the candidate locus is in linkage disequilibrium with a disease mutation in one subpopulation but not in another. In particular, if the most common mutations are different in each subpopulation, it will be random which allele (if any) at a nearby marker is associated with the disease in each subpopulation. Such a situation might also arise because unlinked factors (either genetic or environmental) modifying the expression of the linked mutation differ across subpopulations, as at the CCR5 locus (Gonzalez et al. 1999). It is difficult, at present, to know how common such situations will be.
Although the TDT is statistically valid in the presence of population structure, notice that, because it pools its observations across subpopulations, it can lack power if the association is not present in all subpopulations—especially if the effects from different subpopulations tend to cancel each other out. In contrast, the STRAT tests for association separately in each subpopulation, which allows it to detect the presence of different effects in each subpopulation.
Discussion
In this paper we have described a statistical method (“STRAT”) for performing association mapping in structured populations. Our approach offers some potential advantages over family-based association methods, such as the TDT, which are also valid in the presence of population structure. Possibly the most important of these is that collecting DNA samples from unrelated cases and controls is often far easier than collecting family members of affected individuals. This feature will be an important consideration in designing large studies to detect or confirm associations of small effect. In addition, the ability to use unrelated controls suggests the possibility of establishing public databases of genotype information that can be shared among studies, particularly if genome screens for association become viable.
The results of our power simulations underline the potential importance of allowing for different effects in different subpopulations, rather than pooling information across populations. This could be particularly important where gene-gene or gene-environment interactions differ across subpopulations. Of course, it would be possible to extend our approach to family-based tests in order to detect this sort of effect in that framework too.
Although we have compared the power of the STRAT test directly with the TDT, there are a number of points which should be borne in mind when interpreting these comparisons. Perhaps the most obvious is that the TDT and STRAT approaches have different genotyping requirements. The TDT generally requires 50% extra genotyping at each candidate locus, whereas STRAT requires a series of unlinked markers to infer population structure. Clearly, if there is just one candidate locus, STRAT requires substantially more genotyping. However, for those studies that consider candidate loci in many genes, this issue can disappear, since the candidates themselves can be used for learning about population structure before testing for association. In particular, STRAT will be well-suited to studies which test for association at many SNPs across the genome, in which case no extra marker loci will be required. Conversely, in studies which have collected large numbers of families for linkage mapping, TDT methods can be used immediately to help confirm regions of suspected linkage. A slightly more subtle difference between the STRAT and the TDT is that, when data from structured populations are being analyzed, the STRAT approach may be more suitable than the TDT for fine-scale mapping. When applied in structured populations, the TDT requires linkage—but not especially tight linkage—between a marker and a disease-causing locus in order to detect association between marker and phenotype. In contrast, since STRAT detects association between a marker and phenotype within “unstructured” subpopulations, and since such associations are likely to occur only for markers tightly linked to a disease locus, STRAT should allow mapping on a finer scale.
In recent work, Devlin and Roeder (1999) describe an alternative method for association mapping in structured populations using genome screen data of biallelic markers. They assume that, under the null hypothesis, allele-frequency differences between cases and controls are drawn from the same distribution at all loci. Their approach does not require explicit modeling of population structure and so is likely to be more robust in situations where our model of population structure is inappropriate. However, it will be liable to false positives if some loci show unusually large population differences—due to selection, for example. Our method is valid even at such loci, provided that we can accurately infer population structure (note the examples with extreme allele-frequency differences shown in table 1).
In describing our method, we outlined a statistical test which is designed to be most powerful against a specific alternative model in which the two alleles carried by an individual have independent effects on the risk of disease. However, it would be quite easy to modify this test to handle other alternative models. For example, one might test instead whether the diploid genotypes are associated with the phenotype. In any particular case, the relative power of such tests will depend on the (usually unknown) mode of transmission of the disease.
Although our approach is based on an assumption that we can infer population structure (i.e., estimate Q) accurately, our simulations show that the distribution of our test statistic under the null hypothesis is reasonably robust to some inaccuracy in the estimation of Q (as with data set B, for example). It is not easy to make general statements in advance about how many loci will be necessary for accurate clustering, as this depends on how divergent the subpopulations are—though our experience suggests that, for realistic problems, this will usually be ⩾100 microsatellite loci, or somewhat larger numbers of biallelic markers.
In practice, one would like to assess whether the accuracy of the estimated Q is adequate. This can be done as follows, assuming that markers that are genuinely associated with the phenotype within subpopulations are rare. Under the null hypothesis, the P values at each locus should be uniformly distributed, but, if the clustering is inadequate, there will be an excess of low P values. Thus, one can test each marker locus for association with the phenotype and then test whether the empirical distribution of P values fits a uniform distribution (e.g., using the Kolmogorov-Smirnov test). Estimates of Q can be improved by providing “learning samples” in the estimation procedure (genotypes of individuals whose subpopulations are known). For example, in performing a case-control study of African Americans, in whom there is often substantial European admixture, it would be sensible to make use of genotype data from West Africans and Europeans for estimating Q, if such data were available.
McKeigue (1998) describes a method for detecting linkage in admixed populations that exploits the fact that, when there has been recent admixture, individuals will inherit “segments” of chromosomes from one subpopulation or another, and, as a result, there will be correlations between the ancestry of linked markers within an individual. Our approach is rather different, concentrating on association within subpopulations, rather than these correlations in ancestry. Indeed, our model of population structure completely ignores these correlations, treating the ancestry of all loci as being independent. Explicit modeling of the correlations should improve our method. First, it should improve accuracy of the estimated Q. Second, markers near to the candidate locus should provide additional information about the origin of each individual’s candidate alleles, which might allow greater power. Unfortunately, the fact that haplotype phase information is typically absent in a case-control study may reduce the utility of this sort of information (and makes the implementation more difficult). Absence of haplotype information also reduces the ease and efficiency with which SNPs can be used for fine mapping purposes. For these reasons, collecting DNA from relatives—even offspring—may be useful in case-control studies, as it would allow haplotypes to be at least partially reconstructed.
In combination with Pritchard and Rosenberg (1999), we have now described a complete approach for use of case-control studies to test for association. Pritchard and Rosenberg (1999) provided a simple diagnostic test for whether a particular study was liable to spurious associations. If that diagnostic test indicates that spurious associations are indeed a potential problem, the present paper provides an alternative method for testing for association. Further, when there is population structure, but the sampling of cases is not biased towards particular subpopulations, and so spurious associations are not a problem, tests of association may lack power for the reasons described above. The method described here will be useful in these situations too.
Software Availability
The software used for this study will be made available at http://www.stats.ox.ac.uk/~pritch/home.html.
Acknowledgments
This work was supported by a Hitchings-Elion fellowship from Burroughs-Wellcome Fund (to J.K.P.), by grants from the University of Oxford and the Wellcome Trust, reference 057416 (to M.S.), by a National Defense Science and Engineering graduate fellowship (to N.A.R.), and by grants GR/M14197 and 43/MMI09788 from the Engineering and Physical Sciences Research Council (EPSRC) and the Biotechnology and Biological Sciences Research Council (BBSRC), respectively (to P.D.). We wish to thank A. Kong and M. Frigge, for helpful discussions, and two anonymous reviewers, for thoughtful comments on the manuscript.
References
- Boehnke M, Langefeld CD (1998) Genetic association mapping based on discordant sib pairs: the discordant-alleles test. Am J Hum Genet 62:950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G, Amos W, Bellamy R, Siddiqui MR, Frodsham A, Hill A, Rubinsztein D (1999) An empirical exploration of the (δμ)2 genetic distance for 213 human microsatellite markers. Am J Hum Genet 65:1125–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood estimation from incomplete data via the EM algorithm (with discussion). J R Stat Soc B 39:1–38 [Google Scholar]
- Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004 [DOI] [PubMed] [Google Scholar]
- Ewens WJ, Spielman RS (1995) The transmission/disequilibrium test: history, subdivision, and admixture. Am J Hum Genet 57:455–464 [PMC free article] [PubMed] [Google Scholar]
- Feldman MW, Kumm J, Pritchard JK (1999) Mutation and migration in models of microsatellite evolution. In: Goldstein D, Schlotterer C (eds) Microsatellites: evolution and applications. Oxford University Press, Oxford, pp. 98–115 [Google Scholar]
- Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW (1995) Genetic absolute dating based on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA 92:6723–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Bamshad M, Sato N, Mummidi S, Dhanda R, Catano G, Cabrera S, et al (1999) Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc Natl Acad Sci USA 96:12004–12009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR (1990) Gene genealogies and the coalescent process In: Futuyma D, Antonovics J (eds) Oxford surveys in evolutionary biology. Oxford University Press, Oxford, p. 1–44 [Google Scholar]
- Jorde LB, Bamshad MJ, Watkins WS, Zenger R, Fraley AE, Krakowiak PA, Carpenter KD, et al (1995) Origins and affinities of modern humans: a comparison of mitochondrial and nuclear genetic data. Am J Hum Genet 57:523–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Rogers AR, Bamshad M, Watkins WS, Krakowiak R, Sung S, Kere J, Harpending HC (1997) Microsatellite diversity and the demographic history of modern humans. Proc Natl Acad Sci USA 94:3100–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048 [DOI] [PubMed] [Google Scholar]
- Lazzeroni LC, Lange K (1998) A conditional inference framework for extending the transmission/disequilibrium test. Hum Hered 48:67–81 [DOI] [PubMed] [Google Scholar]
- McKeigue PM (1997) Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am J Hum Genet 60:188–196 [PMC free article] [PubMed] [Google Scholar]
- ——— (1998) Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am J Hum Genet 63:241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, et al (1998) Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet 63:1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–513 [PMC free article] [PubMed] [Google Scholar]