RAPID DEGRADATION OF BIM BY THE UBIQUITIN-PROTEASOME PATHWAY MEDIATES SHORT-TERM ISCHEMIC TOLERANCE IN CULTURED NEURONS (original) (raw)
. Author manuscript; available in PMC: 2006 Mar 17.
Published in final edited form as: J Biol Chem. 2006 Jan 23;281(11):7429–7436. doi: 10.1074/jbc.M512138200
Abstract
A previous exposure to a non-harmful ischemic insult (preconditioning) protects the brain against subsequent harmful ischemia (ischemic tolerance). In contrast to delayed gene-mediated ischemic tolerance, little is known about the molecular mechanisms that regulate rapid ischemic tolerance, which occurs within one hour following preconditioning. Here we investigate the degradation of the pro-apoptotic Bcl-2 family member Bim as a mechanism of rapid ischemic tolerance. Bim protein levels were reduced 1 hour following preconditioning and occurred concurrent with an increase in Bim ubiquitination. Ubiquitinated proteins are degraded by the proteasome, and inhibition of the proteasome with MG132 (a proteasome inhibitor) prevented Bim degradation and blocked rapid ischemic tolerance. Inhibition of p42/ p44 mitogen-activated protein kinase activation by U0126 reduced Bim ubiquitination, Bim degradation and blocked rapid ischemic tolerance. Finally, inhibition of Bim expression using antisense oligonucleotides also reduced cell death following ischemic challenge. Our results suggest that following preconditioning ischemia, Bim is rapidly degraded by the ubiquitin-proteasome system resulting in rapid ischemic tolerance. This suggests that the rapid degradation of cell death promoting proteins by the ubiquitin-proteasome pathway may represent a novel therapeutic strategy to reduce cell damage following neuropathological insults e.g. stroke.
Keywords: Bcl-2, stroke, U0126, PD98059, MAPK, preconditioning
Abbreviations: p42/ p44 MAPK; p42/ p44 mitogen-activated protein kinase; MEK, MAPK kinase; PI3 kinase, phosphatidylinositol 3 kinase; Bim, Bcl-2 interacting mediator of cell death; Bcl, B cell lymphoma; BH, Bcl-2 homology domain; NGF, Nerve growth factor; P62 UBA, 62 kDa ubiquitin-associated domain; OGD, oxygen and glucose deprivation
Tolerance is the phenomenon whereby previous exposure to a sub-toxic insult conditions the cell or organism against a more severe toxic challenge (1). Tolerance appears to be a conserved endogenous phenomenon and has been described in multiple organisms from simple cells to more complex systems (1,2). Ischemic tolerance has been observed in the brain; a brief preconditioning ischemic insult results in a reduced activation of cell death pathways that occur following a harmful ischemic event (1,3–6).
Two types of ischemic tolerance have been reported for the brain: delayed and rapid ischemic tolerance. Delayed ischemic tolerance develops over 1–3 days in vivo (7,8) or 24 hours in vitro (9–11), is mediated by a gene-based mechanism and requires new protein synthesis (1,6,12). In contrast, rapid ischemic tolerance is protein synthesis-independent and occurs within 1 hour of the preconditioning ischemia (13–17). Rapid ischemic tolerance can be blocked by adenosine A1 receptor antagonists (17,18) and mimicked by KATP channel openers (13,19,20). The above suggests that different molecular mechanisms contribute to each type of tolerance.
The Bcl-2 family of genes regulate cell death processes and consist of two groups: the pro-cell death family members, for example Bax, and the pro-survival members, for example, Bcl-2 (21). The ratio between pro-apoptotic and pro-survival members of the Bcl-2 family determines the cell’s fate (22,23). Bim (also called Bod) is a member of the pro-apoptotic Bcl-2 family only containing a BH3 domain (e.g. Bim, Bad and Bid) (24). Bim is expressed as at least 3 isoforms (BimEL, BimL BimS) that determine its cell death potency (24–26); BimS is most potent. Bim plays a critical role as a mediator of neuronal cell death (27). Raising Bim levels increases cell death, where as Bim deficient mice are resilient to many cell death-promoting stimuli (28).
Interleukin-3 and nerve growth factor (NGF) regulate Bim protein levels, via a raf/ p42/ p44 mitogen-activated protein kinase (p42/ p44 MAPK)-dependent mechanism, but not via an Akt-dependent mechanism (29,30). Phosphorylation of Bim by p42/ p44 MAPK targets Bim for degradation by the ubiquitin-proteasome system (31–33). Since Bim is an important mediator of neuronal cell death, we investigated the regulation of Bim protein levels via the ubiquitin-proteasome system in an in vitro model of rapid ischemic tolerance.
EXPERIMENTAL PROCEDURES
Materials and methods
The proteasome inhibitor MG-132 (Z-Leu-Leu-Leu-CHO) was purchased from Calbiochem, (San Diego, CA); cycloheximide, U0126 (1,4-Diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene) and PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) from Sigma (St Louis, MO); anti-caspase 3, anti-cleaved caspase 3, anti-phosphorylated p42/ p44 MPAK and native p42/ p44 MAPK from Cell Signaling (Beverley, CA); anti-Bim from Stressgen (Victoria, BC, Canada); anti-Bid from R & D Systems (Minneapolis, MN); anti-Bax and anti-α-tubulin from Santa Cruz Biotechnology (Santa Cruz, CA); p62 UBA agarose beads from Affiniti (Exeter, UK), antisense oligonucleotides were synthesized by Sigma-Genosys (The Woodlands, TX).
Cortical cell culture
Sprague-Dawley rat pups were used to prepare cortical neuronal cultures, as previously described (12). Briefly, cortices were dissected from 10–12 rat pups (P1-2) and enzymatically dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA). Cells were plated out at a density of 400,000 cells per coverslip for cell viability assays, or 5,000,000 cells per 10 cm culture dish (Primara; Becton Dickinson, San Jose, CA, USA) for immuno blotting or immunoprecipitation in Neurobasal-A/ B27 media (Invitrogen, Carlsbad, CA, USA) for 7–10 days.
Ischemic tolerance
Oxygen and glucose deprivation (OGD) was performed by washing the cells with phosphate buffered saline (PBS) (NaCl (1.37 mM), KCl (2.7 mM), Na2HPO4 (10 mM), KH2PO4 (1.7 mM); pH 7.4) supplemented with 0.5 mM CaCl2, 1.0 mM MgCl2 and placing culture dishes in an anaerobic chamber for 30 or 120 min (Forma Scientific, Marjetta, OH, USA)(85 % N2, 5% H2, 10 % CO2; 35 ºC) as previously described (34,35). Anaerobic conditions in the chamber were monitored using Gaspack anaerobic indicator strips (Becton Dickinson, San Jose, CA, USA). OGD was terminated by removing cells from the anoxia chamber, replenishing with Neurobasal A media and replacing them back into the normoxic incubator.
Cultured cells were subject to the following treatments: 1) Control cells were washed with PBS and maintained in Neurobasal A media (without B27 supplement), 2) Some cells were preconditioned with 30 min OGD and then recovered for 24 hours in Neurobasal A media, 3) Some cells were subject to 120 min OGD (harmful ischemia) followed by 24 hours recovery in Neurobasal A media, 4) Some cells were preconditioned with 30 min OGD and recovered for 1 hour before being subject to a harmful 120 min OGD ischemia. Cell death was performed 24 hours following the final OGD challenge. In preliminary experiments the time interval between preconditioning ischemia and harmful ischemia was varied from 1–24 hours (see Fig 1a). For rapid tolerance experiments the interval between preconditioning and harmful ischemia was 1 hour.
Figure 1. Rapid ischemic tolerance in vitro.
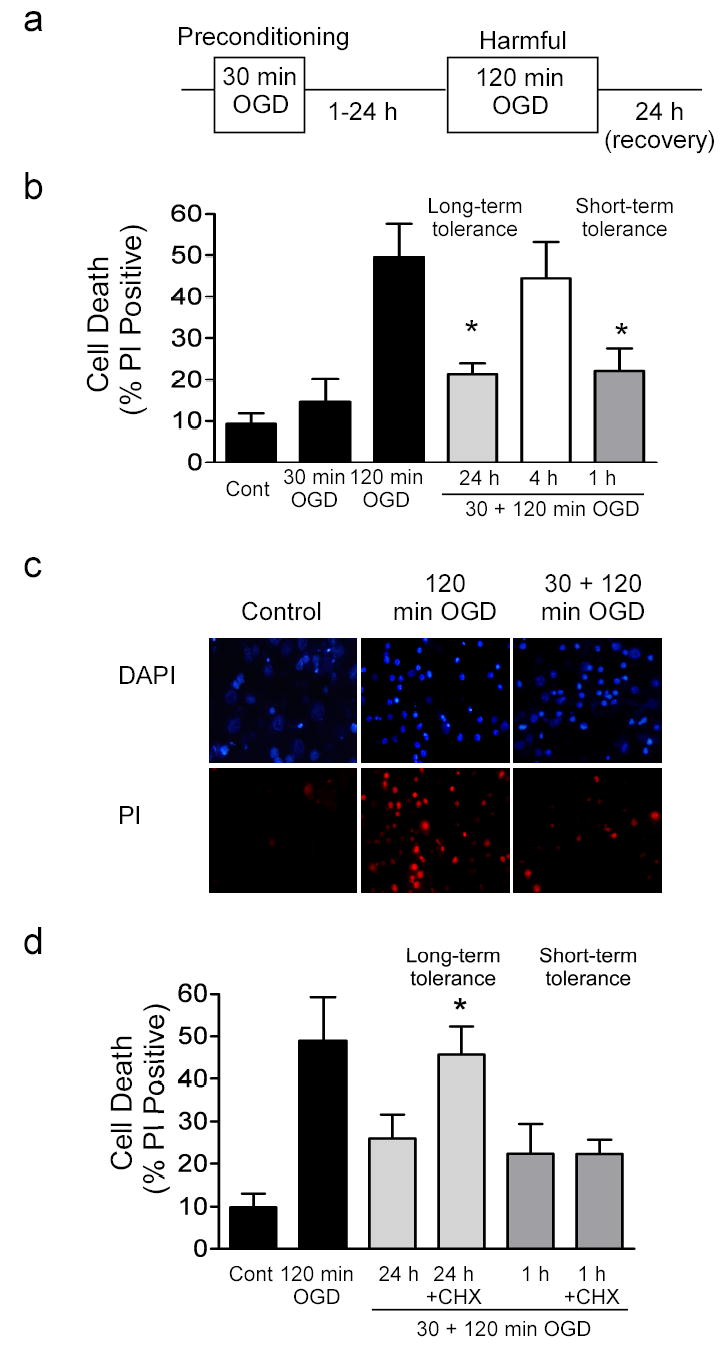
a) Outline of oxygen and glucose deprivation (OGD) protocol. Cells are subject to 30 min OGD (preconditioning), recovered for variable time periods (1–24 hours) and then subject to 120 min OGD (harmful). For rapid ischemic tolerance an interval of one hour was used between preconditioning ischemia and harmful ischemia. Cell death was assessed by propidium iodide (PI) staining 24 hours following ischemia. Some cells receive 30 min or 120 min OGD only, or no OGD (control). All drug treatments were applied following 30 min OGD and prior to 120 min OGD. b) Effect of various recovery times following 30 min OGD preconditioning on 120 min OGD -induced cell death. Gray blocks denote preconditioned cells. Data shown are mean ± SEM, (n=5). c) Representative images of PI stained cells following no ischemia (control), 120 min OGD and cells preconditioned with 30 min OGD, and then subject to 120 min OGD 1 hour later. Cells were stained and images were acquired 24 hours following last ischemic insult. Note decreased number of PI positive cells in tolerant cultures compared to cells exposed to harmful ischemia without preconditioning. d) Effect of cycloheximide on rapid ischemic tolerance. Cycloheximide was incubated with the cells for 1 hour or 24 hours following preconditioning (Gray denotes long-term tolerance, hatching denotes short-term tolerance). Data shown are mean ± SEM, (n=4) and analyzed by one-way ANOVA with Bonferroni’s post-hoc test (* denotes P<0.05).
Cell death assay (Propidium Iodide staining)
For cell death assays, coverslips containing cortical cells were incubated with propidium iodide (1.5 μg/ml) for 2 min, washed with PBS and fixed with 4 % formaldehyde. Cells were permeabilized with 0.1 % Triton X-100 and then mounted onto glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA, USA). Cell death was determined as the ratio of propidium iodide-stained cells, to the total number of DAPI-stained cells, visualized using a fluorescence microscope.
Immunocytochemistry and TUNEL
Cultured cells on coverslips were permeablized with 3% triton X-100. The samples were pre-blocked with 2% goat serum and 1% BSA, followed by incubation with primary antibody at 4 °C overnight (anti-caspase 3 from Cell Signaling). The samples were washed three times with PBS and incubated with secondary antibody for 1 hour at 37°C (Jackson Labs). Slides were washed and mounted in a medium containing 4′, 6 diamino-2-phenylindole (DAPI) (Vector Laboratories, Burlington, CA). TUNEL labeling was performed according to manufacturers instructions (Roche, Indianapolis, IN, USA). Immunolabeling was studied using a Leica microscope under Ex/Em wavelengths of 340/425 nm (blue), 500/550 nm (green) and 580/630 nm (red) respectively. Images were collected using an Optronics DEI-750 3-chip camera equipped with a BQ 8000 sVGA frame grabber and analyzed using Bioquant (Nashville, TN).
Immunoblotting
Immunoblotting was performed as previously described (36). Tissue samples were lysed in a nondenaturing buffer containing the protease inhibitors phenylmethylsulfonylfluride (100 μg/ml), aprotinin (1 μg/ml), leupeptin (1 μg/ml), pepstatin (1 μg/ml), NaF (50 mM) Na3VO4 (2 mM) and phosphatase inhibitor cocktail (Sigma, St Louis, MO). Protein concentration was determined by Bradford reagent spectrophotometrically at A595. Protein samples (50 μg) were denatured in a gel-loading buffer at 100 °C for 5 min and then loaded onto 12 % SDS-polyacrylamide gels. Proteins were transferred to polyvinylodene difluoride membranes and incubated with primary antibodies at 4 °C overnight. Membranes were incubated with anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (Cell Signaling Technology, Beverly, MA, USA) followed by chemiluminesence detection (NEN Life Science Products, Boston, MA, USA) and then exposed to Kodak film (Biomax). Images were captured using a Dage 72 camera.
Ubiquitin pull-down assay
Cells were plated out on 10 cm dishes at 5 million cells/dish. Cell lysates were prepared in a RIPA buffer (Tris (10 mM, pH 8.0), NaCl (140 mM), glycerol (5 %), sodium deoxycholate (0.1 %), SDS (0.1 %), Triton X-100 (1 %), protease Inhibitors with EDTA (Roche CompleteTM, Roche, Indianapolis, IN, USA)). Total protein (0.8 mg) was cleared and then incubated with 30 μl P62 UBA agarose beads (Affiniti, Exeter, UK) at 4 °C for 2 hours. Samples were washed using an immunoprecipitation kit (IP50, Sigma). The protein/bead mixture was denatured at 95 °C for 5 min and then loaded onto 10 % SDS-polyacrylamide gels. Blots were subject to immunoblotting as above. Ubiquitin pull down was verified by re-probing blots with anti-ubiquitin antibody (Santa Cruz, CA).
Bim Antisense
Antisense oligonucleotides complementary to the bim cDNA sequence (accession number AF136927) were identified and synthesized by Sigma Genosys (AS 1 5′ CAGAAGGTTGCTTGGCCA3′, AS 2 5′ CCACCTTCTCTGTCACAC3′, AS 3 5′ CCTCCTGAGACTGCCTTA3′, AS 1 5′ CTCTCAGCAGGCTGCAAT 3′). Sense oligonucleotides identical to the coding cDNA sequence were (S1 5′-TGGCCAAGCAACCTTCTG-3′, S2 5′-GTGTGACAGAGAAGGTGG-3′, S3 TAAGGCAGTCTCAGGAGG-3′ S4 5′- ATTGCAGCCTGCTGAGAG-3′). Oligonucleotides were phosphothioate modified on the first and last 3 linkages to reduce degradation. Cortical neuronal cultures were incubated for 48 hours with a cocktail of antisense (4 x 1 μM of each oligo), sense (4 x 1 μM of each oligo) or control scrambled sequence (5′-TCGGCTAGACACCATCGT-3′; 4 μM). Cultures were replenished with oligonucleotides every 24 hours. Cultures were subject to 120 min OGD and cell death assessed by PI staining. Some cultures were incubated with Bim antisense, sense or scrambled control and then subject to immunoblotting for Bim, Bid and α-tubulin, as above.
Data analysis
Data are presented as mean ± SEM of n determinations. Data from cell death assays were analyzed using repeated-measures one-way analysis of variance (ANOVA) with Bonferroni’s post-hoc test (Graphpad Prism, Graphpad Software, San Diego, CA, USA). Significance was accepted at p<0.05.
RESULTS
An in vitro model of rapid ischemic tolerance
We investigated the molecular mechanisms involved in rapid ischemic tolerance using an in vitro model of oxygen and glucose deprivation (OGD). When cells were subject to 120 min OGD we observed approximately 50–60 % increase in PI positive staining of cells, which we interpret as an increase in cell death (Fig. 1b & 1c). In previous studies approximately 60 % of these cells had features of apoptosis (TUNEL positive) (Fig. 2b) (35). Consistent with our previous study of long-term tolerance (34), when the cultures were preconditioned with 30 min OGD, 24 hours prior to a harmful dose of 120 min OGD, PI staining (cell death) was reduced (Fig. 1b). When the interval between preconditioning and harmful ischemia was reduced to 4 hours, the protective effect of preconditioning was lost (Fig. 1b). However, when the interval between preconditioning and harmful ischemia was reduced to 1 hour, we also observed a decrease in PI positive cells, which we interpret as less cell death (rapid/short-term ischemic tolerance) (Fig. 1b & 1c). The total number of cells in the cultures was not affected by either harmful ischemia 120 min OGD or preconditioning ischemia (See Supplementary Fig. 1; also see(34)).
Figure 2. Preconditioning ischemia reduces cleavage of caspase 3 following harmful ischemia.
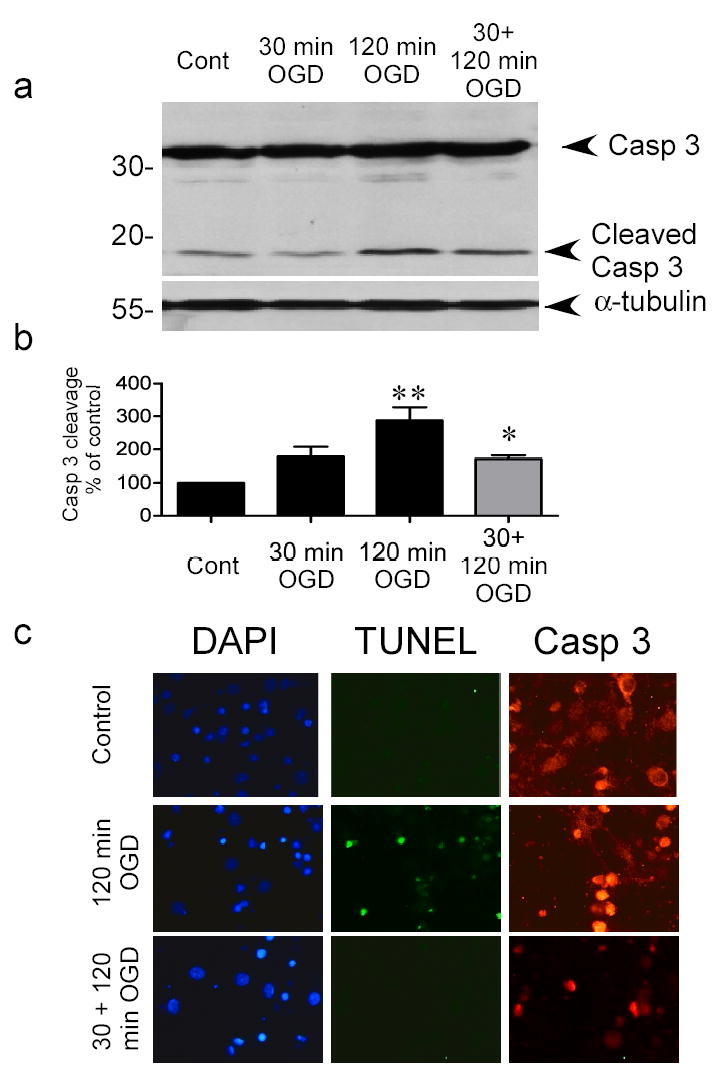
a) Cells were preconditioned with 30 min OGD, 1 hour prior to 120 min OGD. Caspase 3 cleavage was determined 24 hours later by immunoblotting with a cleaved caspase 3 P19/17 fragment specific antibody. Blots were reprobed with α-tubulin to control for loading. Image shown is a representative blot of 5 independent experiments. b) Quantification of western blots. Data shown are mean ± SEM (n=5) and analyzed by one-way ANOVA with Bonferroni’s post-hoc test (** denotes P<0.01 vs. control group and * denotes P<0.05 vs. 120 min OGD group). c) Immunocytochemical detection of caspase 3 in control, ischemia-treated and tolerant cortical cells. Cells were subject to various ischemic treatments and caspase 3 levels determined by immunocytochemistry 24 hours later. DNA breaks were also assessed using TUNEL. Note the increased TUNEL staining in 120 min OGD treated cells, and the more nuclear pattern of caspase 3 staining in cells following ischemia. Images are representative of 2 experiments.
Long term ischemic tolerance requires new protein synthesis and is blocked by protein synthesis inhibitors (10,34). The protective effect of rapid ischemic preconditioning was not reduced when the protein synthesis inhibitor cycloheximide was added to the cells for 1 hour between OGD treatments (Fig. 1d). In contrast the protective effect of delayed ischemic tolerance was reduced when cells were incubated with cycloheximide for 24 hours following preconditioning, which is consistent with previous reports (10,34) (Fig. 1d). These data suggest that rapid ischemic tolerance does not require the synthesis of new proteins.
Reduced cleavage of an executioner caspase in rapid ischemic tolerance
It has been suggested that following preconditioning the activation of cell death pathways is reduced (4,5), accordingly we investigated the cleavage of the quintessential cell death protease caspase 3. Caspase 3 requires proteolytic cleavage for activation (37). Caspase 3 cleavage was increased three-fold 24 hours following 120 min OGD, compared to control cells (Fig 2a & 2b). However, in cells preconditioned with 30 min OGD, 1 hour prior to 120 min OGD, caspase 3 cleavage was reduced (Fig. 2a & 2b). These data suggest that preconditioning reduces caspase-3 mediated cell death pathways following harmful ischemia.
To further examine the location of caspase 3 in tolerant cells we visualized full-length caspase 3 expression using immunocytochemistry. Caspase 3 appeared as a cytosolic staining pattern in control cortical cell cultures (Fig. 2c). The staining was stronger in the nucleus following 120 min OGD as well as being denser in the cytosol (Fig. 2c). In contrast few cells stained positive for caspase 3 following preconditioning, however a small number did show the more dense staining pattern of injured cells (Fig. 2c). Taken together these data suggest that preconditioning ischemia reduces the activation of caspase 3 following a subsequent injurious ischemic insult.
Decreased levels of the pro-apoptotic protein Bim following preconditioning
We investigated the expression of the pro-apoptotic protein Bim following preconditioning. Bim has been shown to play a prominent role in neuronal cell-death pathways (27,28,38). Bim protein was expressed as a 26 kDa protein in control cells (Fig. 3a). Bim protein levels were decreased in cortical cultures 1 hour following 30 min OGD (preconditioning), but recovered to normal levels at 4 hours (Fig. 3a and 3b). In contrast the expression of the pro-apoptotic cell death proteins Bid and Bax did not change (Fig. 3a).
Figure 3. Preconditioning ischemia reduces Bim protein levels.
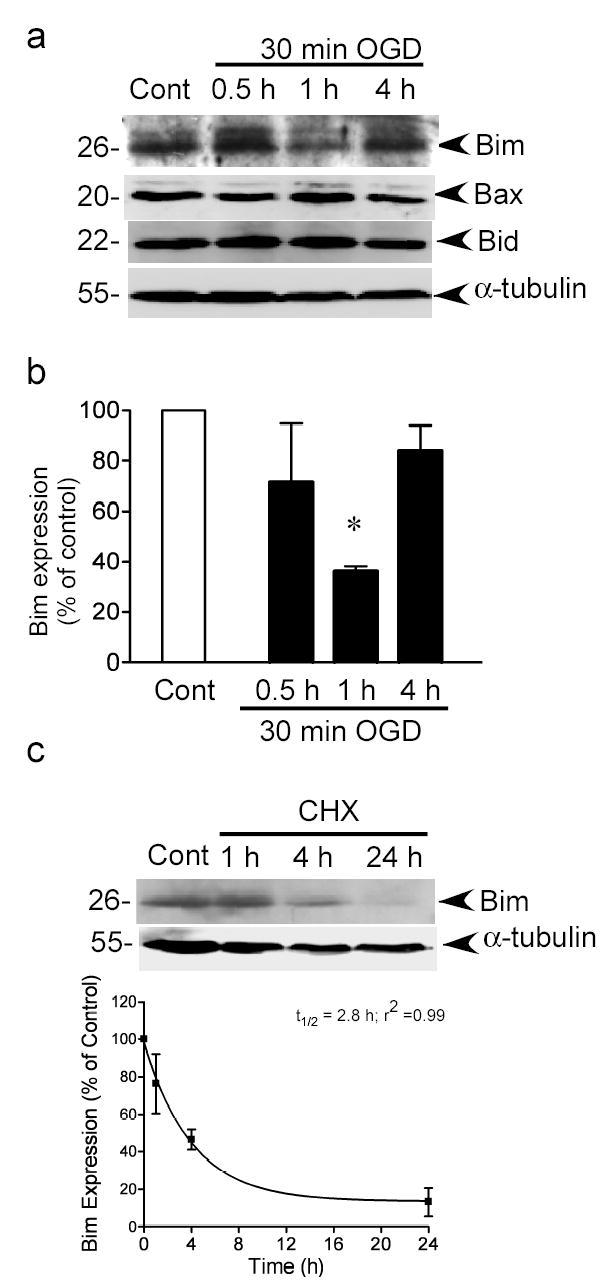
a) Bim, Bid and Bax protein levels were determined by immunoblotting. Blots were re-probed with α-tubulin to control for loading. Image shown is a representative image from 3–4 independent experiments. b) Quantification of Bim immunoblots. Data were analyzed by one-way ANOVA with Bonferroni’s post-hoc test, * denotes P>0.05, n=4). c) Cells were incubated with cycloheximide (10 μM) for 1–24 hours and Bim levels were determined by immunoblot. Blots were reprobed for α-tubulin. The optical densities from 3 independent experiments were plotted and data fitted to a mono-exponential decay curve to determine the half-life of Bim (t½ =2.8h, r2= 0.99). Data shown are mean ± sem (n=3).
In order to test whether changes in Bim expression were due to a blockade in translation, we incubated cells with a high concentration of cycloheximide (10 μM) for 1–24 hours and then measured Bim protein levels by immunoblot. As can be seen in Fig. 3c, the resulting decrease in Bim expression was slower than that following preconditioning ischemia. We also analyzed the density of the Bim signal and determined that the half-life of Bim in neuronal cultures following cycloheximide treatment was 2.8 hours (Fig. 3c), similar to its half-life (approx. 4 hours) in HEK293 cells (32). These data suggest that the decrease in Bim levels following preconditioning are not due to decreased translation rates of the protein, but suggest that Bim is actively degraded.
Bim ubiquitination and proteasomal degradation following preconditioning ischemia
Bim protein levels are regulated by the ubiquitin-proteasome pathway (31,32), hence we investigated whether Bim was ubiquitinated following 30 min OGD preconditioning. Our initial experiments attempted to replicate the Bim ubiquitin immunoprecipitation shown by Akiyama (39), however we were unable to precipitate Bim using their suggested antibody, nor a Stressgen antibody (not shown). This result may be due to differences in cell types used in these experiments; their experiments were performed in mouse cells where ours use rat cortical cells. Hence we sought an alternative approach.
It has been shown that the P62 protein (sequestome-1) binds ubiquitinated proteins and shuttles them to the proteasome for degradation (40–42). Conjugation of the ubiquitin-binding domain (UBA) of P62 to agarose beads has been shown to precipitate/pull-down ubiquitinated tau (40). We applied a similar approach using a commercially available P62 UBA protein conjugated to agarose (Affiniti, UK). Using these P62 UBA beads we performed pull-down assays to determine whether preconditioning increase the association of P62 to Bim. In control untreated cells, we observe variable amounts of Bim pulled down by these beads (Fig. 4a and 5e). Following 30 min OGD and 1 hour recovery there was an increase in Bim pulled-down by P62 UBA beads, which appeared in a ladder pattern with a higher molecular weight than native Bim (Fig. 4a). While not direct evidence of Bim ubiquitination, these data are consistent with Bim being ubiquitinated following preconditioning ischemia (30 min OGD).
Figure 4. Bim is ubiquitinated following preconditioning and inhibition of the proteasome blocks rapid ischemic tolerance and Bim degradation.
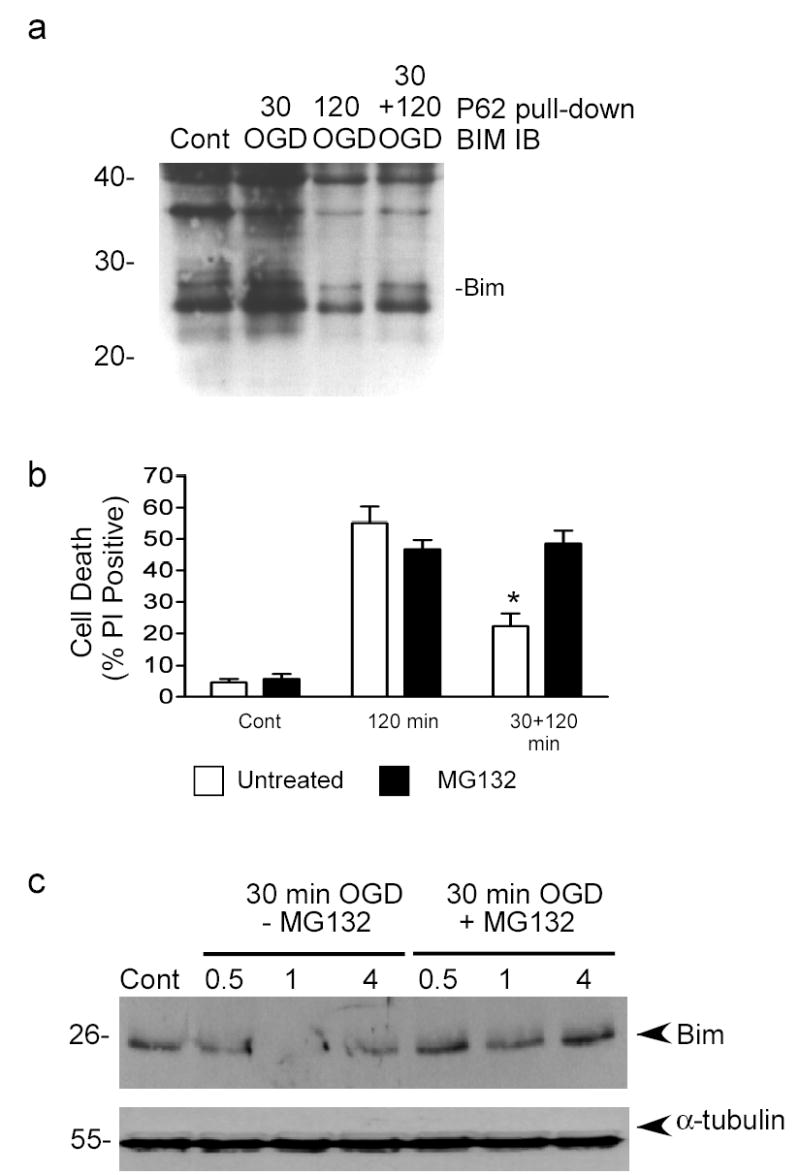
a) Bim ubiquitination was determined by pull-down with a ubiquitin-binding domain conjugated to agarose beads (P62 UBA-agarose). Experiments were run in the presence of MG132 (0.5 μM) to prevent degradation of ubiquitinated Bim. Pulled-down proteins were immunoblotted (IB) for Bim. Image shown is representative of 3 independent experiments. The weight of native Bim is marked. b) Cells were preconditioned with 30 min OGD and then incubated with MG132 (0.5 μM) for 1 hour. Cell death was assessed using propidium iodide (PI) staining. Note that MG132 blocks short term tolerance. Data shown are mean ± SEM (n=6). Data are analyzed by one-way ANOVA, with Bonferroni’s post hoc test (* denotes P<0.01). c) Cells were preconditioned with 30 min OGD and then recovered for 0.5, 1 or 4 hours with or without MG132 (0.5 μM). Bim protein levels were determined by immunoblot. Blots were re-probed with α-tubulin to control for loading. Data shown are representative blot of 3 independent experiments.
Figure 5. p42/ p44 MAPK regulates Bim degradation following preconditioning.
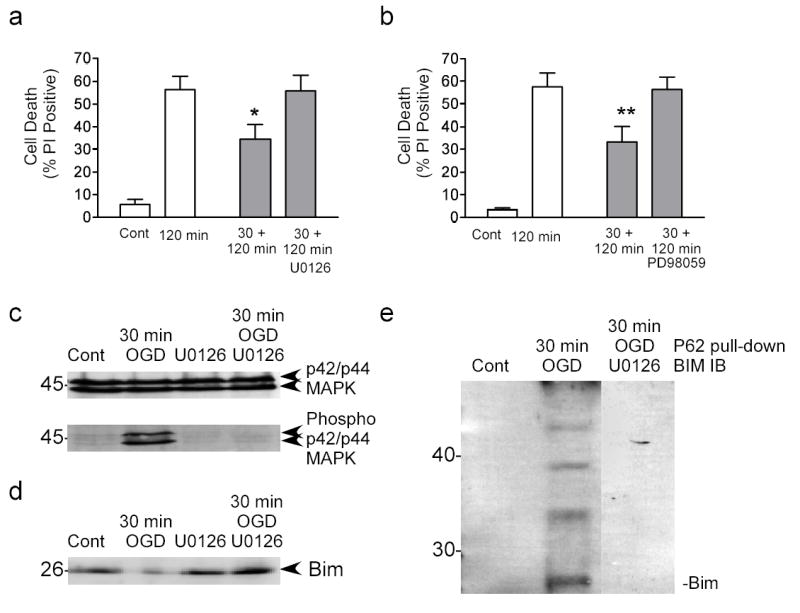
a) Cells were preconditioned with 30 min OGD and then incubated for 1 hour with U0126 (10 μM). Cell death was assessed 24 hour following 120 min OGD using propidium iodide staining. Data shown are mean ± SEM (n=8). Data were analyzed by one-way ANOVA, with Bonferroni’s post-hoc test (* denotes P<0.05). b) Cells were preconditioned with 30 min OGD and then incubated for 1 hour with PD98059 (10 μM). Cell death was assessed 24 hour following 120 min OGD using propidium iodide staining. Note that both U0126 and PD98059 block ischemic tolerance. Data shown are mean ± SEM (n=5). Data were analyzed by one-way ANOVA, with Bonferroni’s post-hoc test (** P<0.01). c) Cells were preconditioned with 30 min OGD and the recovered in the presence of U0126 for 1 hour. p42/ p44 MAPK phosphorylation were determined by immunoblot. Blots were reprobed for total p42/ p44 MAPK expression. d) Cells were subject to preconditioning ischemia and then recovered in the presence of U0126. Bim expression was determined by immunoblot. Note that U0126 blocks tolerance-induced Bim degradation. Data shown are representative of 3 independent experiments. e) Cells were preconditioned with 30 min OGD and then recovered in the presence of U0126 for 1 hour. Bim ubiquitination was assessed by p62 UBA precipitation. Bim levels were then detected by immunoblot. The precipitation of ubiquitinated Bim was reduced by U0126 (composite image).
If Bim is degraded by the ubiquitin–proteasome pathway, then blocking this pathway should block rapid ischemic tolerance and reduce Bim degradation following preconditioning. In order to investigate the effect of transient inhibition of the proteasome, we incubated cells with the irreversible proteasome inhibitor MG132 following preconditioning with 30 min OGD, but prior to harmful 120 min OGD. MG132 (0.5 μM) had no effect on basal cell death, or 120 min OGD -induced cell death (Fig. 4b). However, there was a reduction in the neuroprotective effect of preconditioning in cells treated with MG132 (Fig. 4b), suggesting that blocking the proteasome inhibits rapid ischemic tolerance.
We then further investigated the effect of MG132 on the degradation of Bim using immunoblotting. Bim protein levels were reduced 1 hour following preconditioning ischemia (Fig. 4c). When cells were recovered in the presence of MG132 following preconditioning ischemia, Bim protein levels were not reduced (Fig. 4c). In comparison, α-tubulin levels were not reduced (Fig. 4c). Taken together, these data suggest that inhibition of the proteasome with MG132 prevents both Bim degradation and rapid ischemic tolerance.
p42/ p44 MAPK regulates Bim degradation during rapid ischemic tolerance
The phosphorylation and resultant ubiquitination of Bim is regulated by p42/ p44 MAPK (31,32), hence we determined the effect of inhibition of p42/ p44 MAPK on rapid ischemic tolerance and Bim degradation. Cells were incubated for 1 hour following 30 min preconditioning OGD, with either PD98059 or U0126 (both 10 μM), compounds that prevent p42/ p44 MAPK activation by inhibiting the upstream regulatory MAPK kinase (Mek). Both U0126 and PD98059 blocked the neuroprotective effect of preconditioning (Fig. 4a & 4b, respectively), suggesting that active p42/ p44 MAPK regulates rapid ischemic tolerance. U0126 and PD98059 had no effect on basal cell death or 120 min OGD-induced cell death (Supplementary Fig. 2).
To show that p42/ p44 MAPK activation increases following preconditioning ischemia we used antibodies to phosphorylated p42/ p44 MAPK. There were low levels of p42/ p44 MAPK phosphorylation in control cells, however phosphorylation of p42/ p44 MAPK increased one hour following preconditioning ischemia (Fig. 5c). The increase in p42/ p44 MAPK phosphorylation following preconditioning ischemia was blocked by U0126 (Fig. 5c). In control cells treated with U0126 alone there was no phosphorylation of MAPK (Fig. 5c). As a control we also probed the blot for non-phospho-p42/ p44 MAPK. Total levels of p42/ p44 MAPK did not change following preconditioning ischemia or U0126 treatment (Fig. 5c). These data suggest that p42/ p44 MAPK activity increased following preconditioning ischemia.
We further investigated the effect of U0126 on Bim protein levels following 30 min OGD. We observed a decrease in Bim expression 1 hour following 30 min preconditioning ischemia (Fig. 5d). The decrease in Bim was blocked by U0126 (Fig. 5d). However, U0126 had no effect on Bim protein expression in control treated cells (Fig. 5d). These data suggest that the decrease in Bim levels observed following preconditioning is mediated via the p42/ p44 MAPK system.
Finally we investigated the effect of U0126 on the Bim ubiquitination determined by its interaction with p62 UBA. Ubiquitinated proteins were precipitated using p62 UBA beads and immunoblotted for Bim. U0126 reduced the pull-down of Bim following preconditioning 30 min OGD (Fig. 5e). Taken together these data suggest that p42/ p44 MAPK regulates the ubiquitination and degradation of Bim, resulting in rapid ischemic tolerance.
Reduced expression of Bim reduces ischemia-induced cell death in vitro
In order to show that a reduction in Bim expression increases the survival of neurons exposed to ischemia, we used an antisense approach to block expression of Bim, and hence mimic the neuroprotective effect of preconditioning. Cells were incubated with a cocktail of antisense oligonucleotides (4 oligos x 1μM) to Bim for 48 hours as previously described (38). Control cells received either sense oligonucleotides (4 oligos x 1 μM) or a scrambled oligonucleotide (1 oligo x 4 μM). Bim expression was reduced in the antisense (25 %) but not the sense or scrambled oligonucleotide-treated cells (Fig. 6a & b). In contrast the expression of another BH3-only pro-apoptotic member of the Bcl-2 family, Bid, did not change (Fig. 6a). Bim antisense reduced 120 min OGD-induced cell death in cortical cultures by nearly 50 % (Fig. 6c); in contrast sense and scrambled control oligonucleotides had no effect on 120 min OGD-induced cell death (Fig. 6c). Basal levels of cell death were not effected in these cultures (not shown). Taken together these data suggest that reducing Bim protein levels results in an increased protection of cortical neurons in response to harmful ischemia.
Figure 6. Antisense oligonucleotides reduce Bim expression and ischemia-induced cell death.
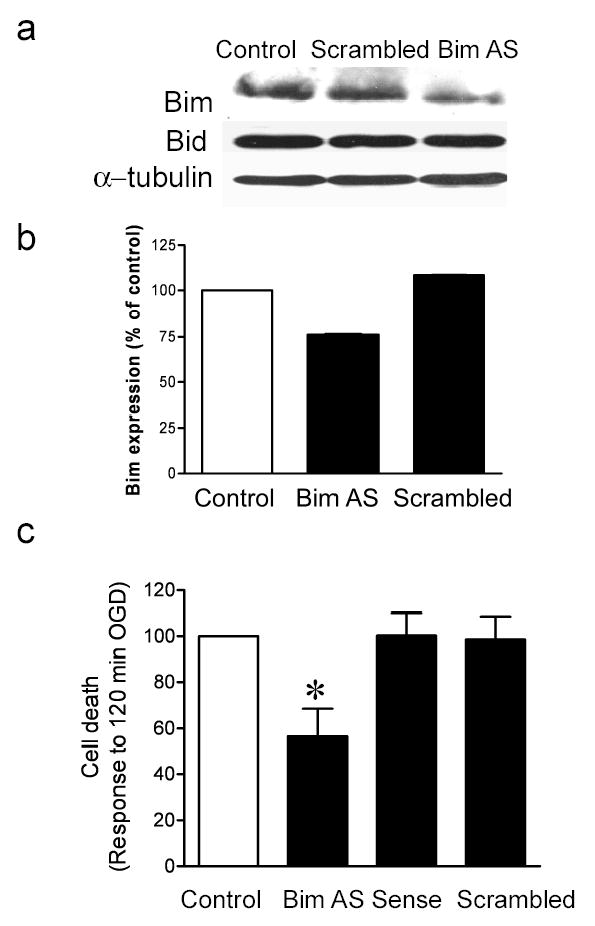
Cells were incubated for 48 hours with Bim antisense (AS) or scrambled control (SC) oligonucleotides. Bim and Bid protein levels were determined by immunoblot. To control for loading, blots were re-probed for α-tubulin. b) Quantification of immunoblots. c) Cells were treated for 48 hours with Bim antisense, sense or scrambled control oligonucleotides prior to 120 min OGD. Cell death was assessed 24 hours later by propidium iodide staining. Note that Bim antisense but not sense or scrambled control blocks 120 min OGD-induced cell death. Data shown are mean ± SEM (n=7). Data are analyzed by one-way ANOVA, with Bonferroni’s post-hoc test (* denotes P<0.05 vs. Control untreated cells).
DISCUSSION
Here we investigated the down-regulation of Bim protein levels by the ubiquitin-proteasome pathway as a potential mechanism of rapid ischemic tolerance. Following preconditioning with 30 min OGD, we observed a decrease in Bim protein levels and a concurrent increase in Bim ubiquitination. The proteasome inhibitor MG132 and inhibitors of p42/44 MAPK reduced the degradation of Bim. Blocking the proteasome and p42/ p44 MAPK activation also blocked rapid ischemic tolerance. Finally, we showed that directly reducing Bim expression protects neuronal cultures from the effect of harmful ischemia. Taken together, these data suggest that rapid ischemic tolerance involves the degradation of Bim protein levels by the ubiquitin-proteasome pathway and is mediated by p42/ p44 MAPK.
Whilst delayed ischemic tolerance has been the focus of many studies in the brain, the mechanisms involved in rapid ischemic tolerance are less well understood. Both forms of tolerance result in the reduced activation of cell death pathway activation (Fig. 1d)(4,5), however the mechanisms regulating each form of tolerance are quite distinct. Delayed ischemic tolerance is protein synthesis-dependent, which suggests that the upregulation of specific genes following preconditioning mediates the neuroprotective effect (12). For example, preconditioning increases the expression of the pro-survival protein Bcl-2 (8,9,34) and blocking the increase in Bcl-2 expression following preconditioning obviates the tolerance effect (8). In contrast, we show that rapid ischemic tolerance is not inhibited by the protein synthesis inhibitor cycloheximide (Fig 1c). Taken together with the fact that tolerance occurs within 1 hour following preconditioning, suggests that rapid biochemical processes mediate the effect.
Rapid ischemic tolerance in the brain can be mediated by adenosine receptors (18) (17), the opening of KATP channels (13,19,20) and possibly protein kinase C (16). Consistent with these observations, we found in preliminary experiments that rapid ischemic tolerance was blocked by the adenosine receptor antagonist 8-cyclopentyl-1,3,dipropyl xanthine (not shown). These mechanisms have also been indicated in the regulation of ischemic tolerance in the heart (43–45). However, it is unclear how these mechanisms would lead to the reduced activation of cell death pathways following ischemia. Here we propose that one mechanism of rapid ischemic tolerance is the selective degradation of programmed cell death pathway components following preconditioning.
Bim is amongst the most potent of all pro-apoptotic Bcl-2 family of cell death regulatory proteins (24) and plays an important role in neuronal cell death (27,46). We observed a rapid decrease in Bim protein levels following preconditioning ischemia (Fig. 3a). In our experiments we were unable to detect the direct ubiquitination of Bim by immunoprecipitation methods, hence we relied on a pull-down assay. P62, also known as sequestome–1, has been shown to bind to ubiquitinated proteins resulting in their targeting to the proteasome for degradation (40,42). A role for the proteasome in degrading Bim following preconditioning ischemia is suggested given the proteasome inhibitor MG132 blocked Bim degradation (Fig 4c). Finally, reduction of Bim expression using antisense oligonucleotides results in neuroprotection. Taken together these data suggest that the decrease in Bim protein expression following preconditioning ischemia is mediated by the ubiquitin-proteasome system, which contributes to the neuroprotective state in rapid tolerance.
Our results suggest that following preconditioning, p42/ p44 MAPK regulates the degradation of Bim (Fig. 5). This finding is consistent with other reports (31–33). Ley et al recently reported that p42/ p44 MAPK is a BimEL kinase and describe the direct interaction between the two proteins (32). Our observations are also consistent with other studies describing the down-regulation of Bim protein levels by NGF or cytokines, via a p42/ p44 MAPK-dependent mechanism (29,30).
The role of Bim phosphorylation in its activation as well as its degradation is unclear. Bim can be phosphorylated on Ser 109 and Thr 110 by NGF via p42/ p44 MAPK, which also inhibits its pro-apoptotic activity (29). Phosphorylation of Ser 65 by p42/ p44 MAPK has been shown to promote Bim degradation (31,33). However, phosphorylation of Bim on Ser 65 by jun N-terminal protein Kinase (JNK) in superior cervical ganglion cells following NGF withdrawal was reported to mediate the cell death promoting activity of Bim (47). (In our own experiments in cortical cultures, attempts to detect endogenous Bim phosphorylation with a Bim Ser 65 antibody were unsuccessful, Meller-unpublished data). Additionally, the role of JNK could not be further investigated because the JNK inhibitor SP600125 (10 μM) (48) inhibited 120 min-induced cell death, hence precluding further study (data not shown). The role of p42/ p44 MAPK in mediating Bim degradation is not in dispute, but rather the consequence of JNK-mediated Bim phosphorylation is unclear within this context. Clearly, further investigation is warranted to determine the role of the different protein kinases in regulating Bim activation and degradation.
While our data strongly support the role of ubiquitination in mediating the degradation of Bim, another potential mechanism could involve decreased Bim transcription and protein translation. Bim expression is regulated by the forkhead transcription factor (38,49,50), which is inversely controlled by its phosphorylation by protein kinases, such as Akt. However, this is an unlikely mechanism, because the rapid time course of the decrease in Bim that we observed is quicker than translational blockade-induced decrease in Bim protein levels (Fig. 2). We determined that the half-life of Bim to be nearly 3 hours, hence the inhibition of Bim expression would take longer than 1 hour to be observed, unless protein stability were affected e.g. increased degradation. Since MG132 blocked the degradation of Bim, our data suggests that the decrease in Bim levels following preconditioning is due to increased proteasomal degradation of Bim, rather than decreased Bim protein expression.
Whilst a number of groups have reported that Bim is degraded by the ubiquitin-proteasome pathway (31,33), it is not clear which protein is the E3 ligase necessary for ubiquitination. Interestingly, in a study by Breitshopf (51) it was shown that the degradation of Bid was not related to its binding to other Bcl-2 family members. This suggests that another Bid interacting protein, and not a Bcl-2 family protein may regulate Bid degradation. It is not yet known which protein or proteins form the E3 ligase for Bim, and whether it is regulated by its interactions with other Bcl-2 family members. Clearly, identification of the Bim E3 ligase could provide novel therapeutic strategies to reduce neurological damage. Of particular translational interest, a strategy that leads to the degradation of Bim would be expected to have relatively rapid therapeutic effects.
In summary, these data show how the ubiquitination and proteasomal degradation of Bim may in part, mediate the endogenous neuroprotective mechanisms of rapid ischemic tolerance in neurons. The rapid time course of Bim degradation and the neuroprotective effect of reducing Bim expression suggest that a novel class of fast acting therapeutics targeted to Bim, may reduce cell damage following neurological insults.
Acknowledgments
This work was supported by the American Heart Association grant 0465430Z (RM), National Institutes of Health grants NS050669 (RM), NS24728 (R.P.S.) and by the Medical Research Foundation of Oregon (RM).
References
- 1.Dirnagl U, Simon RP, Hallenbeck JM. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 2.Ishida T, Yarimizu K, Gute DC, Korthuis RJ. Shock. 1997;8:86–94. doi: 10.1097/00024382-199708000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 4.Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Perez-Pinzon MA. J Cereb Blood Flow Metab. 2001;21:1401–1410. doi: 10.1097/00004647-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Qi S, Zhan RZ, Wu C, Fujihara H, Yamakura T, Baba H, Taga K, Shimoji K. Neurosci Lett. 2001;315:133–136. doi: 10.1016/s0304-3940(01)02368-0. [DOI] [PubMed] [Google Scholar]
- 6.Kirino T. J Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Graham SH, Zhu RL, Simon RP. J Cereb Blood Flow Metab. 1996;16:566–577. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu S, Nagayama T, Jin KL, Zhu L, Loeffert JE, Watkins SC, Graham SH, Simon RP. J Cereb Blood Flow Metab. 2001;21:233–243. doi: 10.1097/00004647-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Bossenmeyer-Pourie C, Daval JL. Brain Res Mol Brain Res. 1998;58:237–239. doi: 10.1016/s0169-328x(98)00123-5. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Proc Natl Acad Sci U S A. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tauskela JS, chakravarthy BR, Murray CL, Wang Y, Comas T, Hogan M, Hakim A, Morley P. Brain Res. 1999;827:143–151. doi: 10.1016/s0006-8993(99)01322-0. [DOI] [PubMed] [Google Scholar]
- 12.Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Pinzon MA, Born JG. Neuroscience. 1999;89:453–459. doi: 10.1016/s0306-4522(98)00560-0. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. J Cereb Blood Flow Metab. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Reshef A, Sperling O, Zoref-Shani E. Brain Res. 1996;741:252–257. doi: 10.1016/s0006-8993(96)00939-0. [DOI] [PubMed] [Google Scholar]
- 16.Reshef A, Sperling O, Zoref-Shani E. Neurosci Lett. 1997;238:37–40. doi: 10.1016/s0304-3940(97)00841-0. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura M, Nakakimura K, Matsumoto M, Sakabe T. J Cereb Blood Flow Metab. 2002;22:161–170. doi: 10.1097/00004647-200202000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Pinzon MA, Mumford PL, Rosenthal M, Sick TJ. Neuroscience. 1996;75:687–694. doi: 10.1016/0306-4522(96)00311-9. [DOI] [PubMed] [Google Scholar]
- 19.Reshef A, Sperling O, Zoref-Shani E. Neurosci Lett. 1998;250:111–114. doi: 10.1016/s0304-3940(98)00458-3. [DOI] [PubMed] [Google Scholar]
- 20.Reshef A, Sperling O, Zoref-Shani E. Adv Exp Med Biol. 2000;486:217–221. [PubMed] [Google Scholar]
- 21.Kroemer G. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 22.Adams JM, Cory S. Trends Biochem Sci. 2001;26:61–66. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- 23.Adams JM, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 26.Miyashita T, Shikama Y, Tadokoro K, Yamada M. FEBS Lett. 2001;509:135–141. doi: 10.1016/s0014-5793(01)03145-3. [DOI] [PubMed] [Google Scholar]
- 27.Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 28.Strasser A, Puthalakath H, Bouillet P, Huang DC, O’Connor L, O’Reilly LA, Cullen L, Cory S, Adams JM. Ann N Y Acad Sci. 2000;917:541–548. doi: 10.1111/j.1749-6632.2000.tb05419.x. [DOI] [PubMed] [Google Scholar]
- 29.Biswas SC, Greene LA. J Biol Chem. 2002;17:17. doi: 10.1074/jbc.M208086200. [DOI] [PubMed] [Google Scholar]
- 30.Shinjyo T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A, Houghton PJ, Look AT, Ozawa K, Inaba T. Mol Cell Biol. 2001;21:854–864. doi: 10.1128/MCB.21.3.854-864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 32.Ley, R., Ewings, K. E., Hadfield, K., Howes, E., Balmanno, K., and Cook, S. J. (2003) J Biol Chem [DOI] [PubMed]
- 33.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 34.Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, Henshall DC, Simon RP. J Cereb Blood Flow Metab. 2005;25:234–246. doi: 10.1038/sj.jcbfm.9600024. [DOI] [PubMed] [Google Scholar]
- 35.Meller R, Stevens SL, Minami M, Cameron JA, King S, Rosenzweig H, Doyle K, Lessov NS, Simon RP, Stenzel-Poore MP. J Cereb Blood Flow Metab. 2005;25:217–225. doi: 10.1038/sj.jcbfm.9600022. [DOI] [PubMed] [Google Scholar]
- 36.Meller R, Skradski SL, Simon RP, Henshall DC. Neurosci Lett. 2002;324:33–36. doi: 10.1016/s0304-3940(02)00166-0. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S. Cell Death Differ. 1999;6:1060–1066. doi: 10.1038/sj.cdd.4400600. [DOI] [PubMed] [Google Scholar]
- 38.Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. J Clin Invest. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, Inaba T, Sanjay A, Baron R, Kawaguchi H, Oda H, Nakamura K, Strasser A, Tanaka S. Embo J. 2003;22:6653–6664. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babu JR, Geetha T, Wooten MW. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 41.Vadlamudi RK, Joung I, Strominger JL, Shin J. J Biol Chem. 1996;271:20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- 42.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miura T, Miki T, Tsuchihashi K, Iimura O. Exs. 1996;76:365–382. doi: 10.1007/978-3-0348-8988-9_22. [DOI] [PubMed] [Google Scholar]
- 44.Ogawa T, Miura T, Shimamoto K, Iimura O. J Am Coll Cardiol. 1996;27:225–233. doi: 10.1016/0735-1097(95)00412-2. [DOI] [PubMed] [Google Scholar]
- 45.Baker JE, Contney SJ, Gross GJ, Bosnjak ZJ. J Mol Cell Cardiol. 1997;29:845–848. doi: 10.1006/jmcc.1996.0361. [DOI] [PubMed] [Google Scholar]
- 46.Harris CA, Johnson EM., Jr J Biol Chem. 2001;276:37754–37760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- 47.Putcha G, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 48.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 50.Gilley J, Coffer PJ, Ham J. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Breitschopf K, Zeiher AM, Dimmeler S. J Biol Chem. 2000;275:21648–21652. doi: 10.1074/jbc.M001083200. [DOI] [PubMed] [Google Scholar]