Accelerated Rejection of Renal Allografts From Brain-Dead Donors (original) (raw)
Abstract
Objective
To define the potential influences of donor brain death on organs used for transplantation.
Summary Background Data
Donor brain death causes prompt upregulation of inflammatory mediators on peripheral organs. It is hypothesized that this antigen-independent insult may influence the rate and intensity of host alloresponsiveness after engraftment.
Methods
The rates of survival of unmodified Lew recipients sustained by kidney allografts from brain-dead, normal anesthetized, and anesthetized ventilated F344 donors were compared. Brain death was induced by gradually increasing intracranial pressure under electroencephalographic control. Tracheotomized brain-dead animals and anesthetized controls were mechanically ventilated for 6 hours before transplant nephrectomy. The rate and intensity of the acute rejection event were examined by histology, immunohistology, and reverse transcriptase–polymerase chain reaction.
Results
Animals bearing kidneys from brain-dead donors died of renal failure secondary to acute rejection at a significantly faster rate than those from anesthetized living controls or anesthetized animals ventilated for 6 hours. Within 3 hours after placement and reperfusion of brain-dead donor grafts, significant neutrophil infiltration was observed, followed by increasing numbers of macrophages and T cells. mRNA of proinflammatory mediators detected in kidneys within 6 hours of brain death and upregulated even before transplantation increased thereafter and appeared to accelerate and amplify host alloresponsiveness, as manifested by the rapid expression of chemokines, cytokines, adhesion molecules, and major histocompatibility complex class II antigens in the engrafted organ. The process evolved in the controls less intensely and at a slower rate.
Conclusions
Donor brain death is a significant risk factor for peripheral organs used for transplantation. The activated state of such organs appears to trigger host immune mechanisms that accelerate the process of acute rejection. The effects of this central injury may explain in part the less satisfactory performance of cadaver organs in human transplantation compared with those from living sources.
Transplantation has evolved as the treatment of choice for many patients with end-stage organ disease. Success has improved progressively, particularly in the short term. However, despite a functional survival rate of most grafts of more than 80% at 1 year, the ultimate goal of providing long-term treatment for an irreversible process has not been achieved; the rate of attrition over time has remained relatively stable throughout the entire experience. 1 Whereas early immune-mediated injury is considered to be primarily responsible for graft dysfunction and failure, the influence of nonimmunologic, antigen-independent events may have been underestimated; there appears to be a continuum between inflammatory changes secondary to an initial nonspecific insult and the onset of alloresponsiveness. This concept has been emphasized by recent pooled data from the United Network of Organ Sharing that show not only that survival rates of kidneys from living-unrelated and one haplotype matched living-related donors are similar, but also that organs from all living donors, regardless of differences in genetic relationship with a given recipient, demonstrate consistently superior results to those from cadavers. 2 Various risk factors that might explain this striking discrepancy include donor age and preexisting conditions such as hypertension or diabetes, the effects of initial ischemia/reperfusion injury, inadequate functioning nephron mass, and viral infections. An additional obvious difference between living and cadaver donor sources is the influence of brain death.
Brain death is a profound central catastrophe uniquely relevant to the cadaver donor, the primary source of solid organs for transplantation. Such persons have suffered sudden, extensive, and irreversible central nervous system damage secondary to trauma, hemorrhage, or infarction. Both the initial and long-term function of engrafted cadaver organs may also depend on the cause of the injury as well as on donor demographics; results are worst when the organ is taken from an older donor who has died of an intracerebral hemorrhage or thrombotic stroke and best from a younger person who has died of acute head injury. 3 Besides age and other variables, the resultant physiologic and structural derangements that may occur during and after brain death and before organ removal and the actual engraftment procedure itself may be different between the two donor populations. In this regard, the use of young healthy living animals as donors in virtually all experimental studies of organ transplantation may not provide an accurate prototype for the study of clinical events involving cadavers.
Brain death has been shown in several experimental models to perturb significantly the function and structure of somatic organs. 4,5 The objectives of the present study are to assess how a controlled and reproducible model of gradual-onset brain death alters renal graft survival after transplantation by influencing the process of acute host alloresponsiveness. The increased immunogenicity of kidneys from brain-dead donors secondary to rapid upregulation of histocompatibility antigens and proinflammatory mediators may explain in part the consistently inferior clinical results of organs from cadaver donors compared with those from living sources.
METHODS
Brain Death Model
All animals in the study were handled in accordance with the Guide for the Care of Laboratory Animals published by the National Institutes of Health and the guidelines of the Harvard Medical Area Standing Committee on Animals. Inbred adult (200–250 g) male Fisher (F344, RT1ivi) rats were used as kidney donors for Lew (RT11) recipients (Harlan Sprague-Dawley, Indianapolis, IN). Donor brain death was induced by slow inflation (200 ± 25 μL saline) of a Fogarty arterial embolectomy catheter (3F; Baxter Healthcare Co., Irvine, CA) placed intracranially through an occipital bur hole. Herniation of the brain stem was confirmed by a flat line on electroencephalography (EEG & Polygraph Data Recording System, model 79D, Grass Medical Instruments, Quincy, MA), apnea, absent reflexes, and maximally dilated and fixed pupils. The rats were tracheotomized and intubated with a #13 blunt-tipped cannula (Luer Stub Adapter, Clay-Adams, Inc., New York, NY) before induction of the central injury, then mechanically ventilated for 6 hours (respiration rate 105/min, tidal volume 2.0 mL; Rodent ventilator model 683, Harvard Instruments, South Natick, MA) before kidney removal for transplantation. Intraarterial blood pressure was monitored continuously with a PE 50 catheter placed into the left femoral artery and attached to a transducer and recorder (Byk Gould, Konstanz, Germany). To avoid peripheral effects of hypotension-associated ischemia, rats with a mean arterial blood pressure of less than 80 mmHg were excluded from the study. Arterial blood gas analyses were performed hourly. Urine production during the 6-hour ventilation period was determined using a a PE 50 catheter placed into the urinary bladder through a small laparotomy incision.
Surgical Techniques
Renal allografts from F344 donors were grafted into unmodified Lew recipients using standard microsurgical techniques. All animals were anesthetized with pentobarbital 30 mg/kg (Nembutal Sodium solution, Abbott Laboratories, Chicago, IL). The nonperfused kidneys were transplanted orthotopically to recipient renal vessels and ureter by end-to-end anastomoses using 10–0 prolene after the left kidney had been mobilized and removed. Right nephrectomy was performed 6 days later. The time between release of the vascular clamps and return of obvious uniform cortical blood flow was defined as reperfusion time after transplantation.
Animal Groups
In the experimental group, kidneys were placed into unmodified hosts 6 hours after induction of donor brain death. As controls, anesthetized living F344 donors (LD) and anesthetized tracheotomized and ventilated (6 hours) living donors (VLD) were used. The time of death of the graft recipients from renal failure secondary to complete and irreversible rejection of their allografts was compared between the brain-dead (n = 12), LD (n = 13), and VLD (n = 9) groups. Late postoperative complications such as hydronephrosis secondary to ureteric obstruction were ruled out by autopsy; animals with such conditions were discarded. Organs were harvested and analyzed serially for histology, reverse transcriptase–polymerase chain reaction (RT-PCR), and immunohistology immediately before transplantation (0 hours) or were removed from representative recipients 3, 6, and 24 hours after placement and 3 and 7 days later (n = 4 per time interval per group). Further histologic assessment was performed at weekly intervals on harvested grafts (n = 3 per group) until they failed. Because the results of LD and VLD controls did not differ appreciably, they were combined except where noted.
Histology and Immunohistology
Kidneys were fixed in 10% buffered formalin. Paraffin sections were stained with hematoxylin and eosin and assessed by light microscopy. For immunohistology and RT-PCR, cortical segments were immediately snap-frozen in liquid nitrogen. Monoclonal antibodies (obtained from Harlan Bioproducts for Science, Indianapolis, IN, except as noted) were directed against all rat leukocytes (CD 45, OX-1), rat T cells (TCR-αβ, R73), natural killer (NK) cells (CD 161, 10/78), B cells (RLN-9D3), mononuclear phagocytes (CD 68, ED1), and neutrophils (RP3; donated by F. Sendo, Yamagata, Japan). Additional antibodies were directed against E-selectin (CD 62 E, BBA-1; Bristol-Myers Squibb, Seattle, WA), major histocompatibility complex (MHC) class II antigens (OX-3), and the cytokines interleukin (IL)-2 (1D10), IL-4 (OX-81), IL-10 (R&D, Minneapolis, MN), interferon γ (DB-1), IL-8, monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, and RANTES (LeukoSite, Inc., Cambridge, MA). Control monoclonal antibodies and secondary antibodies were purchased from Dako (Carpinteria, CA).
Cryostat sections were fixed in paraformaldehyde-lysine-periodate for staining of activation antigens or fixed in acetone for localization of cytokines, and stained by a peroxidase–antiperoxidase method, as previously described. 6 Briefly, sections were incubated overnight with primary antibodies at 4°C, followed by incubation at room temperature with bridging antibodies, peroxidase–antiperoxidase complexes, and diaminobenzidine. Isotype-matched monoclonal antibodies and controls for residual endogenous peroxidase activity were included in each experiment. Numbers of labeled leukocytes in 20 consecutive high-power fields (×40) were determined in three kidneys per group. Expression of cytokines and chemokines in these fields are reported based on semiquantitative assessment. 6
Competitive RT-PCR
Competitive semiquantitative RT-PCR of representative cell products was performed in a standardized fashion. 7–9 Competitive DNA mimics for each factor were constructed using a PCR MIMIC construction kit (Clontech Laboratories, Inc., Palo Alto, CA) for rat MCP-1, tumor necrosis factor-α (TNFα), IL-1β, ICAM-1, and β-actin. The sequences of the primers and annealing temperature were as follows: MCP-1, 5′-ATGCAGGTCTCTGTCACG-3′ and 5′-CTAGTTCTCTGTCATACT-3′, 55°C; TNFα, 5′-TACTGAACTTCGGGGTGATTGGTCC-3′and 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′, 60°C; IL-1β, 5′-TGATGTTCCCATTAGACAGC-3′and 5′-GAGGTGCTGATGTACCAGTT-3′, 55°C; ICAM-1, 5′-AGAAGGACTGCTTGGGGAA-3′and 5′-CCTCTGGCGGTAATAGGTG-3′, 60°C; and β-actin, 5′-TTGTAACCAACTGGGACGATATGG-3′and 5′-GATCTTGATCTTCATGGTGCTAGG-3′, 60°C. Amplification was initiated with incubation at 94°C for 2 minutes followed by amplification cycles as follows: 94°C for 15 seconds, annealing temperature for 30 seconds, 72°C for 1 minute. Densities of competitive mimic and target DNA bands were measured by scanning densitometry using ScanJet 4c (Hewlett Packard, Corvallis, OR) with Adobe PhotoShop software (Adobe Inc., Mountainview, CA). Expression of mRNA for each sample was expressed as a ratio of β-actin used as an internal control. PCR reactions for each factor were repeated twice and were found not to differ appreciably from one another. This method has been deemed as accurate as scintillation counting of radiolabeled products. 10
Statistics
Statistical significance relating to differences in the numbers of infiltrating cells and expression of their products was ascertained using the Student t test and the Mann-Whitney test. The results are expressed as mean ± standard deviation and were considered significant at P < .05. Survival data were calculated both using mean ± SD and median ± standard error. Graft survival was expressed graphically using the Kaplan-Meier survival curve. Statistical differences in survival between the groups were assessed by the log-rank sum test.
RESULTS
Physiologic Changes After Brain Death
During inflation of the Fogarty balloon and the gradual onset of brain death, the animals reacted with sharply increased arterial blood pressure lasting for 15 to 30 minutes (mean arterial pressure 206 ± 38 mmHg at 10 minutes vs. 102 ± 15 before injury; n = 40, P < .0001). After this period of autonomic storm, the rats maintained stable blood pressure (average mean arterial pressure 80–100 mmHg) until the kidney was removed for transplantation after the 6-hour ventilation period. VLD control animals were constantly normotensive (average mean arterial pressure 80–100 mmHg; n = 20). Electroencephalography showed flat-line tracings in brain-dead animals compared with continued physiologic activity in the controls. The mean urine production of allografts from control animals during this preoperative period was 0.9 ± 0.3 mL per 6 hours (n = 20); brain-dead rats showed some diuresis (1.2 ± 0.3 mL per 6 hours; n = 20, P < .005). Blood gas levels remained in the normal range in all animals during the 6 hours of ventilation.
Ischemia/Reperfusion Time
The mean cold ischemic time necessary for the transplant procedure did not differ appreciably between the donor groups (21 ± 3 minutes for controls, 23 ± 3 minutes for brain-dead animals). However, the period of reperfusion between the release of the vascular clamps and complete cortical perfusion (as assessed by a uniform pink color and appropriate turgor of the previously pale and flaccid organ) revealed significant differences between the grafts from brain-dead rats and living controls (18 ± 5 vs. 4 ± 2 minutes, respectively;P < .0001) despite sustained normotension of the donors before engraftment.
Allograft Survival
Recipients of kidney allografts from brain-dead donors had an accelerated rate of acute rejection and died significantly earlier of renal failure than controls (brain-dead donors: 20 ± 15 days; LD, 40 ± 25 days; VLD, 38 ± 18 days;P < .01;Fig. 1). Median survival for the groups was brain-dead donors, 16 ± 3 days; LD, 30 ± 6 days; and VLD, 31 ± 3 days (P = .01).

Figure 1. Survival of unmodified recipients of renal allografts from different donor groups (Kaplan-Meier survival curve). Survival of recipients of grafts from brain-dead donors was significantly shorter than survival of grafts from living donor or ventilated living donor sources (P < .01).
Histology
Although characteristic morphologic changes associated with acute irreversible immunologic rejection were observed in the transplanted allografts of all groups, the rate and intensity of the process were strikingly different between grafts from brain-dead and control donors. Within the first 24 hours, there was margination and pooling of neutrophils in the microvasculature and glomeruli of kidneys from brain-dead animals. By 3 days, relatively large numbers of mononuclear cells were scattered throughout the graft substance and had formed clumps around vessels and tubules. Minimal cellular activity was noted in control kidneys during this early interval. Comparable histologic changes occurring between 10 and 15 days in the experimental group and between 20 and 60 days in the controls included increasing interstitial and endothelial inflammation, and loss of tubules. Finally, massive cellular infiltration, interstitial edema, and necrosis become obvious at the end of the rejection process. The LD and VLD control groups showed no differences in the rate of progression of the acute phenomena.
Immunopathology
No immunohistologic changes were detected in kidneys after 6 hours of ventilation and before transplantation (0 hours), regardless of donor source. Within 3 hours after engraftment and reperfusion of the organs from brain-dead donors, however, neutrophils began to infiltrate the renal tissue. Their peak at 12 hours was followed within 12 to 24 hours by progressive numbers of macrophages and T cells entering the grafts. These latter populations also increased after 24 hours in control kidneys but to a lesser extent than cells in brain-dead donor allografts (Figs. 2 and 3). Numbers of B and NK cells did not vary significantly between the groups and contributed less than 5% of graft cellularity at any point (data not shown).

Figure 2. Infiltration of leukocyte populations into renal allografts from brain-dead and control donors. Cell counts were based on examination of 20 fields per marker and three animals per group and were expressed as cells (mean ± standard deviation) per high-power field (×40).
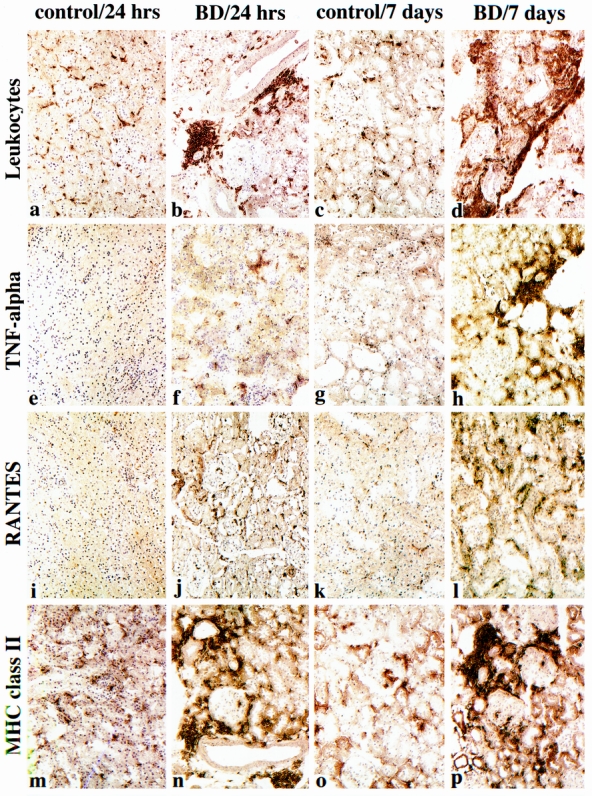
Figure 3. Effects of brain death on intragraft events at 24 hours and 7 days after transplant. Compared with the relatively few CD45+ leukocytes present at 24 hours in control allografts (A), their accumulation in perivascular areas is markedly increased by brain death (B). This enhancement of leukocyte recruitment was further seen at 7 days; control grafts (C) showed only minor increases in cellularity, whereas grafts from brain-dead donors showed large perivascular and interstitial cell aggregates (D). Intragraft expression of tumor necrosis factor-α, evident in conjunction with interstitial macrophages, was increased in kidneys from brain-dead donors but not control grafts at 24 hours (E, F) and further increased at 7 days (G, H). The chemokine RANTES was expressed by 24 hours in grafts from brain-dead donors but not control animals (I, J), increasing thereafter (K, L). MHC class II induction was markedly upregulated at 24 hours in brain-dead donors and by 7 days showed dense expression on leukocytes and tubular cells (N, P). There was little activity in control organs (M, O). (Serial cryostat sections, representative of four animals per group per time point; hematoxylin counterstain, ×200).
Cell surface molecules and inflammatory mediators became apparent within a few hours after transplantation of brain-dead donor kidneys in contrast to control allografts. By 6 hours, there was obvious induction of E-selectin and ICAM-1 and expression of multiple cytokines by mononuclear cells, including TNFα and interferon-γ (Table 1). There was also focal labeling of infiltrating leukocytes and graft endothelial cells for the chemokines and macrophage chemoattractants, MCP-1, MIP-1, and RANTES, as well as the neutrophil chemoattractant IL-8. Further expression of these cell products as well as upregulation of MHC class II on leukocytes and endothelial cells was observed at 24 hours (Fig. 3). By days 3 and 7, the inflammatory response involving cytokine, chemokine, and MHC class II expression was accelerated in the kidneys from brain-dead donors compared with the less advanced changes in the control donor organs (Table 1, Fig. 3).
Table 1. CYTOKINE AND ADHESION MOLECULE EXPRESSION IN KIDNEY GRAFTS

Competitive RT-PCR
mRNA expression in kidneys of brain-dead and control rats was assessed by RT-PCR 6 hours after brain death and before transplantation (0 hours), a period examined specifically because no immunohistologic changes were identified at that time. mRNA levels for the representative inflammatory cytokines TNF-α and IL-1β and ICAM-1, an early target of infiltrating cells, were significantly (P < .05) upregulated in kidneys from brain-dead donors compared with controls, remaining elevated for 12 to 24 hours thereafter before returning to baseline (n = 5 per group;Fig. 4). Expression of MCP-1 was increased in the brain-dead donor group at 1 hour (P < .05). Little mRNA expression was noted in controls.

Figure 4. mRNA expression of representative cell products in kidney tissue from brain-dead and control donors before transplantation (0 hours) and up to 24 hours thereafter.
DISCUSSION
Understanding of the systemic influences of massive central injury on cellular and molecular changes in peripheral tissue remains limited despite several experimental and clinical studies that have elucidated the resultant complex of neurohumoral, functional, hemodynamic, and morphologic alterations in some peripheral organs. 11–14 The practical question to be asked in the context of transplantation is whether the attendant sequelae of brain death affect the quality of the donor organ and influence host alloresponsiveness toward it, both early after placement as well as in the long term. To examine the first question, we investigated the initial cellular and molecular events developing after the engraftment of kidneys from brain-dead and control donors into unmodified recipients. The weak F344→Lew rat strain combination was chosen because the inevitable episode of acute rejection by unmodified hosts does not occur for several weeks, a prolonged interval that would emphasize any donor-associated differences in onset, rate, and intensity of the event. If a strong strain combination had been used in which the acute rejection process is generally complete by 7 to 10 days in the absence of immunosuppression, any variation secondary to donor-associated circumstances would be difficult to determine accurately. Because the purpose of the study was to assess the effects of brain death on acute rejection, no immunosuppression was administered to the recipients. However, in related experiments, transplanted animals received a course of cyclosporine, which prolongs survival between 6 and 12 months before the recipients die of renal failure secondary to chronic rejection. Despite such treatment, kidneys from brain-dead donors underwent an accelerated course of chronic rejection compared with controls, confirming that such antigen-independent effects were not influenced by immunosuppression.
The syndrome of brain death includes immediate, rapid, and chaotic swings in blood pressure, then an early hypertensive phase followed by later normotension or hypotension. This period of autonomic storm, clearly reproduced in our model, has been thought to result from intense sympathetic stimulation either from direct neural activity or from excessive levels of endogenous catecholamines, with associated elevation of vascular resistance and altered perfusion of peripheral tissue. 14–16 The initial vasoconstriction is presumably so severe that flow to organs may be significantly reduced despite markedly increased systemic perfusion pressure. The subsequent normotensive or hypotensive phase is accompanied by a profound reduction in sympathetic outflow resulting in impaired vascular autoregulation with diminished blood supply and oxygen delivery to organs and tissues. 17 These sequential circulatory changes can produce severe ischemic injury to donor organs before their removal, with potentially damaging effects to the transplanted graft. As a result, the incidence of acute tubular necrosis with delayed graft function is often increased in engrafted kidneys harvested from unstable brain-dead donors compared with those from living sources. 18,19 Although the dramatically elevated levels of catecholamines released during explosive brain death secondary to suddenly increased intracerebral pressure appear particularly responsible for hemodynamic and morphologic changes of somatic organs, these may be considerably attenuated after gradual-onset brain death. 20,21 Although the animals in this study remained relatively normotensive after the latter injury, the effects of peripheral ischemia on a microvascular level were apparently still a significant and integral part of the brain death syndrome, as emphasized by the decreased urine output before transplantation and the prolonged interval necessary for complete renal cortical reperfusion after release of the vascular clamps. Presumably, this delay substantially influences the subsequent cellular and molecular changes in the engrafted organ. Unlike larger species, including humans, the 6-hour period between donor brain death and organ removal for transplantation was the maximum that the rat could tolerate; beyond that interval, the rats’ hearts experienced worsening arrhythmias followed by inevitable arrest. Although many organs are removed from human donors 12 to 24 hours after brain death, the model used in these studies precluded a longer delay before engraftment.
Recent experiments have demonstrated the relation between explosive brain death and the rapid infiltration of leukocyte populations in peripheral organs with intense upregulation of their associated products. 20 Even with a gradual-onset central injury, prompt expression of mRNA of cytokines, chemokines, and adhesion molecules developing within 6 hours of the central catastrophe and before transplantation previews that of the corresponding proteins identified by immunohistology thereafter. The cellular and molecular changes presumably occur secondary to the initial activity of catecholamines as well as circulatory cytokines originating from the injured brain, and trigger a significantly faster and more intense rejection process than in controls. Circulating factors have also been identified after brain injury and may contribute to changes in the peripheral organs. 21 In patients, for instance, upregulation of transcriptional levels of TNF-α and IL-6 in serum have been associated with episodes of transient focal cerebral ischemia. 22,23 The events identified in these experiments emphasize the influence of antigen-independent donor-associated influences in the graft on the later development of host alloimmune activity.
In support of these findings, other investigators have noted that during and after brain death, circulating leukocyte traffic through peripheral organs slows, and cells adhere to vascular endothelium and infiltrate into the tissue. 24 These observations were emphasized in the present experiments by the early presence of the adhesion molecules E-selectin and ICAM-1. Similarly, the proinflammatory lymphocyte-associated cytokines TNFα and interferon-γ are upregulated in association with MHC class II expression. Histocompatibility antigens in peripheral tissue have also been shown to increase secondary to other nonspecific endothelial injuries, particularly ischemia/reperfusion. 25 Their presence presumably influences graft immunogenicity by means of the T-cell recognition process, which acts as an important trigger for host alloresponsiveness. The hypothesis that such cell surface changes evolving promptly in peripheral organs after massive central injury may be a result, at least in part, of excessive catecholamine release is supported by observations that short-term administration of these substances to living donor pigs is followed by poor initial function and reduced survival of kidney allografts. 26 In addition, various hormonal substitution therapies to brain-dead donors before transplantation have shown beneficial effects. 27
The intense local vasoconstriction leading to ischemia of the kidney is an important feature of brain death despite continuing donor normotension before organ removal. The early dynamics secondary to ischemia/reperfusion that are potentially involved in later graft changes have become increasingly well defined and include a striking upregulation of the adhesion molecule–cytokine cascade, infiltration of leukocyte populations, and production of their inflammatory products. 28 The effects of this insult on transplanted human kidneys, as shown by pre- and postreperfusion biopsies, have emphasized the experimental findings. 29 In human organs, P-selectin expression and rapid infiltration of cadaver donor organs by neutrophils are important changes that occur quickly after injury; as shown experimentally, these cells then trigger subsequent inflammatory changes that not only may increase the incidence of delayed graft function but also make the organ more immunogenic and prone to subsequent host immunologic attack. 25,28 Later graft dysfunction may result.
Considering that the only difference between the experimental and control groups in these studies was donor brain death, our findings strongly support clinical data that the incidence and severity of initial acute rejection episodes are greater among cadaver organs than among those from living sources. 2 Such data also reinforce the patient-based observation that insults sustained by the organ at the time of transplantation or even before its procurement from the donor may be risk factors for both early and late allograft failure. This assumption would also explain the apparent correlation noted clinically between the synergistic effects of initial delayed graft function secondary to initial nonspecific injury and acute immunologic rejection, insults that are considerably worse in combination than when the kidney experiences only one alone. 30 Further definition of these early alterations suggests that donor-related therapeutic approaches to normalize peripheral organs should be developed and initiated before the transplantation procedure occurs.
Footnotes
Correspondence: Nicholas L. Tilney, MD, Brigham and Women’s Hospital, 75 Francis St., Boston, MA 02115.
Supported by USPHS grant 5 RO1 DK 46190–25.
Dr. Pratschke and Dr. Wilhelm are recipients of Research Fellowship awards (Pr 578/1–1 and Wi 1677/1–1) from the Deutsche Forschungsgemeinschaft, Germany.
E-mail: bhayslett@rics.bwh.harvard.edu
Accepted for publication January 27, 2000.
References
- 1.Cecka JM, Terasaki PI. Clinical transplants. UCLA Tissue Typing Laboratory 1994; 1–18. [Google Scholar]
- 2.Terasaki PI, Cecka JM, Gjertson DW, Takemoto S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med 1995; 333: 333–336. [DOI] [PubMed] [Google Scholar]
- 3.Busson M, Benoit G, N’Doye P, Hors J. Analysis of cadaver donor criteria on the kidney transplant survival rate in 5,129 transplantations. J Urol 1995; 154: 356–360. [DOI] [PubMed] [Google Scholar]
- 4.Novitzky D, Cooper DKC, Morell D, Isaacs S. Change from aerobic to anaerobic metabolism after brain death, and reversal following triiodothyronine therapy. Transplantation 1988; 45: 32–36. [DOI] [PubMed] [Google Scholar]
- 5.Bruinsma GJ, Nederhoff MG, Geertman HJ, et al. Acute increase of myocardial workload, hemodynamic instability, and myocardial histological changes induced by brain death in the cat. J Surg Res 1997; 68: 7–15. [DOI] [PubMed] [Google Scholar]
- 6.Hancock WW, Whitley WD, Tullius SG, et al. Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts. Transplantation 1993; 56: 643–650. [DOI] [PubMed] [Google Scholar]
- 7.Morishita R, Higaki J, Nagano M, et al. Consistent activation of prorenin mRNA in renal hypertensive rats. Can J Physiol Pharmacol 1991; 69: 1364–1366. [DOI] [PubMed] [Google Scholar]
- 8.Kita Y, Takashi T, Iigo Y, et al. Sequence and expression of rat ICAM-1. Biochem Biophys Acta 1992; 1131: 108–110. [DOI] [PubMed] [Google Scholar]
- 9.Scotte M, Hiron M, Masson S, et al. Different expression of cytokine genes in monocytes, peritoneal macrophages and liver following endotoxin- or turpentine-induced inflammation in rat. Cytokine 1996; 8: 115–120. [DOI] [PubMed] [Google Scholar]
- 10.Chehadeh HE, Zerlauth G, Mannhalter JW. Video image analysis of quantitative competitive PCR products: comparison of different evaluation methods. Biotechniques 1995; 18: 26–28. [PubMed] [Google Scholar]
- 11.Powner DJ, Hendrich A, Nyhuis A, Strate R. Changes in serum catecholamine levels in patients who are brain dead. J Heart Lung Transplant 1992; 11: 1046–1053. [PubMed] [Google Scholar]
- 12.Gramm HJ, Meinhold H, Bickel U, et al. Acute endocrine failure after brain death? Transplantation 1992; 54: 851–857. [DOI] [PubMed] [Google Scholar]
- 13.Herijgers P, Leunens V, Tjandra-Maga TB, et al. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation 1996; 62: 330–335. [DOI] [PubMed] [Google Scholar]
- 14.Mertes PM. Physiology of brain death. In Tilney NL, Strom TB, Paul LC, eds. Transplantation Biology: Cellular and Molecular Aspects. Philadelphia: Lippincott; 1996: 275–290.
- 15.Wicomb WN, Cooper DK, Lanza RP, et al. The effects of brain death and 24 hours’ storage by hypothermic perfusion on donor heart function in the pig. J Thor Cardiovasc Surg 1986; 91: 896–909. [PubMed] [Google Scholar]
- 16.Wicomb WN, Novitzky D, Cooper DK, Rose AG. Forty-eight hours hypothermic perfusion storage of pig and baboon hearts. J Surg Res 1986; 40: 276–284. [DOI] [PubMed] [Google Scholar]
- 17.Bittner HB, Kendall SW, Campell KA, et al. A valid experimental brain death organ donor model. J Heart Lung Transplant 1995; 14: 308–317. [PubMed] [Google Scholar]
- 18.Lagiewska B, Pacholczyk M, Szostek M, et al. Hemodynamic and metabolic disturbances observed in brain-dead organ donors. Transplant Proc 1996; 28: 165–166. [PubMed] [Google Scholar]
- 19.Nagareda T, Kinoshita Y, Tanaka A, et al. Clinicopathology of kidneys from brain-dead patients treated with vasopressin and epinephrine. Kidney Int 1993; 43: 1363–1370. [DOI] [PubMed] [Google Scholar]
- 20.Takada M, Nadeau KC, Hancock WW, et al. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation 1998; 65: 1533–1542. [DOI] [PubMed] [Google Scholar]
- 21.Shivalkar B, Van Loon J, Wieland W, et al. Variable effects of explosive or gradual increase of intracranial pressure on myocardial structure and function. Circulation 1993; 87: 230–239. [DOI] [PubMed] [Google Scholar]
- 22.Amado JA, Lopez-Espadas F, Vasquez-Barquero A, et al. Blood levels of cytokines in brain-dead patients: relationship with circulating hormones and acute phase reactants. Metabolism 1995; 44: 812–816. [DOI] [PubMed] [Google Scholar]
- 23.Zhai QH, Futrell N, Chen FJ. Gene expression of IL-10 in relationship to TNF-alpha, IL-1-beta and IL-2 in the rat brain following middle cerebral artery occlusion. J Neurol Sci 1997; 25: 119–124. [DOI] [PubMed] [Google Scholar]
- 24.Okamoto S, Corso CN, Nolte D, et al. Impact of brain death on hormonal homeostasis and hepatic microcirculation of transplant organ donors. Transplant Int 1998; 11: S404–407. [DOI] [PubMed] [Google Scholar]
- 25.Shoskes DA, Parfrey NA, Halloran PF. Increased major histocompatibility complex antigen expression in unilateral ischemic acute tubular necrosis in the mouse. Transplantation 1990; 49: 201–207. [DOI] [PubMed] [Google Scholar]
- 26.Pienaar H, Schwartz I, Roncone A, et al. Function of kidney grafts from brain-dead donor pigs. Transplantation 1990; 50: 580–582. [DOI] [PubMed] [Google Scholar]
- 27.Novitzky D, Cooper DKC, Reichart B. Hemodynamic and metabolic response to hormonal therapy in brain-dead potential organ donors. Transplantation 1987; 43: 852–885. [PubMed] [Google Scholar]
- 28.Takada M, Nadeau KC, Shaw GD, et al. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. J Clin Invest 1997; 11: 2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koo DD, Welsh KI, Roake JL, et al. Ischemia/reperfusion injury in human kidney transplantation: an immunohistochemical analysis of changes after reperfusion. Am J Pathol 1998; 153: 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Troppman C, Gillingham KJ, Benedetti E, et al. Delayed graft function, acute rejection, and outcome after cadaver renal transplantation. Transplant 1995; 61: 1331–1338. [DOI] [PubMed] [Google Scholar]