Identifying gene regulatory elements by genomic microarray mapping of DNaseI hypersensitive sites (original) (raw)
Abstract
The identification of _cis_-regulatory elements is central to understanding gene transcription. Hypersensitivity of _cis_-regulatory elements to digestion with DNaseI remains the gold-standard approach to locating such elements. Traditional methods used to identify DNaseI hypersensitive sites are cumbersome and can only be applied to short stretches of DNA at defined locations. Here we report the development of a novel genomic array-based approach to DNaseI hypersensitive site mapping (ADHM) that permits precise, large-scale identification of such sites from as few as 5 million cells. Using ADHM we identified all previously recognized hematopoietic regulatory elements across 200 kb of the mouse T-cell acute lymphocytic leukemia-1 (Tal1) locus, and, in addition, identified two novel elements within the locus, which show transcriptional regulatory activity. We further validated the ADHM protocol by mapping the DNaseI hypersensitive sites across 250 kb of the human TAL1 locus in CD34+ primary stem/progenitor cells and K562 cells and by mapping the previously known DNaseI hypersensitive sites across 240 kb of the human α-globin locus in K562 cells. ADHM provides a powerful approach to identifying DNaseI hypersensitive sites across large genomic regions.
Deciphering the complexities of vertebrate gene regulation represents one of the major challenges facing biologists in the post-genomic era. Gene expression is determined by _cis_-regulatory elements such as core promoters, enhancers, silencers, and insulators (Kleinjan and van Heyningen 2005; West and Fraser 2005). The scattered location of these critical elements makes them difficult to locate without biological assays, yet identifying the precise location of such functional elements is essential for our understanding of gene regulation in both health and disease.
Within actively transcribed gene loci, specific regions, which are hypersensitive to DNaseI digestion, have been shown to function as _cis_-regulatory elements critical for regulated gene expression (Weintraub and Groudine 1976; Wu 1980; Elgin 1988). Traditional approaches to mapping DNaseI hypersensitive sites typically rely on labor-intensive Southern blotting techniques, with relatively low resolution limited to short stretches of DNA (Cockerill 2000). Although this approach remains the universally accepted method for identifying regulatory elements, the cumbersome nature of the protocol has limited its application to a relatively small number of genes. Several protocols have been developed in recent years with the aim of large-scale mapping of DNaseI hypersensitive sites (Crawford et al. 2004, 2006; Dorschner et al. 2004; Sabo et al. 2004). However, these approaches require either cloning and sequencing (Crawford et al. 2004, 2006; Sabo et al. 2004) or real-time PCR (Dorschner et al. 2004) on a scale not achievable in most laboratories. In principle, the use of genomic arrays for DNaseI hypersensitive site mapping would permit rapid, cost-effective comparative mapping of DNaseI hypersensitive sites of defined genomic regions from multiple cell types, but such an approach has not yet been reported.
Here we describe a new method, array-based DNaseI hypersensitive site mapping (ADHM), that permits precise, large-scale identification of DNaseI hypersensitive sites from small numbers of cells. The Tal1 (formerly known as Scl) locus was used to establish the protocol, which was then further validated by successfully mapping known DNaseI hypersensitive sites across 240 kb of the human α-globin locus. Previous studies have systematically dissected the transcriptional regulation of the mouse Tal1 locus and have identified multiple conserved regulatory elements that direct expression in transgenic mice to subdomains of the Tal1 expression pattern (Göttgens et al. 1997, 2000, 2002b; Sanchez et al. 1999, 2001; Sinclair et al. 1999; Delabesse et al. 2005). Similarly, the regulatory elements within the α-globin locus have been extensively characterized using both biological and bioinformatics approaches (Yagi et al. 1986; Higgs et al. 1990; Vyas et al. 1992; Hughes et al. 2005). Using a genomic tiling path array for the mouse Tal1 locus, we used ADHM to identify correctly the location of multiple functionally characterized hematopoietic regulatory elements in hematopoietic cell lines and, in addition, identify two novel functional elements within the mouse Tal1 locus. As further validation of the method, we mapped DNaseI hypersensitive sites across 250 kb of the human TAL1 locus in CD34+ primary stem/progenitor cells and mapped previously known and novel DNaseI hypersensitive sites across 240 kb of the human α-globin locus in K562 cells. Our results demonstrate that ADHM permits precise, large-scale identification of DNaseI hypersensitive sites from small numbers of cells.
Results
Our approach entails the generation of DNA fragments adjacent to DNaseI hypersensitive sites throughout the whole genome (Fig. 1A). Nuclei are isolated, chromatin is digested with DNaseI, and DNA is extracted. The digested ends are then blunted using T4 polymerase and an asymmetric double-stranded linker then ligated. The DNA template is generated by ligation-mediated primer extension reactions, as detailed in Methods, using a biotinylated linker-specific primer. The biotinylated primer extension products are captured with streptavidin beads and run with agarose gel electrophoresis to confirm that the captured products are between 200 and 450 bp in length. They are then labeled and hybridized against genomic arrays generated as described (Dhami et al. 2005, P. Dhami, P. Couttet, J. Cooper, I.J. Donaldson, R.M. Andrews, C. Langford, A.R. Green, B. Göttgens, and D. Vetrie, in prep.). A single DNaseI digestion protocol was chosen following pilot Southern blotting and real-time PCR experiments that assessed the influence of DNaseI digestion conditions on the degree of enrichment for known regulatory elements.
Figure 1.

Protocol outline and map of the mouse Tal1 locus. (A) Diagrammatic representation of the protocol followed to obtain DNA template representative of whole-genome DNaseI hypersensitive sites. Nuclei are isolated and treated with DNaseI. DNA is then extracted and repaired to blunt ends with T4 DNA polymerase; (gDNA) digested, repaired genomic DNA. Following ligation to an asymmetric double-stranded linker, biotinylated primer extension is used followed by extraction of biotinylated primer-extended DNA using streptavidin beads. This template is then labeled and hybridized to genomic arrays as described in Methods. (B) Diagrammatic map of mouse chromosome 4 showing the organization of the three genes: Stil (Scl/Tal1 Interrupting Locus), Tal1, and Pdzk1ip1. In the middle panel, the direction of transcription is indicated with black arrows, while the locations of known Stil and Tal1 regulatory elements are indicated with arrows in the far right panel.
The location of the known Tal1 regulatory elements and the organization of the mouse locus are shown in Figure 1B. Table 1 summarizes the identification, location, and functional data available for regulatory elements identified to date across the mouse Tal1 locus. The numbering of the elements reflects their positions in kilobases with respect to the start of Tal1 exon 1a. Promoters for each of the genes reside within highly conserved regions of DNA immediately 5′ to the transcriptional start sites (Aplan et al. 1990, 1991). Tal1 has two promoters located 5′ to the gene (promoter 1a and promoter 1b), and a further promoter within exon 4 (+7 relative to mouse promoter 1a) (Courtes et al. 2000). Tal1 enhancers are also highly conserved (Göttgens et al. 2000, 2001, 2002a; Chapman et al. 2004). Two elements, at −4 and +18/19, direct expression of Tal1 to the vast majority of hematopoietic stem cells, together with hematopoietic progenitors and endothelium (Sanchez et al. 1999; Göttgens et al. 2004). Elements at +1 and +3, relative to the mouse exon 1a, have been shown to target the expression of Tal1 to the developing spinal cord in transgenic mice (Sinclair et al. 1999), while the +23 directs expression to specific regions within the brain (Göttgens et al. 2000). The region at −9 functions as an enhancer in hematopoietic cell lines (Göttgens et al. 1997), and the +40 region is active in the primitive erythroid lineage (Delabesse et al. 2005). The mouse +12 region is homologous to the human +14 region, which has been identified as an TAL1 transcriptional repressor (Courtes et al. 2000).
TABLE 1.
Descriptive summary of the regulatory elements identified to date across the mouse Tal1 locus

Mouse Tal1 array
Figure 2A shows the annotation of the Stil (formerly known as Sil), Tal1, and Pdzk1ip1 (formerly known as Map17) genes above the UCSC mouse/rat/human/dog/chicken conservation score derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). Red bars locate known Stil and Tal1 regulatory elements, while blue bars highlight novel DNaseI hypersensitive sites identified and functionally characterized in this study. Figure 2B shows a DNaseI hypersensitive site array profile generated using 416B cells, a CD34+ primitive myeloid cell line that expresses high levels of Stil, Tal1, and Pdzk1ip1 (Supplemental Fig. 1). The data in Figure 2B represent the mean enrichment of four independent array experiments, derived from two independent cell cultures. Each array is spotted in triplicate. Histogram height represents the log2 of the mean net signal obtained from each arrayed PCR fragment, as described in Methods, while histogram width is proportional to the length of the arrayed PCR fragment. Overlapping histograms represent overlapping PCR fragments on the array. The arrays were normalized, and results were plotted as detailed in Methods. Significant enrichments over baseline were calculated using a new algorithm (see Methods). The algorithm accounts for the fluctuation of baseline DNaseI sensitivity observed over relatively long genomic distances. This fluctuation with the significance cutoff is plotted with the array profile for the extended 200 kb of the mouse Tal1 locus in Supplemental Figure 2. The complete data set for all arrays is available at http://hscl.cimr.cam.ac.uk/supplementary_follows06.html, while the significantly enriched sites from the 416B arrays are detailed with _P_-values in Supplemental Table 1.
Figure 2.

DNaseI hypersensitive site profiles across the mouse Tal1 locus. (A) Organization of the mouse Stil, Tal1, and Pdzk1ip1 genes, with wide and narrow bars representing translated and untranslated exons, respectively. The black bars represent the mouse/rat/human/dog/chicken overall conservation score quantified using phastCons (Siepel et al. 2005), derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). (B) DNaseI sensitivity plot from 416B cells across the Tal1 locus. Fold enrichment over non-enriched input is plotted (log2) against genomic position in kilobases. The plots in B are the means of four independent array experiments, each hybridized in triplicate, as detailed in Methods. The width of each bar represents the width of each spotted PCR product on the array, with overlapping bars representing overlapping PCR products. Red bars represent known regulatory elements, while blue bars represent novel elements identified in this study. Significant enrichments (P ≤ 0.05) are indicated with an asterisk or asterisk + bar (at least two adjacent tiles). (0k) Mouse chromosome 4 coordinate 113,334,279 (NCBI build 33). Functional elements are described in Table 1 and the text, and refer to the location in kilobases relative to the start of mouse Tal1 exon 1a.
There are reproducible peaks of enrichment that localize to the genomic positions of previously identified regulatory elements (red bars). Both Stil promoters and all Tal1 promoters and enhancers known to be functional in hematopoietic progenitor cells were identified as significantly enriched (asterisks indicate P ≤ 0.05). These data are consistent with previous DNaseI hypersensitive site mapping of the core region of the mouse Tal1 locus using Southern blotting techniques (Göttgens et al. 1997). Three elements known to direct Tal1 expression to the developing brain (+1, +3, and +23) and the +12 element, homologous to the human +14 TAL1 repressor (Courtes et al. 2000), were not significantly enriched above the regional baseline in 416B cells. Enrichments were highly reproducible between both biological and technical replicates. As examples, the Coefficient of Variance (CV) between the biological and technical replicates for enrichment at the Stil and Tal1 promoters were highly similar: Stil promoter, CV 11.2% (technical) and 9.0% (biological); Tal1 promoter, CV 15.7% (technical) and 15.6% (biological).
Previous systematic analysis of the 50 kb of the mouse Tal1/ Pdzk1ip1 coregulatory domain (−10 kb to +40 kb relative to promoter 1a) has identified multiple Tal1 regulatory elements (Sanchez et al. 1999; Sinclair et al. 1999; Göttgens et al. 2000, 2002a, b; Delabesse et al. 2005). Within this well-characterized region, two novel DNaseI hypersensitive sites were identified as significantly enriched (+29 and +32) and investigated in functional assays. These sites are outside the region previously mapped with Southern blotting, although restriction enzyme accessibility mapping around the Pdzk1ip1 gene in 416B cells has now identified both regions as hypersensitive using conventional Southern blotting approaches (data not shown). Consistent with its location immediately upstream of Pdzk1ip1 exon 1, the +32 element functioned as a promoter in transient and stable reporter assays (Fig. 3A,B), while the +29 element enhanced the activity of the +32 promoter in both transient and stable assays (Fig. 3A,B). The +29 element alone had no activity as a promoter when cloned into a promoter-less construct (data not shown). The +32 and +29 are therefore likely to represent a promoter and enhancer for Pdzk1ip1, respectively. Taken together, these results suggest that ADHM is able to identify regulatory elements with high specificity and sensitivity. Sites outside the central Tal1/ Pdzk1ip1 coregulatory domain were also identified as significantly enriched in 416B cells, including two sites upstream of the Stil promoter and one site within the Stil 10th intron (Fig. 2). These are likely to represent potential Stil regulatory elements.
Figure 3.

Transfection experiments confirm the functional activity of the novel mouse Tal1 elements identified from the DNaseI hypersensitive site profiles. (A) A representative transient transfection experiment in 416B cells with bars representing means of triplicate relative luminescence values of construct activity +SD as detailed in Methods. (B) A representative stable transfection experiment in MDCT cells with bars representing means of quadruplicate relative luminescence values of construct activity +SD.
Human TAL1 array
Figure 4A shows the annotation of known genes across 150 kb of the human TAL1 locus plotted above the UCSC human/chimp/ mouse/rat/dog/chicken/Fugu/zebrafish conservation score derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). Figure 4B shows the DNaseI hypersensitive site profile obtained from primary CD34+ selected cells and K562 cells, a chronic myeloid leukemia (CML) cell line. In all of these cell types, STIL, TAL1, and PDZK1IP1 are expressed (Supplemental Fig. 1). The normal donor CD34+ plot in Figure 4B represents the mean of four independent array experiments, using material derived independently from two normal donors. The CML CD34+ plot in Figure 4B represents the mean of two independent arrays, using material from a single patient, while the K562 profile represents the mean of four independent arrays with material derived from three independent cell cultures. The complete data set for all arrays are available at http://hscl.cimr.cam.ac.uk/supplementary_follows06.html, while the summary of the significantly enriched sites from the human TAL1 locus, including _P_-values, is in Supplemental Table 2. The array profile for the extended 250 kb of the human TAL1 locus detailing the baseline enrichment plot with significance limits is shown in Supplemental Figure 3.
Figure 4.

DNaseI hypersensitive site profiles across the human TAL1 locus. (A) Organization of the human STIL, TAL1, and PDZK1IP1 genes, with wide and narrow bars representing translated and untranslated exons, respectively. The black bars represent the human/chimp/mouse/rat/dog/chicken/ Fugu/zebrafish overall conservation score quantified using phastCons (Siepel et al. 2005), derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). (B) DNaseI sensitivity plot from normal donor and CML CD34+ cells and K562 cell lines across the TAL1 locus. Fold enrichment over non-enriched input is plotted (log2) against genomic position in kilobases. The normal donor and K562 plots represent the mean values of four independent array experiments, while the CML data represent the mean of two separate experiments, each hybridized in triplicate as detailed in Methods. The width of each bar represents the width of each spotted PCR product on the array, with overlapping bars representing overlapping PCR products. Red bars represent known regulatory elements or regions homologous to known mouse regulatory elements. The blue bars locate the human region homologous to the novel mouse regulatory elements identified in this study. Significant enrichments (P ≤ 0.05) are indicated with an asterisk or asterisk + bar (at least two adjacent tiles). (0k) Human chromosome 1 coordinate 47,505,065 (NCBI build 35). Functional elements are described in Table 1 and the text, and refer to the location in kilobases of the conserved elements relative to the start of human exon 1a.
Strikingly similar patterns of DNaseI hypersensitivity were reproducibly obtained from the three different human cell types. Similar to the mouse Tal1 locus, there was increased DNaseI accessibility across the TAL1/PDZK1IP1 coregulatory domain, with reduced DNaseI accessibility both 5′ and 3′ of this core region. With all the human arrays the 5′-end of CMPK, the STIL promoter, the TAL1 promoter 1a/1b, and TAL1 exon 4 promoter were significantly enriched. The CD34+ primary cells also showed significant enrichment of the −4 element, which has been identified as a stem cell enhancer (Göttgens et al. 2004). The +51 element, which is homologous to the mouse +40 “erythroid” element (Delabesse et al. 2005), was significantly hypersensitive in K562 cells, which correlates with the partial erythroid phenotype of these cells. Biological differences between the murine 416B cells, human K562 cells, and human primary CD34+ cells (e.g., PDZK1IP1 transcripts are ~10-fold lower in human cells) (Supplemental Fig. 1) may partially explain the lack of significant enrichment in the human cells over the two human regions (+40 and +43) homologous to the mouse Pdzk1ip1 elements. However, comparing mouse and human enrichments relative to baseline DNaseI accessibility in this region is made more difficult by the addition of ~8 kb of repeat elements (Long Interspersed Nuclear Elements) in the human sequence, which were omitted from the human array.
Human α-globin array
To further validate the ADHM protocol and the statistical algorithm derived from the TAL1 locus, material from K562 cells was hybridized against a human α-globin array. Figure 5A shows the annotation of known genes across 240 kb of the α-globin locus plotted above the UCSC human/chimp/mouse/rat/chicken conservation score derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). The profile in Figure 5B represents the log2 of mean enrichments of seven independent experiments derived from four independent K562 cell cultures. Red bars represent previously identified DNaseI hypersensitive sites (Yagi et al. 1986; Higgs et al. 1990; Vyas et al. 1992; Hughes et al. 2005; D. Higgs, pers. comm.). Red dashed bars represent the three known DNaseI hypersensitive sites not represented on the array. Significant enrichments are indicated with an asterisk, and crosses represent the three previously identified hypersensitive sites that were enriched above baseline, but did not meet statistical criteria for significant enrichment. The complete data set for all arrays is available at http://hscl.cimr.cam.ac.uk/supplementary_follows06.html, while the summary of the significantly enriched sites from the α-globin locus, including _P_-values is in Supplemental Table 3. The enrichment profile for the α-globin locus detailing the baseline enrichment and significance plots is shown in Supplemental Figure 4.
Figure 5.

DNaseI hypersensitive site profile across the human α-globin locus. (A) Organization of the 12 genes across 240 kb, with wide and narrow bars representing translated and untranslated exons, respectively. The black bars represent the human/chimp/mouse/rat/dog/chicken overall conservation score quantified using phastCons (Siepel et al. 2005), derived using the MultiZ species alignment program (http://genome.ucsc.edu) (Hinrichs et al. 2006). (B) DNaseI sensitivity plot from K562 cell lines. Fold enrichment over non-enriched input is plotted (log2) against genomic position in kilobases. The plot represents the mean values of seven separate array experiments, each hybridized in duplicate as detailed in Methods. The width of each bar represents the width of each spotted PCR product on the array, with overlapping bars representing overlapping PCR products. Red bars represent known regulatory elements, while dashed red bars are known elements not represented on the array. (0k) Human chromosome 16 coordinate 17,913 (NCBI build 35). Significant enrichments (P ≤ 0.05) are indicated with an asterisk or asterisk + bar (at least two adjacent tiles), while crosses represent the three known hypersensitive sites that were enriched above baseline, but did not meet statistical criteria for significant enrichment. The “V” indicates the two novel significant sites identified within the region of the locus previously mapped by conventional techniques (Yagi et al. 1986; Higgs et al. 1990; Vyas et al. 1992; Hughes et al. 2005; D. Higgs, pers. comm.). Functional elements refer to kilobases relative to HBZ exon 1.
The 240-kb region of the α-globin locus shown in Figure 5 has previously been mapped for DNaseI hypersensitive sites using Southern blotting techniques (Yagi et al. 1986; Higgs et al. 1990; Vyas et al. 1992; Hughes et al. 2005; D. Higgs, pers. comm.). Of the 18 known K562 hypersensitive sites represented on the array (red bars), 15 were identified as significantly enriched by our algorithm (P ≤ 0.05). The three remaining sites (−90, −55, −46), which were originally identified as constitutive hypersensitive sites, have no known biological function (Hughes et al. 2005), and exhibited relatively weak DNaseI accessibility compared with the known α-globin erythroid enhancers (Yagi et al. 1986; Higgs et al. 1990; Vyas et al. 1992). ADHM identified only one novel hypersensitive site as significantly enriched (+90; P = 0.02; indicated “V” in Fig. 5). The tile corresponding to this site contains a 350-bp region of highly conserved DNA sequence, colocalizes with a peak in histone H3 K9 acetylation reported by the ENCODE consortium (http://genome.ucsc.edu) (Hinrichs et al. 2006), and is therefore highly likely to represent a _cis_-regulatory element. Taken together, the α-globin data demonstrate that ADHM detected the vast majority of known hypersensitive sites in this well-characterized region, suggesting that ADHM is a sensitive and specific tool for high-throughput mapping of _cis_-regulatory elements.
Discussion
We have developed a novel technique, ADHM, that permits large-scale identification of DNaseI hypersensitive sites using genomic tiling path arrays. This method has been developed using the mouse Tal1 locus, and further validated using the human TAL1 and α-globin loci. Both Tal1 and α-globin are paradigm loci for hematopoietic gene regulation, containing multiple conserved and functionally characterized regulatory elements, which make these loci ideal to establish and validate the protocol. In addition, we have identified novel regulatory elements at the mouse Tal1 locus that have been shown to act as transcriptional regulatory elements in functional assays.
ADHM has numerous advantages over traditional Southern blotting techniques, which represent the current gold standard for identifying DNaseI hypersensitive sites. In particular, DNaseI accessibility can be mapped over large genomic regions using small numbers of cells. Furthermore, an array-based method provides in-built positive and negative control regions, not routinely available using Southern blotting. We have confirmed the sensitivity of ADHM by mapping significant enrichment at 25/28 DNaseI hypersensitive sites previously mapped at the mouse Tal1 locus and the human α-globin locus in 416B and K562 cells, respectively. Our results also suggest that ADHM is highly specific. Across the well-characterized core mouse Tal1 locus, ADHM identified only two previously undetected hypersensitive sites. Both of these are in a region not previously mapped with Southern blotting techniques, and we demonstrate here that both represent _cis_-regulatory elements. Furthermore, across the region of the α-globin locus in which DNaseI hypersensitive sites have been systematically mapped, only one novel site (+90) was identified as significantly enriched. This region contains conserved DNA sequences and is acetylated in K562 cells, strongly suggesting that it is also a regulatory element.
It is informative to compare ADHM with the two other approaches to large-scale mapping that have recently been published. The development of high-throughput cloning and sequencing has shown that sequencing DNA libraries generated from DNaseI digested DNA has the potential to identify large numbers of hypersensitive sites (Crawford et al. 2006). Crawford et al. sequenced 230,000 library tags to identify ~5000 clusters of DNA sequences, which, when validated with real-time PCR, had >80% validity as true hypersensitive sites (Crawford et al. 2006). However, with regards to whole-genome identification of hypersensitive sites, the sensitivity was far inferior to the specificity. Indeed, the authors state that by their calculations, to identify 95% of all true DNaseI hypersensitive sites with a minimum of 80% validity, ~2.3 million sequence reads would be required. Such high-throughput cloning and sequencing is only available to a very limited number of laboratories. An inherent problem with the cloning and sequencing approach is separating true “signal” from “noise.” As the investigators point out, despite extensive efforts to reduce random shearing of high-molecular-weight DNA, at least 70% of sequenced fragments were thought to represent noise. In contrast, our array-based approach provides relative enrichment across a whole locus, which permits a statistical assessment of whether a peak is significantly enriched relative to neighboring regions. This also provides a far more focused approach for researchers attempting to dissect the dynamics of transcriptional regulation at defined loci in different cell types.
An alternative approach that permits the analysis of specific loci uses real-time PCR to identify DNaseI hypersensitive sites (Dorschner et al. 2004). A strength of this approach is the excellent sensitivity, with the investigators detecting all of the 23 known DNaseI hypersensitive sites within the region they studied. However, a marked limitation of this method is the large numbers of quantitative PCR reactions required to study even a single locus, which questions whether this approach can be described as “high throughput.” For example, the authors performed 855 quantitative PCR reactions to study 30.4 kb of the MYC locus in a single cell type, which means that experiments to compare the DNaseI accessibility profiles of different cell types across even small loci will require thousands of quantitative PCR reactions. In contrast, constructing a genomic microarray requires an initial set of PCR reactions that can then be used to construct hundreds of array slides, allowing the rapid analysis of DNA template derived from multiple cell types.
With the falling cost of printing array slides, we believe that most molecular biology laboratories will be able to establish ADHM. However, to circumvent the time and resources required to construct PCR arrays, it is of additional interest whether ADHM could be adapted to work with oligonucleotide arrays. These arrays are now readily available from a range of commercial suppliers, and high-density oligonucleotide arrays have the potential to cover significantly more of the genome per hybridization. Indeed, initial hybridizations to a commercially available oligonucleotide array using DNA template generated with our protocol are encouraging, and indicate that ADHM can detect the vast majority of DNaseI hypersensitive sites on such array platforms (Supplemental Fig. 5).
_Cis_-regulatory elements are central to the evolution of biological complexity (Levine and Tjian 2003). Moreover, genetic and epigenetic alterations affecting _cis_-regulatory elements are being implicated in an increasing number of inherited and acquired diseases (Di Croce et al. 2002; Follows et al. 2003; Laws et al. 2003; van Wijk et al. 2003; Das and Singal 2004; Wu et al. 2004). A technique that permits rapid identification of DNaseI hypersensitive sites across large regions of DNA will, therefore, greatly facilitate the characterization of regulatory elements in normal and pathological states.
Methods
Cell culture and sorting
The 416B and K562 cell lines were cultured as previously detailed (Bockamp et al. 1995, 1997). The human primary cells were obtained from consenting donors. The normal cells were sorted from peripheral blood stem cell harvest material obtained from normal donors treated with G-CSF as part of the Cambridge allogeneic transplant program. The chronic myeloid leukemia material was taken from the peripheral blood of a newly diagnosed chronic-phase patient. CD34+ cells from donors and patient were purified to >95%+ using MACS columns and standard protocols.
The material for each hybridization was generated independently from DNA extracted from nuclei preparations. Each nuclei preparation was derived from separate stocks of frozen or primary cells. The 416B arrays were performed with material generated from two independent nuclei preparations. The normal human donor material was obtained from three nuclei preparations made from two batches of CD34+ selected cells from two different donors. The CML hybridization material was obtained from a single nuclei preparation from a single patient. The K562 hybridizations to the human TAL1 array were performed with material generated from three independent nuclei preparations, while the seven α-globin array experiments were performed with material derived from four independent nuclei preparations. Which nuclei preparation corresponds with which raw data set is detailed at http://hscl.cimr.cam.ac.uk/supplementary_follows06. html.
Preparation of DNA template
Nuclei preparations were made according to standard protocols from 5 million cells (Cockerill 2000). Briefly, cells were washed in ice-cold PBS then re-suspended in 0.2% NP-40 nuclei preparation buffer for 5 min on ice (300 mM sucrose, 10 mM Tris at pH 7.4, 15 mM NaCl, 5 mM MgCl, 0.1 mM EGTA, 60 mM KCl, 0.2% NP-40, 0.5 mM DTT, 0.5 μM spermidine, 1 × protease inhibitor [complete, Roche]). The resulting nuclei were isolated and incubated with a range of DNaseI (Ambion) concentrations (0, 40, 60, 80, 120 units) on ice for 1 h in 1 × DNaseI buffer with 2% glycerol. DNA was then extracted as per standard protocols with care to prevent shearing (Cockerill 2000). After RNase A treatment, on average, 20–30 μg of digested DNA could be extracted from 5 million nuclei, with 7.5 μg of extracted DNA then being used for each experiment. The DNA was initially repaired with T4 polymerase (NE Biolabs), using standard protocols to provide double-stranded blunt ends. The DNA was then precipitated and re-suspended for ligation to a double stranded asymmetric linker (LP21–25: LP25, GCGGTGACCCGGGAGATCTGAATTC; LP21, GAATTCAGATCTCCCGGGTCA) at 16°C for 16 h. Following ligation, the DNA was re-precipitated, re-suspended, and primer-extended. Multiple primer extension steps were performed using Vent exo-polymerase (NE Biolabs) and biotinylated linker-specific primer (B-LP25) using the following temperature profile: 95°C for 3 min, (95°C for 30 sec, 61°C for 30 sec, 72°C for 30 sec) for 35 cycles. Primer-extended template was then purified from linker and digested genomic DNA using streptavidin beads (Dynal) and the standard Dynal protocol. Agarose gel electrophoresis confirmed that the template size was between 250 and 450 bp.
Comparative enrichments for the range of DNaseI concentrations at separate genomic loci (Porphobilinogen deaminase promoter (=hydroxymethylbilane synthase promoter, Stil promoter and Cyp4x1 gene control region) were compared using real-time PCR. Primer sets are available on request. This confirmed that maximal enrichments were obtained in all cell types using 120 units of DNaseI/digestion. This was in keeping with pilot Southern blot experiments that confirmed maximal digestion at known regulatory elements with between 80 and 120 units of DNaseI digestion (Supplemental Fig. 6). Template from this condition was then labeled for hybridization with Cyanine 3 dCTP (Perkin Elmer) using Klenow and previously published protocols (Dhami et al. 2005). Non-enriched input control for hybridizations was taken as digested genomic DNA from the same DNaseI treated sample that had not been enriched by ligation-mediated primer extension. This input was labeled with Cyanine 5 dCTP (Perkin Elmer).
Microarray fabrication
The Tal1 and α-globin tiling path microarrays were fabricated as detailed (Dhami et al. 2005, 2006). In brief, primer pairs were designed using the Primer3 software and Web site with Repeat-Masking (Rozen and Skaletsky 2000). All primer pairs and amplicons were compared against the entire human or mouse genome sequence using e-PCR (Schuler 1997) and BLASTN to identify any potential cross-reacting DNA sequences. PCR amplicons were prepared as detailed previously (Dhami et al. 2005), and arrayed onto Codelink slides (Amersham) in triplicate (Tal1) or duplicate (α-globin) using a Microgrid II arrayer (Biorobotics/Genomic Solutions). Slides were processed to generate single-stranded array elements, as described on the Sanger Institute microarray Web pages, and were stored at room temperature until hybridized.
Hybridization
Cy3 and Cy5 labeled samples were pooled and then precipitated with mouse or human cot-1 DNA. Following pre-hybridization of the array slides with herring sperm DNA/cot-1 DNA, the samples were resuspended in hybridization buffer (Dhami et al. 2005), and hybridized to the slides for 45 h using an automated TECAN 400 hybridization station. The details of which hybridizations are technical versus biological replicates are detailed for each mouse and human array at http://hscl.cimr.cam.ac.uk/supplementary_follows06.html. Following hybridization, micro-arrays were scanned using a ScanArray 4000 confocal laser-based scanner (Perkin Elmer). Mean spot intensities from images were quantified using ScanArray Express (Perkin Elmer) with background subtraction. Control hybridizations with template derived from mouse and human genomic DNA digested with DNaseI confirmed no apparent sequence specificity of DNaseI across the Tal1 and α-globin loci (Supplemental Fig. 7).
Five million primary CD34+ cells or 416B/K562 cells were used for each nuclei preparation, which yielded ~25 μg of digested DNA. Each hybridization was performed using material generated independently from 7.5 μg of DNA extracted from DNaseI treated nuclei. Consequently each hybridization was performed with the material obtained from ~1.5 million cells.
Array structure
The mouse Tal1 416B mean array data covers 83% of the core region between −10 kb (113,392,000) and +40 kb (113,445,612), and 66% of the extended region between 113,334,279 and 113,541,031. The median tile size is 427 bp (range 241–599). The normal donor human TAL1 mean array data cover 65% of the core region between −10 kb (47,420,382) and +52 kb (47,353,495), and 44% of the extended region between 47,262,287 and 47,518,762. The median tile size is 484 bp (range 190–664). The K562 α-globin mean array data cover 63% of the region between 17,913 and 257,516. The median tile size is 1023 bp (range 142–1480).
Data presentation
All raw and mean data from the separate hybridizations are available at http://hscl.cimr.cam.ac.uk/supplementary_follows06.html.
Statistical analysis
The triplicate (Tal1 arrays) and duplicate (α-globin array) values for the spot intensities corresponding to each spotted PCR product were used to calculate mean values for each data point. Any spots flagged by the ScanArray software as poor hybridization signals were discarded. A coefficient of variation (CV) for the range of spot intensity between the triplicate/duplicate readings was calculated. Any values with a CV >100% were discarded from further analysis. Median CVs for the separate arrays were 416B Tal1, 9.62, 9.27, 7.22, 8.29; CD34+ normal donor TAL1, 7.25, 7.78, 7.62, 6.10; CD34+ CML Tal1, 8.10, 6.19; K562 Tal1, 11.11, 12.50, 9.36, 6.07; K562 α-globin, 16.22, 16.55, 14.59, 13.16, 5.79, 5.96, 5.89.
The data in Figure 2, as well as the normal donor and K562 data in Figure 4 were calculated as the mean values from the four independent arrays for each cell type, whereas the data in Figure 5 were calculated from seven independent array experiments. The details of biological and technical replicates are included with the complete data set (http://hscl.cimr.cam.ac.uk/supplementary_follows06.html). If an individual data point was missing from more than two of the four arrays (Figs. 2and4) or four of the seven arrays (Fig. 5), that is, had been filtered out because of poor hybridization or wide CV, the data point was omitted from further analysis. If the CV of a data point was greater than the mean from the independent array experiments, the data point was discarded from further analysis. Each array was normalized by dividing by the median intensity for the array. The normalized values were then averaged and the log2 values plotted in Figures 2,4and5
An algorithm was developed to identify significantly enriched peaks, accounting for two factors. Firstly, baseline DNaseI sensitivity varies substantially over relatively long genomic distances (see, e.g., the difference in general enrichment of the Stil and Tal1 genes in Fig. 2). Significance tests for points of hypersensitivity should therefore use local levels of enrichment to set baseline values. Secondly, the enrichment scores are not normally distributed, owing to the relatively high frequency of hypersensitive sites. Therefore, significantly hypersensitive points should be excluded from the determination of local baseline mean and variance enrichment.
The algorithm proceeds iteratively. For each tile on the array, the local sample mean and sample variance are calculated from the log2 (fluorescence intensity ratios) for tiles within the width of a user-defined window, and excluding all tiles identified as significantly enriched on the previous iteration. The sample means and variances need to be corrected for the bias introduced by excluding significant scores. It can readily be demonstrated that the sample variance of the truncated normal distribution underestimates the true variance of the full normal distribution by the following factor:
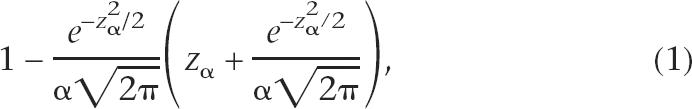
where α is the (one-sided) significance level for exclusion of a point (α = 0.95 here) and z α is the threshold at which the cumulative standard normal distribution has probability α. The sample mean of the truncated normal distribution underestimates the mean of the full normal distribution by the addendum

Therefore, a bias-corrected estimator for the local variance can be generated by dividing the sample variance of the local values (excluding significant values) by the factor in equation 1 above. Similarly, a bias-corrected estimator for the local mean can be calculated by substituting the bias-corrected local variance into equation 2 above and adding to the sample mean. Z scores are calculated from these corrected local means and variances and defined as significant when greater than a threshold z λ, noting that the significance level λ need not be the same as the level for exclusion of points from the calculation of local mean and variance, α. This process is repeated iteratively until no further points are identified as significant (at either α or λ levels).
We have found that the choice of window width for calculating local mean and variance makes little qualitative difference to the results in a range of 5–30 kb. A window of 15 kb was used for the results presented here. A significance level of α = λ = 0.95 for identification of significant peaks and exclusion of peaks from local mean and variance calculations works well, with minimal identification of (presumed) false-positive peaks, and acceptable identification of known regions of hypersensitivity. Q-Q plots of the Z scores excluding statistically significant peaks do demonstrate the normality of their distribution (data not shown), and justify the _P_-values quoted. The algorithm was implemented in S-Plus v6.0 (Insightful Corp.), and the code is available from the authors on request.
Transfection experiments
PCR was used to amplify regions corresponding to 680 bp of +29 and 370 bp of +32. These fragments were cloned upstream of the luciferase reporter gene in pGL2 basic (Promega) and assayed in transfection assays. Primer sequences are available on request. Transient and stable transfections were performed in 416B and MDCT cell lines, respectively, using standard protocols, available on request. Independent triplicate (transient) and quadruplicate (stable) transfection experiments were performed using two independently derived plasmid preparations for each construct. Renilla luciferase cotransfection was used to control for transient transfection efficiency, and data were plotted as fold luminescence relative to pGL2 basic. The stable transfections were performed in quadruplicate and plotted as fold luminescence relative to pGL2 basic. Data in Figure 3, AandB, are representatives of a triplicate and a quadruplicate experiment, respectively. Up to five independent triplicate (transient) or quadruplicate (stable) transfection experiments were performed with each construct.
ACKNOWLEDGMENTS
G.A.F. is a Leukaemia Research Fund Bennett Fellow. Work in A.R.G.’s laboratory is funded by grants from the Leukaemia Research fund, Medical Research Council, and the Wellcome Trust. Work in the laboratories of I.D. and D.V. laboratories is funded by the Wellcome Trust. We are grateful to Jim Hughes and Doug Higgs for their help with locating DNaseI hypersensitive sites across the human α-globin locus.
Footnotes
REFERENCES
- Aplan P.D., Lombardi D.P., Ginsberg A.M., Cossman J., Bertness V.L., Kirsch I.R., Lombardi D.P., Ginsberg A.M., Cossman J., Bertness V.L., Kirsch I.R., Ginsberg A.M., Cossman J., Bertness V.L., Kirsch I.R., Cossman J., Bertness V.L., Kirsch I.R., Bertness V.L., Kirsch I.R., Kirsch I.R. Disruption of the human SCL locus by “illegitimate” V-(D)-J recombinase activity. Science. 1990;250:1426–1429. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- Aplan P.D., Lombardi D.P., Kirsch I.R., Lombardi D.P., Kirsch I.R., Kirsch I.R. Structural characterization of SIL, a gene frequently disrupted in T-cell acute lymphoblastic leukemia. Mol. Cell. Biol. 1991;11:5462–5469. doi: 10.1128/mcb.11.11.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockamp E.O., McLaughlin F., Murrell A.M., Göttgens B., Robb L., Begley C.G., Green A.R., McLaughlin F., Murrell A.M., Göttgens B., Robb L., Begley C.G., Green A.R., Murrell A.M., Göttgens B., Robb L., Begley C.G., Green A.R., Göttgens B., Robb L., Begley C.G., Green A.R., Robb L., Begley C.G., Green A.R., Begley C.G., Green A.R., Green A.R. Lineage-restricted regulation of the murine SCL/TAL-1 promoter. Blood. 1995;86:1502–1514. [PubMed] [Google Scholar]
- Bockamp E.O., McLaughlin F., Göttgens B., Murrell A.M., Elefanty A.G., Green A.R., McLaughlin F., Göttgens B., Murrell A.M., Elefanty A.G., Green A.R., Göttgens B., Murrell A.M., Elefanty A.G., Green A.R., Murrell A.M., Elefanty A.G., Green A.R., Elefanty A.G., Green A.R., Green A.R. Distinct mechanisms direct SCL/tal-1 expression in erythroid cells and CD34 positive primitive myeloid cells. J. Biol. Chem. 1997;272:8781–8790. doi: 10.1074/jbc.272.13.8781. [DOI] [PubMed] [Google Scholar]
- Bockamp E.O., Fordham J.L., Göttgens B., Murrell A.M., Sanchez M.J., Green A.R., Fordham J.L., Göttgens B., Murrell A.M., Sanchez M.J., Green A.R., Göttgens B., Murrell A.M., Sanchez M.J., Green A.R., Murrell A.M., Sanchez M.J., Green A.R., Sanchez M.J., Green A.R., Green A.R. Transcriptional regulation of the stem cell leukemia gene by PU.1 and Elf-1. J. Biol. Chem. 1998;273:29032–29042. doi: 10.1074/jbc.273.44.29032. [DOI] [PubMed] [Google Scholar]
- Chapman M.A., Donaldson I.J., Gilbert J., Grafham D., Rogers J., Green A.R., Göttgens B., Donaldson I.J., Gilbert J., Grafham D., Rogers J., Green A.R., Göttgens B., Gilbert J., Grafham D., Rogers J., Green A.R., Göttgens B., Grafham D., Rogers J., Green A.R., Göttgens B., Rogers J., Green A.R., Göttgens B., Green A.R., Göttgens B., Göttgens B. Analysis of multiple genomic sequence alignments: A Web resource, online tools, and lessons learned from analysis of mammalian SCL loci. Genome Res. 2004;14:313–318. doi: 10.1101/gr.1759004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill P.N. Identification of DNaseI hypersensitive sites within nuclei. Methods Mol. Biol. 2000;130:29–46. doi: 10.1385/1-59259-686-x:29. [DOI] [PubMed] [Google Scholar]
- Courtes C., Lecointe N., Le Cam L., Baudoin F., Sardet C., Mathieu-Mahul D., Lecointe N., Le Cam L., Baudoin F., Sardet C., Mathieu-Mahul D., Le Cam L., Baudoin F., Sardet C., Mathieu-Mahul D., Baudoin F., Sardet C., Mathieu-Mahul D., Sardet C., Mathieu-Mahul D., Mathieu-Mahul D. Erythroid-specific inhibition of the tal-1 intragenic promoter is due to binding of a repressor to a novel silencer. J. Biol. Chem. 2000;275:949–958. doi: 10.1074/jbc.275.2.949. [DOI] [PubMed] [Google Scholar]
- Crawford G.E., Holt I.E., Mullikin J.C., Tai D., Blakesley R., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Holt I.E., Mullikin J.C., Tai D., Blakesley R., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Mullikin J.C., Tai D., Blakesley R., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Tai D., Blakesley R., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Blakesley R., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Bouffard G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Young A., Masiello C., Green E.D., Wolfsberg T.G., Masiello C., Green E.D., Wolfsberg T.G., Green E.D., Wolfsberg T.G., Wolfsberg T.G., et al. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc. Natl. Acad. Sci. 2004;101:992–997. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford G.E., Holt I.E., Whittle J., Webb B.D., Tai D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Holt I.E., Whittle J., Webb B.D., Tai D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Whittle J., Webb B.D., Tai D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Webb B.D., Tai D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Tai D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Davis S., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Margulies E.H., Chen Y., Bernat J.A., Ginsburg D., Chen Y., Bernat J.A., Ginsburg D., Bernat J.A., Ginsburg D., Ginsburg D., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.M., Singal R., Singal R. DNA methylation and cancer. J. Clin. Oncol. 2004;22:4632–4642. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- Delabesse E., Ogilvy S., Chapman M.A., Piltz S.G., Göttgens B., Green A.R., Ogilvy S., Chapman M.A., Piltz S.G., Göttgens B., Green A.R., Chapman M.A., Piltz S.G., Göttgens B., Green A.R., Piltz S.G., Göttgens B., Green A.R., Göttgens B., Green A.R., Green A.R. Transcriptional regulation of the SCL locus: Identification of an enhancer that targets the primitive erythroid lineage in vivo. Mol. Cell. Biol. 2005;25:5215–5225. doi: 10.1128/MCB.25.12.5215-5225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami P., Coffey A.J., Abbs S., Vermeesch J.R., Dumanski J.P., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Coffey A.J., Abbs S., Vermeesch J.R., Dumanski J.P., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Abbs S., Vermeesch J.R., Dumanski J.P., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Vermeesch J.R., Dumanski J.P., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Dumanski J.P., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Woodward K.J., Andrews R.M., Langford C., Vetrie D., Andrews R.M., Langford C., Vetrie D., Langford C., Vetrie D., Vetrie D. Exon array CGH: Detection of copy-number changes at the resolution of individual exons in the human genome. Am. J. Hum. Genet. 2005;76:750–762. doi: 10.1086/429588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami P., Couttet P., Cooper J., Donaldson I.J., Andrews R.M., Langford C., Green A.R., Göttgens B., Vetrie D., Couttet P., Cooper J., Donaldson I.J., Andrews R.M., Langford C., Green A.R., Göttgens B., Vetrie D., Cooper J., Donaldson I.J., Andrews R.M., Langford C., Green A.R., Göttgens B., Vetrie D., Donaldson I.J., Andrews R.M., Langford C., Green A.R., Göttgens B., Vetrie D., Andrews R.M., Langford C., Green A.R., Göttgens B., Vetrie D., Langford C., Green A.R., Göttgens B., Vetrie D., Green A.R., Göttgens B., Vetrie D., Göttgens B., Vetrie D., Vetrie D. Characterisation of regulatory events at the human and murine SCL (TAL1)loci using chromatin immunoprecipitation in combination with genomic microarrays. 2006 (in press) [Google Scholar]
- Di Croce L., Raker V.A., Corsaro M., Fazi F., Fanelli M., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Raker V.A., Corsaro M., Fazi F., Fanelli M., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Corsaro M., Fazi F., Fanelli M., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Fazi F., Fanelli M., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Fanelli M., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Faretta M., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Fuks F., Lo Coco F., Kouzarides T., Nervi C., Lo Coco F., Kouzarides T., Nervi C., Kouzarides T., Nervi C., Nervi C., et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- Dorschner M.O., Hawrylycz M., Humbert R., Wallace J.C., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Hawrylycz M., Humbert R., Wallace J.C., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Humbert R., Wallace J.C., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Wallace J.C., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P.J., Mack J., Hall R., Goldy J., Sabo P.J., Hall R., Goldy J., Sabo P.J., Goldy J., Sabo P.J., Sabo P.J. High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods. 2004;1:219–225. doi: 10.1038/nmeth721. [DOI] [PubMed] [Google Scholar]
- Elgin S.C. The formation and function of DNase I hypersensitive sites in the process of gene activation. J. Biol. Chem. 1988;263:19259–19262. [PubMed] [Google Scholar]
- Follows G.A., Tagoh H., Lefevre P., Hodge D., Morgan G.J., Bonifer C., Tagoh H., Lefevre P., Hodge D., Morgan G.J., Bonifer C., Lefevre P., Hodge D., Morgan G.J., Bonifer C., Hodge D., Morgan G.J., Bonifer C., Morgan G.J., Bonifer C., Bonifer C. Epigenetic consequences of AML1-ETO action at the human c-FMS locus. EMBO J. 2003;22:2798–2809. doi: 10.1093/emboj/cdg250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordham J.L., Göttgens B., McLaughlin F., Green A.R., Göttgens B., McLaughlin F., Green A.R., McLaughlin F., Green A.R., Green A.R. Chromatin structure and transcriptional regulation of the stem cell leukaemia (SCL) gene in mast cells. Leukemia. 1999;13:750–759. doi: 10.1038/sj.leu.2401420. [DOI] [PubMed] [Google Scholar]
- Göttgens B., McLaughlin F., Bockamp E.O., Fordham J.L., Begley C.G., Kosmopoulos K., Elefanty A.G., Green A.R., McLaughlin F., Bockamp E.O., Fordham J.L., Begley C.G., Kosmopoulos K., Elefanty A.G., Green A.R., Bockamp E.O., Fordham J.L., Begley C.G., Kosmopoulos K., Elefanty A.G., Green A.R., Fordham J.L., Begley C.G., Kosmopoulos K., Elefanty A.G., Green A.R., Begley C.G., Kosmopoulos K., Elefanty A.G., Green A.R., Kosmopoulos K., Elefanty A.G., Green A.R., Elefanty A.G., Green A.R., Green A.R. Transcription of the SCL gene in erythroid and CD34 positive primitive myeloid cells is controlled by a complex network of lineage-restricted chromatin-dependent and chromatin-independent regulatory elements. Oncogene. 1997;15:2419–2428. doi: 10.1038/sj.onc.1201426. [DOI] [PubMed] [Google Scholar]
- Göttgens B., Barton L.M., Gilbert J.G., Bench A.J., Sanchez M.J., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Barton L.M., Gilbert J.G., Bench A.J., Sanchez M.J., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Gilbert J.G., Bench A.J., Sanchez M.J., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Bench A.J., Sanchez M.J., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Sanchez M.J., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Bahn S., Mistry S., Grafham D., McMurray A., Vaudin M., Mistry S., Grafham D., McMurray A., Vaudin M., Grafham D., McMurray A., Vaudin M., McMurray A., Vaudin M., Vaudin M., et al. Analysis of vertebrate SCL loci identifies conserved enhancers. Nat. Biotechnol. 2000;18:181–186. doi: 10.1038/72635. [DOI] [PubMed] [Google Scholar]
- Göttgens B., Gilbert J.G., Barton L.M., Grafham D., Rogers J., Bentley D.R., Green A.R., Gilbert J.G., Barton L.M., Grafham D., Rogers J., Bentley D.R., Green A.R., Barton L.M., Grafham D., Rogers J., Bentley D.R., Green A.R., Grafham D., Rogers J., Bentley D.R., Green A.R., Rogers J., Bentley D.R., Green A.R., Bentley D.R., Green A.R., Green A.R. Long-range comparison of human and mouse SCL loci: Localized regions of sensitivity to restriction endonucleases correspond precisely with peaks of conserved noncoding sequences. Genome Res. 2001;11:87–97. doi: 10.1101/gr.153001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttgens B., Barton L.M., Chapman M.A., Sinclair A.M., Knudsen B., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Barton L.M., Chapman M.A., Sinclair A.M., Knudsen B., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Chapman M.A., Sinclair A.M., Knudsen B., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Sinclair A.M., Knudsen B., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Knudsen B., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Grafham D., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Gilbert J.G., Rogers J., Bentley D.R., Green A.R., Rogers J., Bentley D.R., Green A.R., Bentley D.R., Green A.R., Green A.R. Transcriptional regulation of the stem cell leukemia gene (SCL)–comparative analysis of five vertebrate SCL loci. Genome Res. 2002a;12:749–759. doi: 10.1101/gr.45502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttgens B., Nastos A., Kinston S., Piltz S., Delabesse E.C., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Nastos A., Kinston S., Piltz S., Delabesse E.C., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Kinston S., Piltz S., Delabesse E.C., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Piltz S., Delabesse E.C., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Delabesse E.C., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Stanley M., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Sanchez M.J., Ciau-Uitz A., Patient R., Green A.R., Ciau-Uitz A., Patient R., Green A.R., Patient R., Green A.R., Green A.R. Establishing the transcriptional programme for blood: The SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. EMBO J. 2002b;21:3039–3050. doi: 10.1093/emboj/cdf286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttgens B., Broccardo C., Sanchez M.J., Deveaux S., Murphy G., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Broccardo C., Sanchez M.J., Deveaux S., Murphy G., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Sanchez M.J., Deveaux S., Murphy G., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Deveaux S., Murphy G., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Murphy G., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Gothert J.R., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Kotsopoulou E., Kinston S., Delaney L., Piltz S., Kinston S., Delaney L., Piltz S., Delaney L., Piltz S., Piltz S., et al. The scl +18/19 stem cell enhancer is not required for hematopoiesis: Identification of a 5′ bifunctional hematopoietic-endothelial enhancer bound by Fli-1 and Elf-1. Mol. Cell. Biol. 2004;24:1870–1883. doi: 10.1128/MCB.24.5.1870-1883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs D.R., Wood W.G., Jarman A.P., Sharpe J., Lida J., Pretorius I.M., Ayyub H., Wood W.G., Jarman A.P., Sharpe J., Lida J., Pretorius I.M., Ayyub H., Jarman A.P., Sharpe J., Lida J., Pretorius I.M., Ayyub H., Sharpe J., Lida J., Pretorius I.M., Ayyub H., Lida J., Pretorius I.M., Ayyub H., Pretorius I.M., Ayyub H., Ayyub H. A major positive regulatory region located far upstream of the human α-globin gene locus. Genes & Dev. 1990;4:1588–1601. doi: 10.1101/gad.4.9.1588. [DOI] [PubMed] [Google Scholar]
- Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Diekhans M., Furey T.S., Harte R.A., Hsu F., Furey T.S., Harte R.A., Hsu F., Harte R.A., Hsu F., Hsu F., et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.R., Cheng J.F., Ventress N., Prabhakar S., Clark K., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., Cheng J.F., Ventress N., Prabhakar S., Clark K., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., Ventress N., Prabhakar S., Clark K., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., Prabhakar S., Clark K., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., Clark K., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., Anguita E., De Gobbi M., de Jong P., Rubin E., Higgs D.R., De Gobbi M., de Jong P., Rubin E., Higgs D.R., de Jong P., Rubin E., Higgs D.R., Rubin E., Higgs D.R., Higgs D.R. Annotation of cis-regulatory elements by identification, subclassification, and functional assessment of multispecies conserved sequences. Proc. Natl. Acad. Sci. 2005;102:9830–9835. doi: 10.1073/pnas.0503401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan D.A., van Heyningen V., van Heyningen V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws S.M., Hone E., Gandy S., Martins R.N., Hone E., Gandy S., Martins R.N., Gandy S., Martins R.N., Martins R.N. Expanding the association between the APOE gene and the risk of Alzheimer's disease: Possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J. Neurochem. 2003;84:1215–1236. doi: 10.1046/j.1471-4159.2003.01615.x. [DOI] [PubMed] [Google Scholar]
- Levine M., Tjian R., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Rozen S., Skaletsky H., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sabo P.J., Humbert R., Hawrylycz M., Wallace J.C., Dorschner M.O., McArthur M., Stamatoyannopoulos J.A., Humbert R., Hawrylycz M., Wallace J.C., Dorschner M.O., McArthur M., Stamatoyannopoulos J.A., Hawrylycz M., Wallace J.C., Dorschner M.O., McArthur M., Stamatoyannopoulos J.A., Wallace J.C., Dorschner M.O., McArthur M., Stamatoyannopoulos J.A., Dorschner M.O., McArthur M., Stamatoyannopoulos J.A., McArthur M., Stamatoyannopoulos J.A., Stamatoyannopoulos J.A. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries. Proc. Natl. Acad. Sci. 2004;101:4537–4542. doi: 10.1073/pnas.0400678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M., Göttgens B., Sinclair A.M., Stanley M., Begley C.G., Hunter S., Green A.R., Göttgens B., Sinclair A.M., Stanley M., Begley C.G., Hunter S., Green A.R., Sinclair A.M., Stanley M., Begley C.G., Hunter S., Green A.R., Stanley M., Begley C.G., Hunter S., Green A.R., Begley C.G., Hunter S., Green A.R., Hunter S., Green A.R., Green A.R. An SCL 3′ enhancer targets developing endothelium together with embryonic and adult haematopoietic progenitors. Development. 1999;126:3891–3904. doi: 10.1242/dev.126.17.3891. [DOI] [PubMed] [Google Scholar]
- Sanchez M.J., Bockamp E.O., Miller J., Gambardella L., Green A.R., Bockamp E.O., Miller J., Gambardella L., Green A.R., Miller J., Gambardella L., Green A.R., Gambardella L., Green A.R., Green A.R. Selective rescue of early haematopoietic progenitors in Scl−/−mice by expressing Scl under the control of a stem cell enhancer. Development. 2001;128:4815–4827. doi: 10.1242/dev.128.23.4815. [DOI] [PubMed] [Google Scholar]
- Schuler G.D. Sequence mapping by electronic PCR. Genome Res. 1997;7:541–550. doi: 10.1101/gr.7.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Clawson H., Spieth J., Hillier L.W., Richards S., Spieth J., Hillier L.W., Richards S., Hillier L.W., Richards S., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair A.M., Göttgens B., Barton L.M., Stanley M.L., Pardanaud L., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Göttgens B., Barton L.M., Stanley M.L., Pardanaud L., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Barton L.M., Stanley M.L., Pardanaud L., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Stanley M.L., Pardanaud L., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Pardanaud L., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Klaine M., Gering M., Bahn S., Sanchez M., Bench A.J., Gering M., Bahn S., Sanchez M., Bench A.J., Bahn S., Sanchez M., Bench A.J., Sanchez M., Bench A.J., Bench A.J., et al. Distinct 5′ SCL enhancers direct transcription to developing brain, spinal cord, and endothelium: Neural expression is mediated by GATA factor binding sites. Dev. Biol. 1999;209:128–142. doi: 10.1006/dbio.1999.9236. [DOI] [PubMed] [Google Scholar]
- van Wijk R., van Solinge W.W., Nerlov C., Beutler E., Gelbart T., Rijksen G., Nielsen F.C., van Solinge W.W., Nerlov C., Beutler E., Gelbart T., Rijksen G., Nielsen F.C., Nerlov C., Beutler E., Gelbart T., Rijksen G., Nielsen F.C., Beutler E., Gelbart T., Rijksen G., Nielsen F.C., Gelbart T., Rijksen G., Nielsen F.C., Rijksen G., Nielsen F.C., Nielsen F.C. Disruption of a novel regulatory element in the erythroid-specific promoter of the human PKLR gene causes severe pyruvate kinase deficiency. Blood. 2003;101:1596–1602. doi: 10.1182/blood-2002-07-2321. [DOI] [PubMed] [Google Scholar]
- Vyas P., Vickers M.A., Simmons D.L., Ayyub H., Craddock C.F., Higgs D.R., Vickers M.A., Simmons D.L., Ayyub H., Craddock C.F., Higgs D.R., Simmons D.L., Ayyub H., Craddock C.F., Higgs D.R., Ayyub H., Craddock C.F., Higgs D.R., Craddock C.F., Higgs D.R., Higgs D.R. Cis-acting sequences regulating expression of the human α-globin cluster lie within constitutively open chromatin. Cell. 1992;69:781–793. doi: 10.1016/0092-8674(92)90290-s. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Groudine M., Groudine M. Chromosomal subunits in active genes have an altered conformation. Science. 1976;193:848–856. doi: 10.1126/science.948749. [DOI] [PubMed] [Google Scholar]
- West A.G., Fraser P., Fraser P. Remote control of gene transcription. Hum. Mol. Genet. 2005;14:R101–R111. doi: 10.1093/hmg/ddi104. [DOI] [PubMed] [Google Scholar]
- Wu C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286:854–860. doi: 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- Wu X., Zhao H., Suk R., Christiani D.C., Zhao H., Suk R., Christiani D.C., Suk R., Christiani D.C., Christiani D.C. Genetic susceptibility to tobacco-related cancer. Oncogene. 2004;23:6500–6523. doi: 10.1038/sj.onc.1207811. [DOI] [PubMed] [Google Scholar]
- Yagi M., Gelinas R., Elder J.T., Peretz M., Papayannopoulou T., Stamatoyannopoulos G., Groudine M., Gelinas R., Elder J.T., Peretz M., Papayannopoulou T., Stamatoyannopoulos G., Groudine M., Elder J.T., Peretz M., Papayannopoulou T., Stamatoyannopoulos G., Groudine M., Peretz M., Papayannopoulou T., Stamatoyannopoulos G., Groudine M., Papayannopoulou T., Stamatoyannopoulos G., Groudine M., Stamatoyannopoulos G., Groudine M., Groudine M. Chromatin structure and developmental expression of the human α-globin cluster. Mol. Cell. Biol. 1986;6:1108–1116. doi: 10.1128/mcb.6.4.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]