Immuno-LCM: Laser Capture Microdissection of Immunostained Frozen Sections for mRNA Analysis (original) (raw)
Abstract
Microdissection of routinely stained or unstained frozen sections has been used successfully to obtain purified cell populations for the analysis of cell-specific gene expression patterns in primary tissues with a complex mixture of cell types. However, the precision and usefulness of microdissection is frequently limited by the difficulty to identify different cell types and structures by morphology alone. We therefore developed a rapid immunostaining procedure for frozen sections followed by laser capture microdissection (LCM) and RNA extraction, which allows targeted mRNA analysis of immunophenotypically defined cell populations. After fixation, frozen sections are immunostained under RNAse-free conditions using a rapid three-step streptavidin-biotin technique, dehydrated and immediately subjected to LCM. RNA is extracted from captured tissue, DNAse I treated, and reverse transcribed. Acetone-, methanol-, or ethanol/acetone-fixed sections give excellent immunostaining after 12 to 25 minutes total processing time. Specificity, precision, and speed of microdissection is markedly increased due to improved identification of desired (or undesired) cell types. The mRNA recovered from immunostained tissue is of high quality. Single-step PCR is able to amplify fragments of more than 600 bp from both housekeeping genes such as β-actin as well as cell-specific messages such as CD4 or CD19, using cDNA derived from less than 500 immunostained, microdissected cells. Immuno-LCM allows specific mRNA analysis of cell populations isolated according to their immunophenotype or expression of function-related antigens and significantly expands our ability to investigate gene expression in heterogeneous tissues.
The analysis of gene expression is a valuable tool for the assessment of normal or pathologically altered cell and tissue differentiation. The identification of tumor-specific gene expression patterns could lead to the establishment of genetic fingerprints of neoplasms as a versatile tool for diagnostic and prognostic purposes. However, the variable and often significant admixture of non-neoplastic cells can compromise the usefulness of mRNA extracted from bulk tumor tissue and can make the assignment of expressed genes to a specific cell population very difficult.
Microdissection from stained tissue sections and related techniques allow the isolation of morphologically identified cell populations down to the single-cell level for genetic analysis and thus can circumvent or ameliorate the problem of tissue heterogeneity. 1-8 Microdissected tissue as a source of genomic DNA was used successfully for the detection of heterozygosity in primary tumors, detection of tumor-specific genetic alterations in preneoplastic changes adjacent to invasive tumors, 9-13 and the assignment of the malignant cells of Hodgkin’s disease to the B cell lineage. 14,15 More recently, tissue fragments obtained by microdissection of routinely stained frozen sections have also been used for the isolation of mRNA amenable to reverse transcription polymerase chain reaction (RT-PCR), the generation of expression libraries, and related techniques. 8,16-20
However, routinely stained frozen sections without coverslip as necessary for microdissection show greatly reduced cellular detail, which diminishes the ability to distinguish and isolate specific cell populations from complex lesions with an intimate mixture of morphologically similar cell types, such as lymphomas or carcinomas with a diffuse growth pattern. Immunohistochemical staining of frozen sections could help to identify and isolate specific cell populations, even of identical morphology, according to their antigen expression, allowing a more precise microdissection. However, standard immunohistochemical staining protocols usually require several hours of incubation in aqueous media, resulting in significant degradation and loss of RNA through activation of tissue RNAses and other factors.
We therefore established a rapid immunostaining technique for frozen sections, which, in combination with laser capture microdissection (LCM), 3,21 allows very fast, technically easy and precise isolation of specific cell populations for mRNA analysis.
Materials and Methods
Immunohistochemistry
Snap-frozen tissue blocks of normal and neoplastic lymph node, breast, and prostate were chosen from the tissue bank of the Laboratory of Pathology, National Cancer Institute. Four- to eight-micron serial frozen sections were cut on a standard cryostat with a clean blade, and two sections each were mounted on Superfrost Plus (Fisher Scientific, Pittsburgh, PA) or poly-l-lysine-precoated (C to C Laboratory Supplies, Chicago, IL) glass slides. The unfixed sections were immediately stored at −80°C.
The frozen sections were thawed at room temperature for 30 to 60 seconds without drying and immersed immediately in one of the following fixatives: cold acetone (5 minutes), methanol (5 minutes), 4% paraformaldehyde (5 minutes), 70% ethanol (15 to 30 seconds), ethanol/formalin (3:1, 1 minute), and ethanol 70% (15 seconds) followed by acetone (5 minutes). After fixation, the slides were rinsed briefly in 1X phosphate-buffered saline (PBS), pH 7.4, and subjected to immunostaining. The immunohistochemical staining was performed with the DAKO Quick Staining kit (DAKO Corp., Carpinteria, CA), a three-step streptavidin-biotin technique with prediluted mono- or polyclonal (rabbit) primary antibodies optimized for very short staining times. The slides were incubated at room temperature with the primary and secondary antibodies and the tertiary reagent for 90 to 120 seconds each and briefly rinsed with 1X PBS between each step. After color development with diaminobenzidine (DAB) for 3 to 5 minutes and counterstaining with hematoxylin for 15 to 30 seconds, the sections were dehydrated in graded alcohols (15 seconds each) and xylene (twice for 2 minutes each) and air dried. The primary antibodies used are listed in Table 1 ▶ . For primary antibodies other than the prediluted DAKO Quick Staining kit antibodies, the dilutions were determined individually (Table 1) ▶ , and the incubation time was prolonged to 5 to 10 minutes. Placental RNAse inhibitor (Perkin Elmer, Branchburg, NJ) was added to the primary antibody and the DAB solution in a concentration of 200 to 400 U/ml. Negative controls included omission of the primary antibody and incubation with an irrelevant antiserum. One ethanol-fixed section of each run was stained with Mayer’s hematoxylin (30 seconds) and eosin (10 seconds) and dehydrated as described above. All solutions were prepared with DEPC-treated water.
Table 1.
Primary Antibodies Used in This Study
| Name/clone (CD) | Reactivity | Source/dilution |
|---|---|---|
| CLA (CD45) | Pan-leucocyte | DAKO Quickstain |
| L26 (CD20) | B cells | DAKO Quickstain |
| UCHL1 (CD45RO) | T cells | DAKO Quickstain |
| Keratin polyclonal | Cytokeratins, broad spectrum | DAKO Quickstain |
| PSA | Prostate-specific antigen | DAKO Quickstain |
| EMA | Epithelial membrane antigen | DAKO Quickstain |
| Leu1 (CD5) | T cells, B subset | Becton Dickinson (1:20) |
| KP1 (CD68) | Macrophages | DAKO (1:25) |
| MIB1 | Ki67 antigen | DAKO (1:10) |
| BerH2 (CD30) | Ki1 antigen | DAKO (1:10) |
Laser Capture Microdissection
After immunostaining and microscopic control of staining quality and tissue preservation, one of the two sections on the slide was scraped off with a clean razor blade for RNA extraction as described below. The remaining section was used for microdissection using a PixCell laser capture microscope with an infrared diode laser (Arcturus Engineering, Santa Clara, CA) as described previously with modifications. 3,21 In brief, the dehydrated tissue section is overlaid with a thermoplastic membrane mounted on optically transparent caps, and cells are captured by focal melting of the membrane through laser activation. After visual control of the completeness of dissection, the captured tissue was immersed in denaturation solution. Caps briefly placed onto the section without laser activation were used as negative controls.
RNA Extraction, DNAse Treatment and Reverse Transcription
RNA was obtained from both microdissected as well as scraped tissue sections with the Micro RNA isolation kit (Stratagene, La Jolla, CA) as described previously. 3,22 The RNA pellet was redissolved in water treated with sterile diethylpyrocarbonate and incubated with 20 U of DNAse I (GenHunter Corp., Nashville, TN) for 2 hours at 37°C. After re-extraction of RNA, reverse transcription was performed using 12 μl of RNA, 2.5 μmol/L random hexamers, 25 μmol/L dNTPs, and 100 U of MMLV reverse transcriptase (GenHunter). For each sample, a mock reaction without addition of reverse transcriptase was performed.
cDNA Amplification
The primers, their sources, and the expected sizes of the cDNA amplification products are listed in Table 2 ▶ . A total of 35 to 40 cycles of amplification were performed using either conventional PCR or hot-start PCR employing Ampli_Taq_ Gold LD (Perkin Elmer) in conjunction with the _Taq_Start antibody (Clontech, Palo Alto, CA). Appropriate negative controls including amplification of the mock RT reaction product were performed in each run. The PCR products were separated on a 2% agarose gel stained with ethidium bromide.
Table 2.
Primers Used in This Study
| Name | Product size (bp) | Co-amplification of genomic DNA | Source |
|---|---|---|---|
| Actin* | 220 | Yes | Clontech |
| Actin* | 479 | Yes | Clontech |
| Actin* | 650 | Yes | Clontech |
| Actin* | 838 | Yes | Clontech |
| Actin | 630 | No | Raff et al 27 |
| CD4 | 438 | No | Clontech |
| CD19 | 424 | No | ** |
Results
Depending on the duration of fixation, immunostaining according to our protocol required between 12 and 25 minutes from the beginning of fixation until the stained section was ready for LCM. Except for paraformaldehyde, all fixatives rendered satisfactory to excellent staining results and preservation of morphology if the sections were allowed to thaw briefly at room temperature. The quality of staining did not differ significantly between the different antibodies, and excellent staining results were also obtained with primary antibodies other than the prediluted Dako Quick Staining antibodies, with slightly longer incubation times (Table 1) ▶ . In general, the results of immunostaining with either technique were comparable or superior to conventional frozen section immunohistochemistry both in signal intensity and specificity (Figure 1) ▶ .
Figure 1.
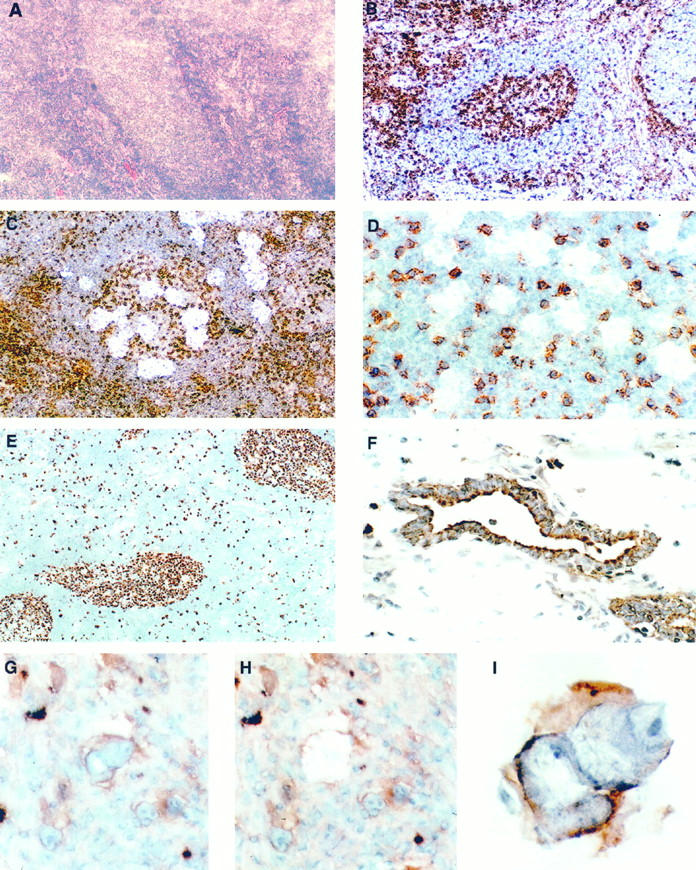
Frozen sections of a reactive lymph node with a central follicle stained with H&E (A) and immunostained for CD45RO (B to D). The identification of the follicle is difficult in the routinely stained section, whereas immunostaining highlights T cells within the germinal center and in the perifollicular area and makes architectural features more evident. Magnification, ×100. Microdissection from germinal centers with spot sizes of 50 to 60 μm (C) and 15 to 20 μm (D) in diameter as seen during LCM, without coverslip. The smaller spot size allows avoidance of interspersed T cells. Magnification, ×100 and ×400. E: Lymph node stained for the proliferation-associated antigen Ki67, using MIB1 antibody (dilution, 1:10) in conjunction with the Quickstain kit. Magnification, ×100. F: Prostate immunostained for cytokeratin. Stromal tissue has been removed by LCM, leaving behind the positively stained glands. Magnification, ×400. G to I: Microdissection of a single Reed-Sternberg cell stained for CD30 before (G) and after (H) removal and the isolated cell adherent to the membrane (I). Magnification, ×400 and ×1000.
Due to the strong contrast of the DAB staining, immunohistochemically stained frozen sections showed a significantly better optical resolution and allowed much faster and more precise identification of stained cell populations, down to the single-cell level. This was extremely valuable for complex lesions without easily distinguishable architectural features, such as benign and malignant lymphoid tissues or tissues with a strong inflammatory component.
LCM was performed successfully with all tested tissue types, and the efficiency and specificity of the capture of selected areas was virtually independent from the fixatives, antibodies, or staining procedures used (Figure 1) ▶ . The use of charged or poly-l-lysine-coated slides did not interfere with LCM, and nonspecific capture and transfer due to detachment of tissue surrounding the area adherent to the membrane was virtually absent. However, any drying of the sections before fixation prevented LCM almost completely, probably due to increased tissue adhesion to the pretreated slides.
The factors influencing recovery and quality of mRNA are listed in Table 3 ▶ . Acetone, methanol, or ethanol/acetone fixation allowed amplification of actin cDNA fragments by RT-PCR from as little as 10 shots (Figure 2) ▶ , corresponding to approximately 200 cells, whereas ethanol and paraformaldehyde fixation rendered significantly less or no amplifiable cDNA (Figure 3) ▶ . Shorter products were amplified more efficiently than longer products, but β-actin fragments of 650 bp were successfully amplified from larger amounts of microdissected tissue. No signals were observed after amplification of the negative RT control without addition of reverse transcriptase. However, reduction of the DNAse I treatment frequently led to the appearance of products of similar length with the actin primer set in the negative RT control, probably due to processed pseudogenes present in contaminating genomic DNA. In addition to these housekeeping genes, cell-specific transcripts such as CD4 and CD19 were successfully amplified from microdissected, immunostained slides from less than 1000 captured cells. Selective dissection of B cells (Figure 1D) ▶ , T cells, or nonlymphoid cells from immunostained sections yielded significantly enriched populations, resulting in predominant amplification of the mRNA specific for the targeted cell type (Figure 4) ▶ .
Table 3.
Immuno-LCM: Factors Influencing Success of Procedure
| Drying steps | Fixative | Staining time/ procedure | Counterstain | RNAse inhibition | |
|---|---|---|---|---|---|
| Morphology | ++ | +++ | + | +++ | 0 |
| Immunostain | ++ | ++ | +++ | 0 | 0 |
| Transfer | ++++ | 0 | ++ | ++ | 0 |
| RNA quality | 0 | ++++ | +++ | 0 | +++ |
Figure 2.

RT-PCR of a 479-bp β-actin fragment from microdissected tissue fragments obtained from an immunostained lymph node after acetone fixation. The estimated total number of dissected cells is indicated above the lane. Approximately 1/20 of the total cDNA volume obtained was used per reaction. Forty cycles of amplification were used.
Figure 3.

RT-PCR amplification of a 479-bp β-actin fragment from immunostained frozen sections of a reactive lymph node showing the influence of different fixatives on RNA recovery. Lane 1, molecular weight marker; lane 2, acetone; lane 3, 4% paraformaldehyde; lane 4, 70% ethanol; lane 5, formalin/ethanol; lane 6, methanol; lane 7, water control. Thirty-five cycles of amplification were used.
Figure 4.

RT-PCR amplification of β-actin (A), CD19 (B), and CD4 (C) from a reactive lymph node immunostained for CD45RO. Microdissectates were obtained randomly (lane 1), from B cell areas (lane 2), and T cell areas (lane 3). Lane 4, negative control. The weak CD4 signal in lane 2 probably results from contaminating T cells or CD4-expressing macrophages not identified on the CD45RO stain.
Discussion
The isolation of immunophenotypically identified cell populations from tissue sections for mRNA analysis significantly expands our ability to investigate gene expression patterns at the microscopic level. With immuno-LCM, cell populations of similar or identical morphology but different phenotype, such as B and T cells, can be identified and isolated from complex tissues lacking easily identifiable architectural features. Furthermore, the use of antibodies directed against proteins indicative of the functional state of the cell, such as proliferation markers, activation antigens, or secretory products makes the functional dissection of a specific cell population possible.
The feasibility of mRNA extraction from microdissected, routinely stained, or unstained frozen sections for various applications has been demonstrated by several groups, using a variety of microdissection techniques and RNA isolation protocols. 3,8,16-20 Good RNA quality was obtained with short staining times for routine histological stains, such as H&E, in conjunction with precipitating fixatives, such as ethanol or acetone, and conventional protection against RNAses. Although PCR analysis of genomic DNA has been performed successfully from microdissected, immunostained frozen as well as paraffin sections, 15,23 the long periods of tissue immersion in aqueous media required for conventional immunohistochemistry is deleterious for RNA, mainly through activation of endogenous RNAses.
Therefore, the adaptation of immunohistochemistry for immuno-LCM requires a significant departure from conventional immunostaining protocols. With our method, excellent staining results were obtained despite the significant reduction in staining time and other modifications to avoid degradation by RNAses. Although some primary antibodies requiring longer incubation times may not be suitable for immuno-LCM, our limited testing showed that the majority of primary antibodies will give good immunostaining if their concentration is adjusted accordingly. The fixatives used, the staining time, and the addition of RNAse inhibitor 24 were the most crucial factors determining RNA quality. Precipitating agents, especially acetone and methanol, proved superior to cross-linking reagents such as paraformaldehyde, which led to a significant decline in amplifiable RNA. Currently, efforts are under way in our laboratory to use low-temperature paraffin embedding in conjunction with the fixatives mentioned above to combine the superior morphology of paraffin-embedded tissue with good preservation of RNA.
For the successful performance of LCM after immunostaining, careful control of tissue adhesion to the slide is necessary to avoid detachment of sections during the staining procedure without compromising laser capture. Although pretreated glass slides were well suited for immuno-LCM, even short drying periods before fixation or storage of stained slides for several days greatly diminished tissue capture. Another critical factor is complete dehydration of the sections after immunostaining. Rapid processing of stained slides allowed complete and specific transfer of selected cells over a wide range of laser pulse energy and spot size and in a variety of tested tissues.
With the commercially available laser capture microscope used in our study, the smallest routinely attainable spot sizes were between 10 and 20 μm. A new prototype currently under development allows spot sizes of less than 5 μm, equivalent to single-cell microdissection. 21 Although the intimate intermingling of different cell types in many tissues sets limits to the specificity of LCM, especially concerning cytoplasmic RNA, the easy identification of immunostained cells allows a faster and more precise sampling procedure with sufficient enrichment of the desired cell type. The successful amplification of cell-specific transcripts with fragment sizes of more than 400 bp from less than 1000 cells without employment of more sensitive techniques, such as nested PCR, demonstrates that immuno-LCM-derived RNA can likely be used for applications such as cDNA library construction or hybridization to cDNA microarrays. 16,25 In comparison with micromanipulator-based techniques, immuno-LCM is orders of magnitude faster and allows procurement of a large number of cells in a short period of time, which increases the yield of high quality RNA and dilutes out contaminating cells. This might help to reduce artifacts introduced by the use of very small numbers of cells, such as loss of RNA through cell sectioning, contamination, or biased template amplification through a high number of cycles.
Depending on the type of RNA analysis desired, our procedure for RNA extraction might be exchanged for simpler protocols. 17,20,24,26 However, we consider the DNAse I treatment an indispensable step for obtaining DNA-free RNA. In our experience, any reduction in the time of digestion or in the concentration of the enzyme to levels recommended in the literature frequently revealed the presence of contaminating DNA in the mock RT control, underlining the necessity for stringent methods of RNA purification.
In conclusion, we demonstrate that immuno-LCM allows recovery of high quality mRNA from immunophenotypically defined cell populations in complex primary tissues. The technique is fast, easy to perform, and versatile. mRNA obtained by immuno-LCM may be used for the generation of expression libraries or the screening of cDNA arrays, thus allowing in vivo analysis of tissue-, cell-, and function-specific gene expression patterns.
Acknowledgments
The technical assistance of Dr. Lynn Sorbara and Loryn Blum is gratefully acknowledged. We thank Dr. Wing C. Chan and Dr. Robert Bonner for helpful discussions.
Footnotes
Address reprint requests to Dr. Mark Raffeld, Hematopathology Section, Laboratory of Pathology, National Cancer Institute, NIH, Building 10, Room 2N110, 9000 Rockville Pike, Bethesda MD 20892. E-mail: mraff@box-m.nih.gov.
Supported in part by a grant from the Austrian Science Funds to F. Fend (Erwin-Schroedinger Stipendium J1402 MED).
References
- 1.Whetsell L, Maw G, Nadon N, Ringer PD, Schaefer FV: Polymerase chain reaction microanalysis of tumors from stained histological slides. Oncogene 1992, 7:2355-2361 [PubMed] [Google Scholar]
- 2.Going JJ, Lamb RF: Practical histological microdissection for PCR analysis. J Pathol 1996, 179:121-124 [DOI] [PubMed] [Google Scholar]
- 3.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui R, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA: Laser capture microdissection. Science 1996, 274:998-1001 [DOI] [PubMed] [Google Scholar]
- 4.Böhm M, Wieland I, Schütze K, Rübben H: Microbeam MOMenT: non-contact laser microdissection of membrane-mounted native tissue. Am J Pathol 1997, 151:63-67 [PMC free article] [PubMed] [Google Scholar]
- 5.Shibata D, Hawes D, Li Z-H, Hernandez AM, Spruck CH, Nichols PW: Specific genetic analysis of microscopic tissue after selective ultraviolet radiation fractionation and the polymerase chain reaction. Am J Pathol 1992, 141:539-543 [PMC free article] [PubMed] [Google Scholar]
- 6.Zhuang Z, Bertheau P, Emmert-Buck MR, Liotta LA, Gnarra J, Lineham WM, Lubensky IA: A microdissection technique for archival DNA analysis of specific cell populations in lesions <1 mm in size. Am J Pathol 1995, 146:620-625 [PMC free article] [PubMed] [Google Scholar]
- 7.Küppers R, Zhao M, Hansmann ML, Rajewsky K: Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO J 1993, 12:4955-4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schütze K, Lahr G: Identification of expressed genes by laser-mediated manipulation of single cells. Nature Biotechnol 1998, 16:737-742 [DOI] [PubMed] [Google Scholar]
- 9.Radford DM, Fair K, Thompson AM, Ritter JH, Holt M, Steinbrueck T, Wallace M, Wells SA, Donis-Keller HR: Allelic loss of chromosome 17 in ductal carcinoma in situ of the breast. Cancer Res 1993, 53:2947-2950 [PubMed] [Google Scholar]
- 10.Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS: Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 1996, 274:2057-2059 [DOI] [PubMed] [Google Scholar]
- 11.Vocke CD, Pozzatti RO, Bostwick DG, Florence CD, Jennings SB, Strup SE, Duray PH, Liotta LA, Emmert-Buck MR, Lineham WM: Analysis of 99 microdissected prostate carcinomas reveals a high frequency of allelic loss on chromosome 8p12–21. Cancer Res 1996, 56:2411-2416 [PubMed] [Google Scholar]
- 12.Zhuang Z, Merino MJ, Chuaqui R, Liotta LA, Emmert-Buck MR: Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive breast cancer. Cancer Res 1995, 55:467-471 [PubMed] [Google Scholar]
- 13.Zhuang Z, Vortmeyer AO, Mark EJ, Odze R, Emmert-Buck MR, Merino MJ, Moon H, Liotta LA, Duray PH: Barrett’s esophagus: metaplastic cells with loss of heterozygosity at the APC gene locus are clonal precursors to invasive adenocarcinoma. Cancer Res 1996, 56:1961-1964 [PubMed] [Google Scholar]
- 14.Küppers R, Rajewsky K, Zhao M, Simons G, Lauman R, Fischer R, Hansmann ML: Hodgkin’s disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci USA 1994, 91:10962-10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanzler H, Küppers R, Hansmann ML, Rajewsky K: Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 1996, 184:1495-1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krizman DB, Chuaqui RF, Meltzer PS, Trent JM, Duray PH, Lineham WM, Liotta LA, Emmert-Buck MR: Construction of a representative cDNA library from prostatic intraepithelial neoplasia. Cancer Res 1996, 56:5380-5383 [PubMed] [Google Scholar]
- 17.Hiller T, Snell L, Watson P: Microdissection RT-PCR analysis of gene expression in pathologically defined frozen tissues. Biotechniques 1996, 21:38-44 [DOI] [PubMed] [Google Scholar]
- 18.Luqmani YA, Lymboura M: Subtraction hybridization cloning of RNA amplified from different cell populations microdissected from cryostat tissue sections. Anal Biochem 1994, 222:102-109 [DOI] [PubMed] [Google Scholar]
- 19.Chuaqui RF, Englert CD, Strup SE, Vocke CD, Zhuang Z, Duray P, Bostwick DG, Linehan WM, Liotta LA, Emmert-Buck MR: Identification of a novel transcript up-regulated in a clinically aggressive prostate carcinoma. Urology 1997, 50:302-307 [DOI] [PubMed] [Google Scholar]
- 20.To MD, Done SJ, Redston M, Andrulis IL: Analysis of mRNA from microdissected frozen tissue sections without RNA isolation. Am J Pathol 1998, 153:47-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S, Liotta LA: Laser capture microdissection: molecular analysis of tissue. Science 1997, 278:1481-1483 [DOI] [PubMed] [Google Scholar]
- 22.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 23.d’Amore F, Stribley JA, Ohno T, Wu G, Wickert RS, Delabie J, Hinrichs SH, Chan WC: Molecular studies on single cells harvested by micromanipulation from archival tissue sections previously stained by immunohistochemistry or nonisotopic in situ hybridization. Lab Invest 1997, 76:219-224 [PubMed] [Google Scholar]
- 24.Taylor GP, Troyer DA, Giambernardi TA, Klebe RJ: Extraction of RNA from single frozen sections. J Pathol 1998, 184:332-335 [DOI] [PubMed] [Google Scholar]
- 25.DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM: Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nature Genet 1996, 14:457-460 [DOI] [PubMed] [Google Scholar]
- 26.von Eggeling F, Ballhausen W: Freezing of isolated cells provides free mRNA for RT-PCR amplification. BioTechniques 1995, 18:408-411 [PubMed] [Google Scholar]
- 27.Raff T, van der Giet M, Endemann D, Wiederholt T, Paul M: Design and testing of β-actin primers for RT-PCR that do not co-amplify processed pseudogenes. BioTechniques 1997, 23:456-460 [DOI] [PubMed] [Google Scholar]