The discovery of signal transduction by G proteins. A personal account and an overview of the initial findings and contributions that led to our present understanding (original) (raw)
. Author manuscript; available in PMC: 2008 Apr 1.
Published in final edited form as: Biochim Biophys Acta. 2006 Oct 4;1768(4):756–771. doi: 10.1016/j.bbamem.2006.09.027
Abstract
The realization that there existed a G-protein coupled signal transduction mechanism developed gradually and was initially the result of an ill fated quest for uncovering the mechanism of action of insulin, followed by a refocused research in many laboratories, including mine, on how GTP acted to increase hormonal stimulation of adenylyl cyclase. Independent research into how light-activated rhodopsin triggers a response in photoreceptor cells of the retina and the attendant biochemical studies joined midway and, without the left hand knowing well what the right hand was doing, preceded classical G protein research in identifying the molecular players responsible for signal transduction by G proteins.
1. Introduction
Although I participated in expanding the field of transmembrane signaling during my whole scientific career, my major contributions happened at the beginning, during the mechanism of action of insulin phase. It focused on hormonal activation of adenylyl cyclase and led to the demonstration that receptors are separate molecules from the adenylyl cyclase proper – then called adenyl cyclase – that adenylyl cyclase-activating receptors acted through a transduction mechanism involving changes in adenylyl cyclase’s sensitivity to ambient Mg2+, and that there existed a role for GTP in the activation of the liver enzyme by glucagon, the binding of which was also changed by GTP. Unwittingly, this made the glucagon receptor the first G-protein coupled receptor – GPCR – to be identified on the basis of changing behavior upon addition of GTP, even though at that time the G-protein as the site of action of the GTP, was still unknown. What became the G protein was first conceptualized as a transducing function – referred to as transducer – that could or could not be executed by a molecule separate from receptor and adenylyl cyclase.
2. It all began in Martin Rodbell’s laboratory
My role began the day Martin Rodbell took me from his home to his laboratory in Building 10 of the NIH Campus in Bethesda. I had arrived the evening before from Argentina with a brand new PhD degree in Biochemistry, and Marty – as we all called him – had offered me to stay in his house until I found housing of my liking. I had come to Rodbell’s laboratory because he had recently published work on the isolation of free fat cells and their exquisite sensitivity to insulin, including that of insulin’s effect to antagonize the lipolytic actions of catabolic hormones such as epinephrine, glucagon and adrenocorticotropin, ACTH. Sutherland’s group had shown that these hormones induce lipolysis using cAMP as a second messenger (a discovery for which he was to receive the 1971 Nobel Prize in Medicine), and that in adipose tissue, insulin reduced the levels of cAMP that had been elevated by lipolytic hormones (shown for epinephrine in Fig. 1 — adapted from Butcher et al., 1966 [1]). It was Rodbell’s hypothesis that insulin antagonized lipolysis by inhibiting the adenylyl cyclase, and this would be my project in his laboratory. Knowing that I would have to measure adenylyl cyclase I had, before leaving Argentina, set up the assay used by Sutherland’s group to quantify cAMP. This assay used an enzymatic reaction cascade in which the cAMP formed by the enzyme stimulated phosphorylase-_b_-kinase kinase, the activated phosphorylase-_b_-kinase phosphorylated muscle phosphorylase b and the activated phosphorylase (phosphorylase a) converted glucose-1-phosphate to glycogen with formation of inorganic phosphate, which was visualized with a colorimetric Fiske–Subarow reaction. As sources of phosphorylase-_b_-kinase and its kinase (now known as cAMP-dependent protein kinase or PKA), I used a crude glycogen preparation isolated from rabbit skeletal muscle which I knew came with glycogen metabolizing enzymes adsorbed to it. The phosphorylase b in the assay came from a batch of rabbit muscle enzyme I had crystallized during my thesis work guided by Micheal Appleman, who was doing his postdoctoral work with Luis Leloir at the Institute for Biochemical Research “Fundación Campomar” in Buenos Aires. Leloir, who with Héctor Torres had co-signed my PhD thesis, directed the Campomar Foundation, and would later receive the 1970 Nobel Prize in Chemistry for the discovery of sugar nucleotides a few years before I had come to the Campomar laboratories. It is difficult to describe my surprise, when on that first day, Rodbell showed me a brand new assay for measuring adenylyl cyclase based on conversion of [α-32P]ATP – 3 million cpm per assay tube; purchased from ICN – to [32P]cAMP, that could be completed in an hour or so. The assay gave a blank of 150–200 cpm, a basal activity of 300–350 cpm and an ACTH-stimulated activity of 450–500 cpm. The method to isolate the [32P]cAMP was easy, fast, and vastly superior to the Sutherland assay, and consisted of a two-step separation of the reaction product from the substrate and its breakdown products. This separation was based on Gopal Krishna’s observations that cAMP was retarded on a Dowex50-H+ column, eluting after the bulk of the unconverted [32P]ATP and its hydrolytic products [32P]ADP and [32P]AMP, and that 32P-labeled products that still contaminated the [32P]cAMP in the Dowex 50 eluates could be adsorbed onto a nascent mixture of BaSO4/Zn(OH)2. The [32P] cAMP formed by the enzyme was recovered in the supernatant after a short spin in a table top clinical centrifuge and quantified by liquid scintillation counting. For the next months I worked side by side with Martin Rodbell and his technician, Ann Butler, on the final validation steps of the assay, improving conditions, reducing variability and on increasing our assay capacity to more than the 20 columns that Rodbell was handling when I first came. The NIH Machine Shop made racks that held the Dowex columns in 5 rows of 10; we purchased a Sorval GLC-1 benchtop centrifuge in which we could spin the BaSO4/Zn (OH)2 suspensions, 48 tubes at a time, and added higher levels of unlabeled ATP, theophylline, EDTA, and an ATP regenerating system to improve linearity of cAMP production as a function of time. Authentic [3H]cAMP was added at the end of the assays to monitor recovery in the BaSO4/Zn(OH)2 supernatant.
Fig. 1.
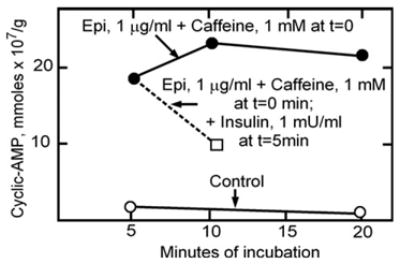
Epinephrine increases and insulin opposes and reverses the epinephrine-induced increase in cAMP levels in isolated fat pads. (Adapted from Butcher et al., 1966 [1]).
Sometimes addition of insulin appeared to reduce stimulation of the fat cell ghost adenylyl cyclase by ACTH or epinephrine, but this was highly variable.
I had arrived on a stormy January 31st, 1967. On July 1st, Rodbell left for what turned out to be a 16-month sabbatical stay in Albert Renold’s Institute of Clinical Biochemistry at the University of Geneva, to investigate other effects of insulin on fat cells. Left alone with a solid training in enzymology from my graduate student time in Leloir’s institute, and a capacity to run up to 100-tube assays per day, I continued characterizing the adenylyl cyclase from an enzymological view point. A typical day started at 9 am with the arrival of 20 male Sprague–Dawley rats from which I dissected epididymal fat pads, which were digested in a medium with collagenase, to then prepare free fat cells by passing the digested tissue through silk screen and floating the cells away from remaining debris by a short 15-s spin in the benchtop centrifuge. I then suspended the fat cells in a hypotonic medium and lysed them by gently inverting the tubes with the cells 10 times, as Rodbell had taught me. Upon lysis the cells released their fat, as well as most of their contents, resealing immediately thereafter, and yielding sacs that Martin Rodbell called “fat cell ghosts”, and I often referred to as “anemic fat cells”. They had lost about 80–90% of their cytoplasmic content, retaining nuclei, some mitochondria, and an active hormone-sensitive adenylyl cyclase system. These membrane sacs were collected as a pellet in the benchtop centrifuge.
I generated time courses at low and high ATP, and pH curves, plus and minus insulin. I assayed activity at low and high Mg2+, without and with EDTA in the assay, plus and minus insulin. I tested the effect of different monovalent cations at low and high Mg2+, in the absence and presence of EDTA or EGTA, plus and minus insulin. I always tested for basal and ACTH- and fluoride-stimulated activities. Fluoride had been shown by Sutherland’s group to stimulate adenylyl cyclase activity by a mechanism equally obscure as the one by which hormones stimulated activity, but unrelated to fluoride’s ability to inhibit ATPases that hydrolyze the substrate. I discovered that EGTA, but not EDTA, abolished stimulation by ACTH and documented a requirement for Ca2+ in the action of ACTH, but not in that of glucagon or epinephrine, the other two hormones I was often testing for their ability to stimulate adenylyl cyclase. Sporadically there seemed to be a reduction in cAMP formed in the presence of insulin. But these effects were never reproducible. I wrote weekly letters to Rodbell — by hand, on thin airmail paper. Once I thought that I had it, and wrote that definitely there seemed to be an effect of insulin if I used very low Mg2+, but that he should not tell anybody until I could confirm it better. He promptly mentioned it at that year’s Laurentian Hormone Conference – August 1967 – and it appears in the record of the discussion that followed his presentation. Marty made me a coauthor on his chapter for this contribution (Rodbell et al., 1968 [2]).
Spurred by ACTH’s requirement for Ca2+, I wondered whether ACTH was acting on the same enzyme as epinephrine and glucagon. Sutherland’s group had proposed hormone-sensitive adenylyl cyclases to be a family of single molecules, each hormone interacting with its adenylyl cyclase (Fig. 2, adapted from Robison et al., 1967 [3])1. If so, the effects of epinephrine and ACTH should have been additive. They were not. Moreover, propranolol, a beta-adrenergic receptor blocker, inhibited only the effect of epinephrine, leaving those of ACTH and glucagon untouched. It became clear that the hormone binding sites, the receptors, were separate from adenylyl cyclase, a theme we would explore in more detail after Rodbell’s return from Europe.
Fig. 2.
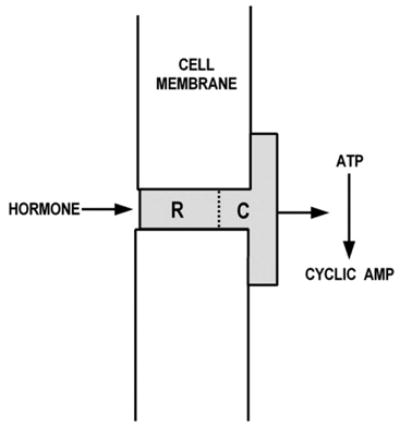
Adenylyl cyclase as an epinephrine receptor model. (Adapted from Robison et al., 1967 [3]).
The relationship between ATP and Mg ion baffled me. High ATP was inhibitory at low Mg2+ but not at high Mg2+, high Mg2+ would stimulate absolute activity more in the absence than the presence of hormone. I eventually designed a matrix experiment, in which I varied both variables simultaneously. It took me about a week to plan the experiment and to calculate mixtures in which I varied both, unlabeled and radioactive ATP, so as to economize the use of the labeled compound. Labeled ATP was expensive — $240/mCi, and I must have used my weekly allowance of 5 mCi for this single experiment. Calculations were done by hand and slide rule —the programmable Wang calculator would arrive 2 years later; Neil Armstrong had not yet set foot on the Moon. The day before the assay, I numbered the 10×75 mm test tubes I would use, which I had washed the previous day by overnight submersion into chromosulfuric cleaning solution. In fact, I washed tubes every day, leaving them drying overnight for use on the next day, while the new batch was soaking in the red-brown sulfuric acid mix for rinsing the next day. Nowadays, disposable tubes are clean and can be used directly. The day of the matrix experiment, I started very early, pipetted the reaction mixes and placed them on ice before starting the fat cell-fat cell ghost preparation. Although normally I had no access to technical help, for this experiment, I had the help of Ann Butler: I started the reactions by adding 10-μl aliquots of ghost suspension to 40 μl of pre-pipetted mix and placed the tube into the 30 °C Dubnoff shaking incubation bath, and she stopped the reaction 10 min later with a “stop” solution containing cold ATP, carrier cAMP, labeled [3H]cAMP and sodium dodecyl-sulfate, all at 15 s intervals.
The results from the matrix experiment were important (Fig. 3A). For one, they ordered and made sense of the many previous experiments where I had measured the ATP-dependence of the enzyme at one or another Mg2+ concentration, or measured the effect of varying Mg2+ at one or another concentration of ATP. Moreover, it gave me the first good insight into the fact that the mechanism by which fluoride and hormone stimulated activity should basically be the same, or if not, at least very similar. What I learned was that the dependence of activity on ATP followed pretty much Michealis–Menten kinetics with an apparent _K_m of 0.1–0.2 mM, when assayed in the absence of hormone or fluoride. In contrast, for both hormone- and fluoride-stimulated activities, the dependence on ATP showed some cooperativity and appeared not to saturate until above 3–5 mM. Hence, relative stimulations by hormone and fluoride were higher at high than at low ATP. The dependence on Mg2+ (in excess of ATP plus EDTA in the assay), was clearly cooperative for all three activities, basal, hormone- and fluoride-stimulated, with a Hill coefficient of 2 for all three, but differing in that hormone and fluoride activities were left-shifted along the Mg2+ concentration axis with respect to basal (Fig. 3B). The activation mechanism, I concluded, was one of lowering the enzyme’s requirement for Mg2+. The fact that the apparent _K_m for ATP was right shifted for hormone and fluoride-stimulated activities with respect to basal, was a puzzle for which the explanation became clear only after we had learned about the GTP effect on glucagon binding.
Fig. 3.
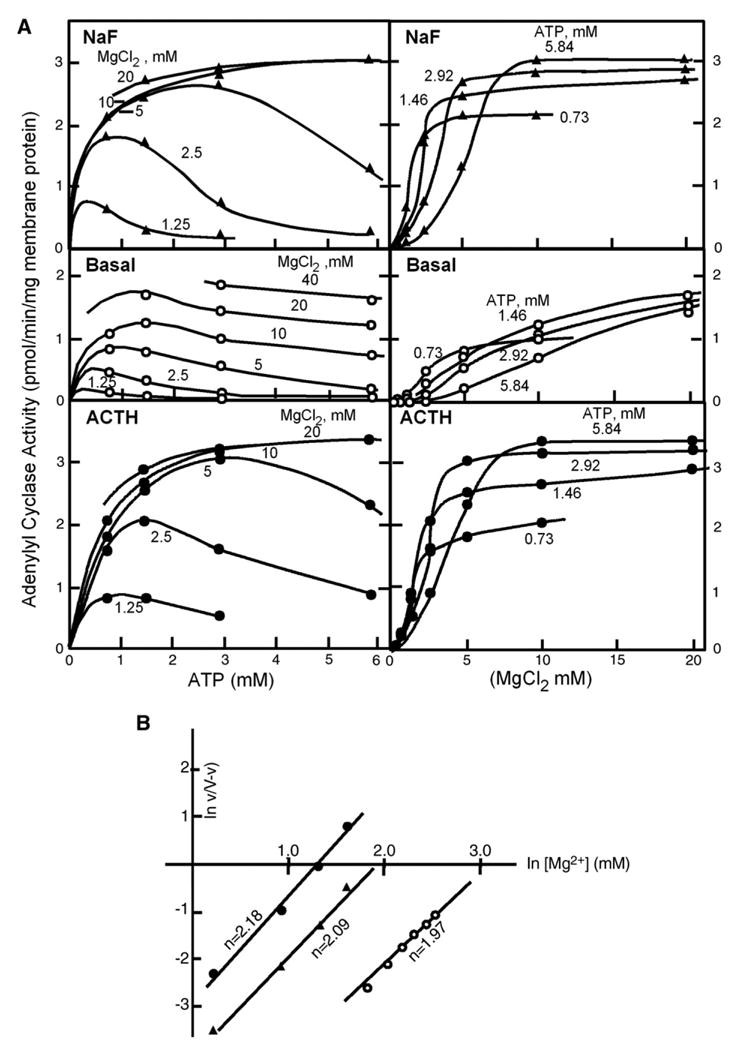
(a) The Matrix Experiment: Dependence of basal (middle panels), fluoride- (top panels), and ACTH- (bottom panels) stimulated adenylyl cyclase activities in fat cell ghosts on the concentrations of ATP (left panels) at the indicated MgCl2 concentrations, and on the concentration of MgCl2 (right panels) at the indicated concentrations of ATP. A, direct plot of data. (b) The Matrix Experiment: B, replot of the fitted curves according to Hill. (From Birnbaumer at al., 1969 [4]). ○, basal activity; ▲, fluoride-stimulated activity; ●, ACTH-stimulated activity; ACTH, adrenocorticotropin.
I did not run the full matrix experiment without and with insulin, but with the results at hand, I tested for an effect of insulin at critical points along the ATP–Mg2+ continuum, and eventually settled concluding that there was no effect. Now we know that the reduction of cAMP levels observed by Sutherland’s group had been due to activation of the phosphodiesterase — I had been looking at the wrong enzyme.
After the matrix experiment, I re-focused on the interdependence of the stimulation of the ghost adenylyl cyclase by hormones. From Mathys Staehelin at CIBA, Rodbell received an ACTH analog with four D-amino acids substituting for their corresponding L-amino acids. It proved to be a competitive inhibitor that did not affect stimulation by either glucagon or epinephrine. Later, we tested a glucagon homologue, des-His1-glucagon, given to us by Finn Sundby from the Novo Research Institute in Denmark, and found it to block the action of glucagon without affecting those of ACTH and epinephrine. It became clear to us that receptors had to be molecules that were separate from adenylyl cyclase. Fat cell adenylyl cyclase and lipolysis was also stimulated by LH, a glycoprotein hormone, and we proposed the model shown in Fig. 4: several hormone receptors affect a single adenylyl cyclase by changing its interaction with Mg2+.
Fig. 4.
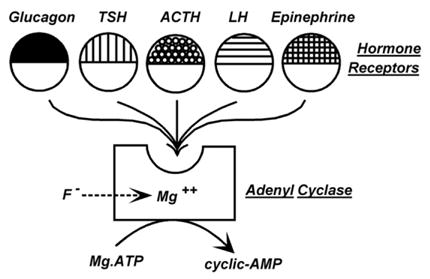
1969 conceptualization of an adenylyl cyclase system stimulated by multiple hormones, each interacting with a separate receptor having two domains: a ligand binding domain responsible for hormone specificity and a common adenylyl cyclase domain allowing each to interact with a common adenylyl cyclase where they affected the dependence of the enzyme on Mg. Fluoride is viewed as acting by bypassing the hormone receptors, but effecting the same kinetic parameter. (From Rodbell et al.,1970b [7]).
Eventually, Martin Rodbell returned from his sabbatical in Europe. This had two consequences. I had an ear for my thoughts and the size of the group expanded to include Steve Pohl, a research associate, who arrived 2 months before Marty’s return, and Michiel Krans, a Dutch fellow, who arrived a few months after Rodbell. Tom Demar joined us as well to take care of us and make sure we had clean materials to work with. We also expanded from the single 200 sq. ft laboratory in which Rodbell and Ann Butler had shown me the adenylyl cyclase assay, to a second 200-sq. ft laboratory on the 8D corridor of Building 10. Steve helped me to bring the data on the kinetics of the fat cell adenylyl cyclase to a publishable form, and in February 1969 we submitted two papers to the J. Biol. Chem., one on the fat cell enzyme properties including the matrix experiment (Birnbaumer et al, 1969 [4]), and the other on the existence of separate hormone receptors acting on a common adenylyl cyclase system (Birnbaumer and Rodbell, 1969 [5]). When secretin, purified by Victor Mutt and Erik Jorpes at the Karolinska Institute in Stockholm, was shown to be lipoplytic, we tested its effect on the fat cell adenylyl cyclase and found it too to stimulate through a receptor site distinct form those mediating the effects of epinephrine, ACTH and glucagon (Rodbell et al., 1970a [6]).
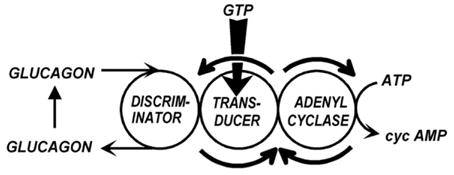
We began thinking of a transduction mechanism and of Mg2+ as an integral part of this mechanism. We began speaking of discriminators, transducers and amplifiers (Fig. 5), which now correspond to GPCRs, G-proteins and effectors, but then were just concepts that facilitated communication. The role of GTP still had to be discovered and our thoughts were more on the side of phospholipids playing key roles in transduction, as different detergents had distinct and non-linear effects (Rodbell et al., 1970b [7]).
Fig. 5.
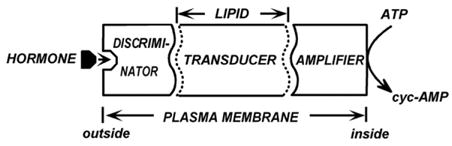
Late 1969 representation of the hormone sensitive adenylyl cyclase as a system formed of three functional elements: Discriminator, Transducer and Amplifier. (From Rodbell et al., 1970b [7]).
In spite of the beauty of the adenylyl cyclase system in fat cell ghosts, it was not very amenable to biochemical study. Fat cell ghosts could not be frozen and thawed with preservation of hormone-stimulated adenylyl cyclase activity. This required us to prepare fresh fat cells every day. The original work by Sutherland and colleagues leading to the identification of cAMP as a second messenger had been done with dog liver homogenates, in which they found that glucagon and epinephrine were able to activate phosphorylase. They had gone on to show that the hormones interacted with the particulate fraction to generate a heat stable factor – cAMP –that then caused the activation of phosphorylase in the supernatant in which the hormones were unable to promote phosphorylase activation (Berthet et al., 1957 [8]). From David Neville at the National Institute of Mental Health (NIMH), we obtained a highly purified preparation of liver plasma membranes which Steve Pohl tested for presence of epinephrine and glucagon responsive adenylyl cyclase. Even though the liver membrane adenylyl cyclase was not stimulated by epinephrine, it had a robust response to glucagon that was the same in fresh and frozen membranes (Pohl et al., 1969 [9]). We taught Tom Demar to prepare Neville membranes, as we called them, and had him make industrial quantities that went into tubes stored in liquid nitrogen. This allowed us to pull out what we needed when we needed, without having to worry about their replenishment. Steve and I began studying the properties of the liver plasma membrane system and to establish similarities and differences between liver and fat cell adenylyl cyclases (Pohl et al., 1971 [10]; Birnbaumer et al., 1971 [11]). In parallel, Rodbell with Michiel Krans began to work on labeling glucagon with 125I and to develop an assay to measure the binding of glucagon to liver membranes with the intention of characterizing the liver glucagon receptor.
At that time this was the logical next step in researching the mechanism of action of hormones, and we were not the only ones to take this course of action. In fact several studies of this type were being done quite near to us. But who was doing what, was not always known to me. I knew that Pierre Freychet, a visiting fellow from France with Jessie Roth, had labeled insulin with 125I in a laboratory around the corner of ours and was validating an insulin receptor binding assay that laid the foundations of the present understanding of insulin’s interaction with its receptor (Freychet et al., 1971a [12]; and b [13]). But I did not know that Kevin Catt, who had experience in setting up radioimmunoassays, and hence in hormone labeling techniques, was beginning to study binding of 125I-labeled hCG to testis membranes at the other end of Building 10 (De Kretser et al., 1971 [14]; Catt et al., 1971 [15]). I also did not know that Robert Lefkowitz, who worked in Ira Pastan’s laboratory and off and on used one of our two liquid scintillation counters — ‘Hi’ he would say ‘I am Bob Lefkowitz. I work in Ira Pastan’s lab. Can I use your scintillation counter tonight?’ — was working on an ACTH binding assay with adrenal membranes. Maybe this was because I was too encapsulated in my own world. This world had 1000% of my attention. Marty and Michiel, after several trials, had settled on labeling glucagon with 125I using low concentrations of chloramine T as catalyst. I set up ISCO’s preparative polyacrylamide gel electrophoresis system to which we applied the full iodination reaction, containing 1 mCi of 125I. I gave the fractions from the electrophoresis to Michiel and Marty to test for binding to liver membranes. Glucagon was very sensitive to damage by oxidation and the reaction mix had several labeled peaks, of which one bound to the membranes with the expected characteristics: specific for glucagon, as it was not prevented by excess ACTH or secretin, but was prevented by unlabeled glucagon. Moreover, the binding occurred over a concentration range similar to that over which glucagon stimulated the liver adenylyl cyclase activity (Rodbell et al., 1971a [16]). The binding assay was simple: membranes were incubated with 125I-labeled glucagon in the presence of 1% albumin, a Tris buffer, 1 mM EDTA, and additives such as unlabeled glucagon, ions, etc. The reaction was stopped by layering the reaction mixture on top of a 10% sucrose solution in a Beckman polyethylene microfuge tube, followed by a 5 min spin in the cold room — I remember Michiel rushing from lab to cold room (exactly across from the lab), starting the microfuge, hurrying back to the lab, where Marty was removing the next five samples from the incubation bath, overlaying them on sucrose and giving the tubes to Michiel for centrifugation. All done at 5 min intervals. The tube tips (bottoms) with the washed and pelleted membranes were then cut off with a scalpel and 125I retained by the membranes was quantified in a gamma ray scintillation counter.
We thought we had a receptor assay, and presented some of our results at a colloquium on “The Role of Adenyl Cyclase and 3′5′-cyclic AMP in Biology”, held in November 1969 at the Fogarty Center on the NIH Campus (Rodbell et al., 1970b [7]). At this same meeting, Bob Lefkowitz presented some of his data on preparation of iodinated ACTH and its interaction with adrenal membranes, published soon thereafter (Lefkowitz et al., 1970 [17]). Before submitting our data for publication, we still performed a time course study which however gave an unexpected result. At 5 nM of 125I-glucagon, the binding reaction took about 20 min to reach equilibrium. And, while addition of glucagon at time zero prevented 125I-glucagon from binding, post-addition at 20 min resulted in very slow release of the labeled hormone (Fig. 6). Functional studies with the same membranes in which we stimulated adenylyl cyclase activity with 4–5 nM labeled or unlabeled glucagon, a concentration that caused an activation that was about 50% of maximal, gave time courses of cAMP accumulation, taken at 30 s intervals, that extrapolated to the time of glucagon addition (Fig. 7) and did not show the lag-time predicted by the time course of “receptor” occupancy. Moreover, post-addition of des-His1-glucagon, the competitive inhibitor of glucagon’s ability to activate adenylyl cyclase, stopped the activation by glucagon with at most a 30 s delay (Fig. 7), an effect that was predicted by the binding study to be quasi irreversible (Fig. 6).
Fig. 6.
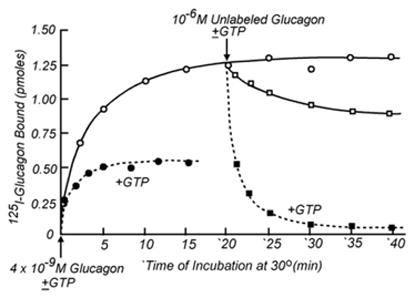
The binding of 125I-glucagon to liver plasma membranes: Effect of GTP to accelerate the interaction rates. (Adapted from Rodbell et al., 1970c).
Fig. 7.
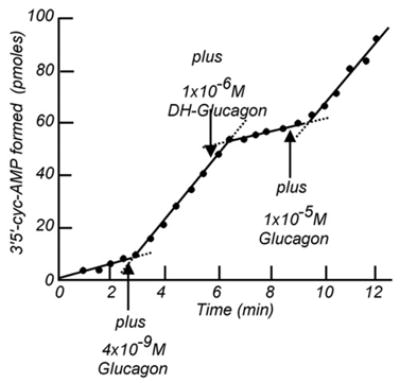
Rapid activation of liver adenylyl cyclase by glucagon and its fast reversal. (From Birnbaumer et al., 1972 [78]).
Since there was a clear difference in the incubation conditions used for measuring adenylyl cyclase and those at which we were measuring glucagon binding, we decided to carry out the binding assay under adenylyl cyclase assay conditions, i.e., in the presence of ATP, MgCl2 and the ATP regenerating system (creatine kinase and creatine phosphate). The result was again unexpected: in a 15 min binding assay, addition of adenylyl cyclase assay reagents decreased the amount of 125I-glucagon bound by about 50% (Rodbell et al., 1971b [18]). Little did we know that we had just found the “GTP effect” and that this finding would be the beginning of the “signal transduction by G proteins” era. In fact the term signal transduction did not yet exist, though no doubt it would evolve from the transducer function we were proposing to intervene between hormone binding and adenylyl cyclase activation.
Of the reagents added, we rapidly learned that it was the ATP that reduced binding. As to specificity, GTP and ATP, but not UTP or CTP, were effective, and, to our surprise, GTP was effective at 1000 times lower concentrations than ATP. Among different guanine nucleotides, the only other form able to reduce binding was GDP (Rodbell et al., 1971b [18]). Nelson Goldberg, who had developed an assay for cGMP based on its hydrolysis to 5′GMP and subsequent enzymatic conversion to GTP, and depended on the purity of ATP used to phosphorylate GMP to GTP, had recently reported that commercially available ATP was contaminated with GTP, varying from a low of 0.1% up to 5% and even 10%. It became clear therefore that I had never measured adenylyl cyclase activity without adding GTP. The possibility arose that GTP might be necessary for hormonal activation of adenylyl cyclase. This might be the reason that high levels of ATP were needed to observe robust hormonal stimulations relative to basal activity in the matrix experiment.
We split tasks. I looked for a functional correlate to the GTP effect on binding at the level of adenylyl cyclase activity, using the assay I had been using, while Marty Rodbell sought to develop an alternate substrate for adenylyl cyclase: AMP-PNP, now AMP-P(NH)P (Rodbell et al., 1971c [19]).
Rodbell synthesized [α-32P]AMP-P(NH)P from [α-32P]ATP and P(NH)P, which he obtained from Ralph Yount at Washington State University (Pulman, WA). Yount had shown that AMP-PNP could replace ATP in the allosteric regulation of the muscle actomyosin ATPase, without being a substrate for the ATPase. Rodbell’s idea was that if AMP-P(NH)P was a substrate for adenylyl cyclase he might be able to test for regulatory effects of GTP and ATP that might not be mimicked by AMP-P(NH)P.
On my side, I found what I already knew, i.e., that at very low ATP I could not obtain a robust stimulation by glucagon, and that addition of either ATP or (and this was new) GTP, led to robust stimulation of adenylyl cyclase by glucagon. Rodbell’s approach proved to be more definitive. Using an enzymatic approach based on the three-step synthesis of aminoacyl tRNAs, in which an intermediate of the form [amino acyl~tRNA~AMP] is formed from ATP and amino acid, that can decay to back to amino acid, tRNA and ATP, if excess PPi is added, he drove the intermediate to form AMP-P(NH)P by adding excess P(NH)P and made [α-32P]AMP-P(NH)P from [α-32P]ATP and P(NH)P. [α-32P]AMP-P(NH)P proved to be a substrate for adenylyl cyclase, which could now be assayed for hormonal stimulation under conditions that would not lead to transphosphorylations among GTP and ATP. Addition of glucagon still stimulated activity when added to the incubation at the same time as liver membranes. But, addition of glucagons 10–15 min after the cyclase reaction had been started, yielded no increase in activity unless GTP was also added (Fig. 8).
Fig. 8.
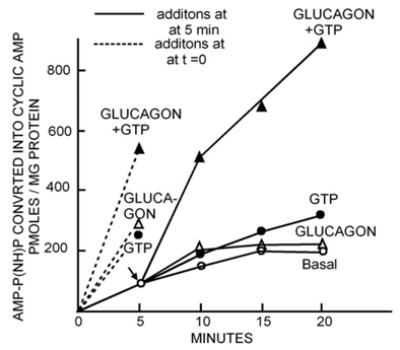
Essential role for GTP in the activation of liver adenylyl cyclase by glucagon. (From Rodbell et al., 1991c [19]).
At the level of 125I-glucagon binding, addition of GTP was found to accelerate both, dissociation and association. Binding was now reversible, which it was only very poorly in the its absence; addition of GTP to membranes with pre-bound 125I-glucagon caused it to dissociate (Fig. 6). We also determined that the reduction of 125I-glucagon-binding observed when we had added adenylyl cyclase assay reagents – now GTP –resulted from the fact that the rate of the dissociation was increased more than that of association, so that binding as a function of 125I-glucagon concentration was right shifted by a factor of about 3- to 5-fold. Accordingly, at equilibrium the occupancy of the binding site was lower in the presence of GTP than in its absence (Fig. 9)2.
Fig. 9.
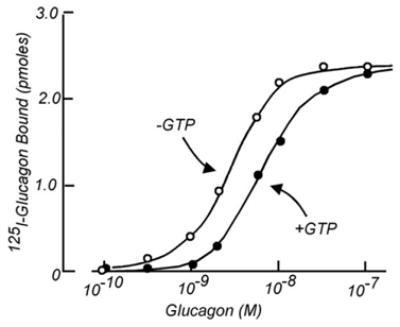
The effect of GTP to decrease the apparent affinity of the liver glucagon receptor for its ligand. (From Rodbell et al., 1971c [19]).
We speculated that actions of hormones could be regulated from within the cell and were not necessarily an obligatory consequence of simple changes in the hormone’s circulating levels. We began thinking of the transducer as being the site of action of GTP. In March 1970, we drew the model shown in Fig. 10, in which rapid binding of glucagon had a driving force operating on the transducer which then acted on adenylyl cyclase increasing its activity. Whether or not the GTP-binding transducer was a molecule separate from either receptor or adenylyl cyclase, was not addressed (Rodbell et al., 1970c [20]).
Fig. 10.
Late 1970 model of the glucagon-sensitive liver membrane adenylyl cyclase viewed as an information transfer system, incorporates the effects of GTP and ascribes a role for GTP in signal transduction.
Evidence for the existence of a separate molecule with a transducer function came from three quite independent research lines. Two were logical extensions of the line initiated by the GTP effects on glucagon binding and its role in glucagon receptor-mediated activation of the adenylyl cyclase system. The third, while prompted by the nucleotide requirement in hormonal activation of adenylyl cyclase, focused on biochemical changes occurring in rod photoreceptor cells upon illumination.
3. Paths to G proteins
By mid 1971, Steve Pohl and I had left Martin Rodbell’s laboratory. Michiel Krans had also left, returning to the University of Leiden where he had an assistant professorship waiting for him in the Division of Metabolism and Diabetes of the Department of Medicine. Bob Lefkowitz had also left the NIH to take a position at the Massachusetts General Hospital, where he began working on a beta-adrenergic receptor binding assay using [3H]norepinephrine as a ligand. After us, Rodbell filled his laboratory with a new research team, his second, as I had been his first postdoctoral fellow. Among the members of this new team was a very tall man, Martin Low, who had come from the Karolinska Institute to spend a sabbatical with Rodbell. Low took the fat cell system to the next step and prepared highly purified membranes in which the cyclase system became stable. And with stability came absence of stimulation by hormones –previously seen also by Leonard Jarett (Jarett et al., 1971 [21]) –but in contrast to Jarett, Low was now able to restore hormonal activation by addition of GTP. Changing assay conditions then showed that GTP could have both positive and negative effects – a preview of Gs and Gi (Harwood et al., 1973 [22]) – and that this applied to both hormonal as well as fluoride stimulation (Harwood and Rodbell, 1973 [23]). In my new lab, at Northwestern University in Chicago, I made similar observations about dual regulation of adenylyl cyclase, but in renal membranes where GTP and ATP improved stimulation by vasopressin, while stimulation by PGE1 was reduced by GTP (Birnbaumer, 1973 [24]). And Gopal Krishna, then an investigator in B.B. Brodie’s Biochemical Pharmacology Branch of the National Heart Institute, found that the human platelet prostaglandin-stimulated adenylyl cyclase behaved exactly the same as the rat’s liver membrane adenylyl cyclase. It required GTP or ATP to show stimulation by PGE1 when, by now commercially available, [α-32P]AMP-P(NH)P was used as substrate (Krishna et al., 1972 [25]). This in fact was the first of many reports on a role for GTP or ATP in hormonal stimulation or inhibition of adenylyl cyclase.
4. Activation of adenylyl cyclase by hormones involves a GDP/GTP nucleotide exchange reaction and its deactivation is coupled to GTP hydrolysis: the hormone-stimulated GTPase
As interesting as these results may have been, the next breakthrough in our – the field’s – way of thinking came from two different sides. One was Rodbell’s invention in 1974, of GMP-P(NH)P, (or “_gimpimp_” as he called it). This non-hydolyzable GTP analogue proved to be a “super-stimulator” of adenylyl cyclases (Londos et al., 1974 [26]): while GTP itself increased basal activity only minimally, compared to its effect on hormonal stimulation, GMP-P(NH)P was by itself as effective in stimulating activity as the combination of hormone and GTP. But instead of being a rapid activator of adenylyl cyclase, activation by GMP-P(NH)P exhibited a lag. This lag was abolished by addition of hormone, and the resulting activation became irreversible — resistant to washing (Schramm and Rodbell, 1975 [27]). In turkey erythrocytes, this lag was so extended that in a standard 10 min incubation, GMP-P(NH)P showed essentially no stimulatory effect unless hormone (isoproterenol) was added. Clearly hormones were accelerating the rate at which GMP-P(NH)P acted.
Could it be that instead of a requirement for GTP in the action of hormones, there was a requirement for hormone in the action of GTP? The importance of this question is that it shifted the emphasis from asking how hormone acted to how GTP acted. GMP-P(NH)P had become an essential tool to address this question. In his article on GMP-P(NH)P being a “super” GTP, Rodbell raised the question whether the persistence of the activated state of the adenylyl cyclase might be related to GMP-P(NH)P not being susceptible to hydrolysis and speculated that the faster action and lower effect of GTP on basal activity might imply the existence of a GTPase closely associated with adenylyl cyclase (Londos et al., 1974 [26]).
The next advance in our understanding of the hormone-stimulated adenylyl cyclase came from the finding in Zvi Selinger’s laboratory of a hormone stimulated GTPase. Working with turkey erythrocyte membranes, Selinger and his graduate student, Dan Cassel, set out to test Rodbell’s GTPase idea and discovered an isoproterenol-stimulated, low _K_m GTPase (Cassel and Selinger, 1976 [28]). Activation of the GTPase was independent of adenylyl cyclase activity, as stimulation of the GTPase by isoproterenol was unaffected in membranes in which adenylyl cyclase had been inactivated with p-chloromercuribenzoate. Cassel and Selinger also studied the mechanism by which cholera toxin activated adenylyl cyclase. Adenylyl cyclase activation by cholera toxin had been reported by Geoffry Sharp and Sixtus Hyenie (Sharp and Hyenie, 1971 [29]), who in turn had followed up observations by Martha Vaughan and William Greenough that choleragen activated lipolysis – a cAMP stimulated process – in fat cells (Vaughan et al., 1970 [30]), and by Nelson Goldberg that cholera toxin elevated cAMP levels in intestinal mucosa (Schafer et al., 1970 [31]), where cAMP promoted water secretion. Cassel and Selinger confirmed that cholera toxin activated adenylyl cyclase, and found that cyclase activation went hand in hand with loss of hormone-stimulated GTPase activity (Fig. 11). It appeared that cholera toxin by inhibiting GTP hydrolysis, converted this nucleotide in a super-stimulator akin to GMP-P (NH)P. They proposed a mechanism in which adenylyl cyclase existed in two forms, an active form with GTP bound to it, and an inactive form resulting from hydrolysis of GTP to GDP plus Pi. Activation by cholera toxin was viewed as the result of inhibiting the active to inactive conversion — the GTPase reaction. Hormone [receptor] was initially viewed as stimulating the binding of GTP which led to the inactive to active adenylyl cyclase transition (Cassel and Selinger, 1977 [32]). In another study, Cassel and Selinger showed that the adenylyl cyclase, irreversibly activated by incubation with GMP-P(NH)P and isoproterenol, could be deactivated by a second incubation with isoproterenol and GMP, and that this correlated with release of prebound [3H]GMP-P(NH)P (Cassel and Selinger, 1978 [33]). Moreover, incubation of turkey membranes with [3H]GTP and isoproterenol, followed by washing, resulted in the retention of [3H]GDP, which could be released by further incubation with isoproterenol (Cassel and Selinger, 1978 [33]). Thus, Cassel and Selinger were the first to completely grasp and propose that the mechanism by which hormones activate adenylyl cyclase involves the activation of a nucleotide exchange reaction that causes the release of GDP and the binding of GTP (Fig. 12). The hormone-stimulated GTPase activity is therefore merely the reflection of the increased cycling rate between inactive and active states. The confirmation that Cassel and Selinger’s model was correct took 5 years, required reconstitution of the hormone-stimulated GTPase from separate components and involved an independent research team, working on another continent with a different set of tools, including the purified, yet to be discovered regulatory component of adenylyl cyclase, now known as Gs (Brandt et al., 1983 [34]).
Fig. 11.
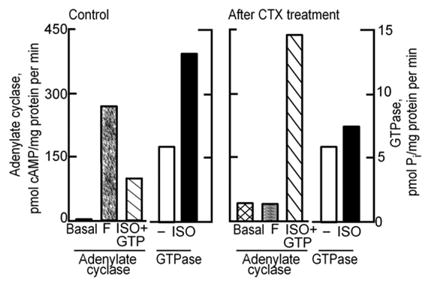
Cholera toxin treatment results in activation of adenylyl cyclase while at the same time it reduces hormone-stimulated GTPase in turkey erythrocyte membranes. (From Cassel and Selinger, 1977 [32]).
Fig. 12.
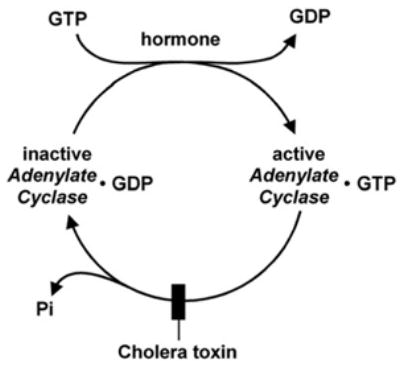
Cassel and Selinger’s 1978 model of the mechanism of hormonal activation of adenylyl cyclase incorporating the role of hormone [receptor] as a nucleotide exchange factor and an intrinsic GTPase responsible for deactivation of the system, and the effect of cholera toxin to activate the system by blocking the GTPase, the adenylyl cyclase turn-off reaction. (From Cassel and Selinger, 1978 [33]).
5. The catalytic activity and the regulatory guanine nucleotide binding site of adenylyl cyclase reside on separate molecules
The introduction of GMP-P(NH)P as a potent activator and stabilizer of adenylyl cyclase activity led to the testing of other modified GTP analogues, notably GMP-P(CH2)P and GTPγS, of which GTPγS proved to be equivalent to GMP-P(NH)P. The stabilizing effect prompted the German biochemist Thomas Pfeuffer in Wuerzburg to attempt the solubilization and purification of the activated enzyme, following both its enzymatic activity and its guanine nucleotide binding activity (Pfeuffer and Helmreich, 1975 [35]; Pfeuffer and Eckstein, 1976 [36]). These studies revealed that the binding component and the enzyme could be separated either by differential extraction, or by size fractionation techniques (Pfeuffer and Helmreich, 1975 [35]). Eventually these studies led Pfeuffer to develop a GTP-affinity matrix, to which GTP was covalently bound by a phenylenediamine bridge attaching to GTP’s γ-phosphate. After membrane solubilization this affinity matrix retained the GMP-P(NH)P-binding component suspected of being responsible for adenylyl cyclase activation, allowing adenylyl cyclase to flow through. Cyclase activity in the flow through had a much diminished response to GMP-P(NH)P or fluoride and washes with GTP had no measurable adenylyl cyclase activity, but, when combined with the flow through, restored the ability of fluoride to stimulate activity (Pfeuffer, 1977 [37]). Thus, the GTP-binding component appeared to also be the component responsible for fluoride stimulation (Fig. 13). Polyacrylamide gel electrophoresis showed the GTP binding component to have an apparent molecular weight of 42,000 Da.
Fig. 13.
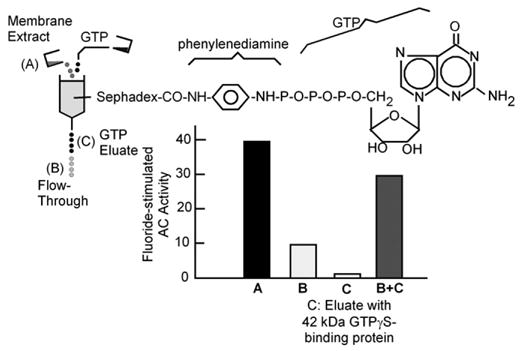
A GTP binding protein mediates activation of adenylyl cyclase by fluoride. (Adapted from Pfeuffer, 1977 [37]).
By an interesting coincidence the 1977 Oct 25 issue of the Journal of Biological Chemistry carried two landmark papers on the same subject. One was Thomas Pfuffer’s full length article on the reconstitution of fluoride-stimulated activity by a guanine nucleotide-binding component (Pfeuffer, 1977 [37]). The other was a short communication by Elliott Ross and Alfred Gilman, then at the University of Virginia, Charlottesville, on the resolution of a hormone and fluoride stimulated adenylyl cyclase system into an enzyme proper and a stimulatory regulatory component mediating the activation by guanine nucleotides and fluoride (Ross and Gilman, 1977 [38]). This discovery would later earn Alfred Gilman the sharing with Martin Rodbell of the 1994 Nobel Prize in Medicine for the discovery of the G protein mediated signal transducing systems.
Ross and Gilman’s discovery had been made possible by the creation in 1975 by Gordon Tompkins’ research group at UCSF in California, of a S49 mouse thymoma cell mutant resistant to the killing (i.e., apoptotic) effect of cAMP, in which no adenylyl cyclase activity could be measured (Bourne et al., 1975 [39]). Then referred to as AC−, the cell turned into the Rosetta Stone of adenylyl cyclase research as it was to evolve in subsequent years. What Ross and Gilman showed was that a detergent extract of wild type S49 cell membranes, that had been heated to inactivate all measurable adenylyl cyclase activity, was able to reconstitute hormone- and fluoride-stimulated adenylyl cyclase activities in AC− cell membranes. Careful examination of AC− membranes for adenylyl cyclase activity showed that they were, in fact, not negative for adenylyl cyclase activity. When assayed with Mn2+ as the divalent cation, AC− membranes still catalyzed conversion of ATP to cAMP, albeit at a much reduced rate, which with Mg2+ was so low that it had been missed. The AC− adenylyl cyclase was unresponsive to hormone, GMP-P (NH)P or fluoride. There was no cyclase activity in the heat-treated wild type S49 cell membrane extract, which therefore contributed the missing, more heat stable, regulatory component, that conferred hormonal, nucleotide and fluoride regulation to the system (Ross and Gilman, 1977 [38]).
As published in 1980, Gilman’s laboratory had gone on to use the reconstitution of fluoride-stimulated adenylyl cyclase activity in AC− membranes as an assay to purify the GTP-binding regulatory component of adenylyl cyclase, thus defining in molecular terms what is today referred to as signal transduction by G proteins (Northup et al., 1980 [40]).
6. Cholera and pertussis toxins are covalent modifiers of two regulatory components of adenylyl cyclase
In 1975, the in vitro adenylyl cyclase stimulating activity of cholera toxin (CTX), an A1B5 heterohexamer, was shown by Michael Gill at Harvard to exert its intracellular action via the 29 kDa A subunit and to require NAD+ as a cofactor (Gill, 1975 [41]). The entry of the A subunit into cells had been shown to depend on the attachment of the B pentamer to cell surface gangliosides. By analogy to diphtheria toxin’s action on elongation factor 2, Michael Gill proposed CTX to act as an ADP-ribosyltransferase that covalently modified, in this case a component of adenylyl cyclase. This component was identified as the GTP-binding regulatory component in two 1978 papers, one by Cassel and Pfeuffer, the other by Gill and Meren. (Cassel and Pfeuffer, 1978 [42]; Gill and Meren, 1978 [43]). Using [32P-AMP]NAD+ as an [α-32P]ADP-ribose donor, both laboratories identified the regulatory component as a 42,000 Da protein in SDS-PAGE gels. The regulatory component of adenylyl cyclase purified in 1980 in Gilman’s laboratory from rabbit liver membranes, contained three polypeptides of which two, one of Mr=45,000, the other of Mr=52,000, were ADP-ribosylated by cholera toxin; the third, of Mr=35,000, became the β subunit. The purified protein conferred responsiveness to hormonal, guanine nucleotide and fluoride stimulation to the adenylyl cyclase in membranes of the AC− variant of S49 cells, meanwhile renamed to cyc− S49 cells. Activation of the α β dimer by [35S]GTPγS was then shown to involve dissociation into α-[35S]GTPγS+β, and the α-GTPγS to be the bearer of the adenylyl cyclase stimulating activity (Northup et al., 1983a [44]; 1983b [45]).
Although by 1981 it was well known that adenylyl cyclases were under dual stimulatory and inhibitory control by hormones and that both types of regulation required GTP, the mechanism by which inhibition came about was not clear. Was there a second, inhibitory regulatory component or might inhibitory hormones act on the equivalent of a GTPase-activating protein, a GAP, that reduced the activity of the same process that stimulated activity? The answer to this dilemma came from Michio Ui at the University of Tokyo. Ui was interested in the mechanism by which vaccination against whooping cough with the attenuated etiologic agent of whooping cough, Bordetella pertussis, sporadically elicited severe hypoglycemic responses, sometimes causing death by convulsions. Michio Ui’s studies led him to conclude that pertussis vaccines sometimes came with a residual amount of toxin which provoked massive insulin release with attendant hypoglycemic convulsions, and that the toxin worked by a mechanism not unlike that of phentolamine, an α-adrenergic receptor blocker, which, when infused in humans with elevated adrenaline levels, suppressed an inhibitory α2-adrenergic regulation that is part of the normal control of insulin secretion (Porte, 1967 [46]). Ui and collaborators referred to the toxin, now pertussis toxin or PTX, as “islet activating protein” or IAP. They purified it to homogeneity and determined its subunit composition to be of the A1B5 type (published in 1982: Tanura et al., 1982 [47]; Fig. 14). With his student Toshiaki Katada, Ui then showed that IAP reversed inhibitory effects of epinephrine on insulin secretion from isolated islets and that membrane adenylyl cyclase from IAP treated islets had much diminished inhibitory response to epinephrine acting via an α2-adrenergic- and GTP-dependent mechanism (Katada and Ui, 1980 [48]; 1981 [49]). These authors also showed that IAP was an ADP-ribosyl transferase labeling a membrane protein of Mr=40,000 (Katada and Ui, 1982 [50]).
Fig. 14.
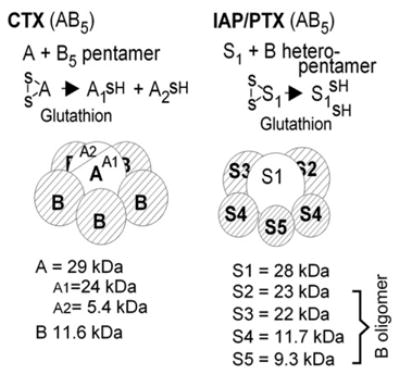
Sructural features of cholera and pertussis toxins. The ADP-ribosyltransferase activity of the A1 (CTX) and S1 (PTX) subunits requires reduction of disulfide bonds by cytosolic glutathione.
Katatda and Ui’s discovery that IAP ADP-ribosylated a distinct 40 kDa protein (Katada and Ui, 1982 [50]), was a key finding that clarified in my mind the mechanisms by which adenylyl cyclase was inhibited by hormones. Hormones inhibited adenylyl cyclase using a GTP-dependent mechanism and IAP had been shown to inhibit negative regulation of adenylyl cyclases. There had to be a Gi! In my laboratory at Baylor College of Medicine in Houston, I had also been working on the purification of the regulatory component of adenylyl cyclase with the help of three postdoctoral fellows: Juan Codina, John Hildebrandt and Ravi Iyengar. We used the general scheme published by Gilman’s laboratory for the purification of rabbit liver Gs, with the exception that we did not stabilize the system with Mg2+- and fluoride-containing buffers, and started from human erythrocytes instead of rabbit liver membranes. At the time of Katada’s and Ui’s publication of the ADP-ribosylation of a 40 kDa protein, we had been staring at an almost pure preparation of Gs (then called Ns by us), which in SDS-PAGE gels had the requisite 42 and 35 kDa bands (human erythrocytes do not have the 52 kDa band), but also an additional 40 kDa band, that by hydroxylapatite chromatography (the last step in Gilman’s purification scheme) was resolved into two fractions, each containing two bands. One fraction had bands of 42 and 35 kDa (Gs), the other fraction had bands of 40k and 35 kDa (??) by hydroxylapatite chromatography, the last step in Gliman’s purification scheme. Independently, John Hildebrandt had established that cyc− membrane adenylyl cyclase, while unresponsive to stimulation by fluoride, was under inhibitory regulation by fluoride — present also in wild type S49 cell membranes.
IAP was not commercially available. I wrote to Ui asking for IAP to test for presence of the 40 kDa protein in cyc− membranes. If he sent it, we would also test the purified fractions for presence of a PTX substrate — possibly the 40 kDa band between the 42 and 35 kDa bands?3. To improve our chances in obtaining the toxin, Ravi Iyengar sent a separate letter to the pharmaceutical company that had purified IAP for Ui. Ui responded that the protein was not pure enough for distribution and that he would be pleased to send IAP to qualified investigators such as me, once he was satisfied with the product. And, he added, his letter should also be taken as an answer to Ravi’s letter to his collaborator in the pharmaceutical company. I next got in touch with Jan Wolff, at the NIH, who had reported at a meeting that he had tested the effect of PTX on inhibitory regulation of TSH-stimulated adenylyl cyclase in thyroid membranes. PTX was more precious than gold and platinum, and he could not give me any. His source had been Richard Manclark, from the FDA laboratories on the NIH campus in Bethesda. I received no reply to my letter to Manclark. I thus, made an appointment to visit him in Bethesda, and flew from Houston with 15 test tubes into which John Hildebrandt had aliquoted wild type and cyc− S49 cell membranes and samples of the Gs purification before and after the hydroxylapatite step, together with reagents for ADP-ribosylation including [32P]NAD+, ready to incubate with the toxin. I left the rack with the pipetted tubes on ice in a styrofoam container, on the floor outside of Manclark’s office, before entering to talk him into collaborating. Richard Manclark turned out to be a pleasant man. He had with him a young collaborator, Ron Sekura, who purified PTX in his laboratory and had worked on the A1B5 subunit makeup of the toxin. At one point Ron said “what we should do, is to see if we can ADP-ribosylate cyc− membranes.” To which I replied “Funny you should say this, because I happen to have…” That afternoon Ron added PTX to the tubes, incubated and then froze the tubes. I flew back to Houston, the styrofoam container, now with dry ice, in the cabin in the overhead compartment. John Hildebrandt, precipitated the proteins, took them up into sample buffer and subjected them to SDS-PAGE. He dried the gel, and exposed it to an X-ray film. The very first film we got, we sent to our new collaborators in Bethesda. It showed that cyc− membranes –which lacked cholera toxin substrate – had the 40 kDa PTX substrate, and that the 40 kDa band that had co-purified with Gs was a PTX substrate. If Gs was an αβ dimer, Gi (then Ni) also had to be an αβ dimer. We published that stimulation and inhibition of adenylyl cyclase were mediated by distinct proteins in Nature (Hildebrandt et al., 1983 [51]), and I asked Ed Krebs in Seattle to communicate our identification of Ni as an αβ dimer to the Proceedings of the National Academy of Sciences. We thought that we had caught up with Al Gilman, who had dominated adenylyl cyclase research field since the late 70 s, and were going to beat him. Fat chance, while I tried to get some IAP from Michio Ui, he had recruited Toshiaki Katada, Ui’s student, to work on the purification of the IAP substrate and had an unlimited supply of IAP. Ed Krebs forwarded our paper to the Proceedings office on March 12, 1983 (Codina at al., 1983 [52]). Katada and Gilman’s paper came out in print in the February 28, 1983, issue of the J. Biol. Chem., 5 months ahead of ours (Bokoch et al., 1983 [53]).
7. Paths leading to the discovery and identification of the light activated GTPase as a signal transducing G protein: visual signal transduction
In 1971, upon measuring adenylyl cyclase activity in light and dark adapted retinas, Mark Bitensky, then at Yale, made the interesting and important observation that the activity in dark adapted retinas was ca. 10-fold higher than in retinas prepared under normal illumination or in dark adapted retinas illuminated prior to the adenylyl cyclase assay. (Bitensky et al., 1971 [54]; Miller et al., 1971 [55]; Bitensky et al., 1972 [56]). He also found that the activity in dark adapted retinas could be inhibited by light also in homogenates, but required the integrity of the rod outer segment (ROS) discs known to carry the visual pigment rhodopsin responsible for initiating the visual signal transduction process. Light-induced inhibition of adenylyl cyclase activity was not observed in homogenate of cone photoreceptor cells, which are not organized in discs but in stacked plasma membrane infoldings, and was lost upon disruption of the rod cell discs with digitonin. Importantly, light induced inhibition of disc adenylyl cyclase under cell-free conditions required addition of ATP or GTP, with GTP being ca. 1000-fold more potent than ATP.
By curious coincidence it turned out that, as I had done when looking for an effect of insulin by measuring adenylyl cyclase instead of phosphodiesterase (PDE), also Bitensky was focusing on the wrong enzyme, because light and its transduction mechanism does not operate on adenylyl cyclase, but on a soon to be discovered ROS PDE. It is to Mark’s credit that he discovered his own error, which became clear when he tested for the linearity of cAMP accumulation as a function of time in dark (high activity) and light (low activity) adapted retinas. While accumulation of cAMP was linear with time in homogenates of dark adapted retinas, in illuminated retinas, 90 +% of the cAMP that was being formed was being hydrolyzed by a very potent light activated PDE (Miki et al., 1973 [57]). Direct PDE assays of homogenized retinas showed the enzyme to be activated by light in an ATP or GTP dependent manner, by the same light spectrum that is absorbed by rhodopsin. At physiological cyclic nucleotide concentrations, the retinal PDE preferred cGMP over cAMP by a ratio of 10:1 (Miki et al., 1973 [57]) and was thus different from the then well known brain PDE. Photoreceptor PDE also differed from the brain enzyme in that it was insensitive to a calcium dependent regulator (CDR, now calmodulin). The ROS PDE was found to be tightly bound to bleached rod discs in the presence of Mg2+, but to be released upon Mg2+ removal in low ionic strength buffer. Differential extraction allowed the enzyme to be easily purified to homogeneity. Although, the pure photoreceptor PDE, (an αβ heterodimer), was no longer activated by light (Miki et al., 1975 [58]), it could be activated if mixed with as little as 2% bleached ROS discs. Photoactivation of rhodopsin was then shown to generate an activator, that could be detected in the purified PDE assay (Keirns et al., 1975 [59]). This activator was heat labile, inactivated by p-chloromercuribenzoate, and resistant to mild trypsin and phospholipase C. Its activity required GTP and it hydrolyzed GTP. It was a light activated GTPase (Wheeler et al, 1977 [60]; Wheeler and Bitensky, 1977 [61]).
Addition of GTP to discs from which the PDE had been removed led to separation of the light activated GTPase from the disc membranes. In 1979, the light activated GTPase became the first G protein to be purified. Originally thought to be an αβ heterodimer with subunits of 41 and 37 kDa (Godchaux and Zimmerman, 1979 [62]), it was later found to be a trimer containing in addition a small ca. 10 kDa subunit that stains poorly with Coomassie blue (Kuhn, 1980 [63]; Baehr et al., 1981 [64]). In their article on the purification of the light-activated GTPase, Godchaux and Zimmerman also proposed bleached membranes (activated rhodopsin) to act as nucleotide exchangers, as Cassel and Selinger had done for the erythrocyte adenylyl cyclase-stimulating β-adrenergic receptor (Cassel and Selinger, 1978 [33]), identifying the GTPase as the target. The light-activated GTPase could also be released from dark-adapted discs by washing just with 1 mM EDTA and 1 mM DTT (Shinozawa et al., 1980 [65]). When applied to a DEAE-Sepharose column, this preparation could be separated into two fractions, G and H, neither of which exhibited GTPase activity, but which when combined hydrolyzed GTP in the presence of photoactivated ROS discs. The G fraction had the GTP-binding and PDE-activating activity; the H fraction (now βγ) stood for helper, since it was required for interaction with rhodopsin in bleached discs from which PDE and GTPase had been washed off (Shinozawa et al., 1980 [65]). These and other experiments (Shinozawa and Bitensky, 1981 [66]; Godchaux and Zimmerman, 1979 [62]; Fung et al., 1981 [67]) led Lubert Stryer, to propose the “flow of information” model of the light-triggered cyclic nucleotide cascade of vision, shown in Fig. 15 (Fung et al., 1981 [67]). In this model, which still stands today, the GTPase is the transducing molecule that takes the information from the activated photoreceptor, and, upon binding GTP acquires PDE-activating activity. The activated PDE then hydrolyzes cGMP, reducing its concentration to levels below those required to keep the cGMP-gated cation channel open, with the ensuing turn-off of the dark current and concomitant loss of glutamate secreting activity. In due time, set by the _k_cat intrinsic to the GTPase molecule, it hydrolyzes the bound GTP. This deactivates the PDE-activating function, with attendant accumulation of newly synthesized cGMP and re-activation of the ligand-gated ion channel. The GTPase, a veritable transducer of the light signal into an electrical signal, is now poised to be activated once more by photoactivated rhodopsin. Aptly, Stryer coined the term “transducin” for the light activated GTPase, a name which, like the model, also still stands today. It would later be shown that the visual PDE is an αβγ2 heterotetramer, in which the γ subunits are inhibitory, and that the mechanism by which the α subunit of transducin complexed to GTP (Tα.GTP) activates the PDE is by removal of inhibitory γ subunits from the PDE by formation of Tα. GTP::γ complexes (Deterre et al., 1988 [68]). Hydrolysis to GDP releases γ from Tα, which now can reassociate with the αβ PDE to reform αβγ2, quenching its activity.
Fig. 15.
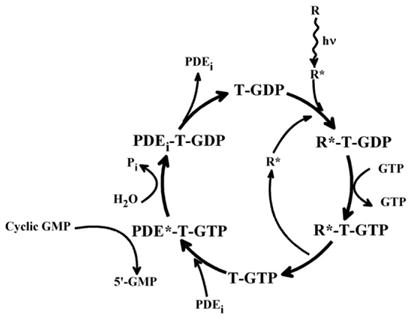
Flow of information in a G protein coupled system as deduced in studies of the light activated rod outer segment cGMP specific phosphodiesterase and summarized by Stryer and collaborators (From Fung et al., 1981 [67]). T, transducin.
8. Gs and Gi are also heterotrimers
Although in retrospect the similarity between the visual and hormonal signal transduction systems should have been obvious, to more than one of us it was not. The fact that Gs and Gi were published to be dimers by both Gilman’s group and ours is proof that we were not looking at the light activated GTPase as a homologue of Gs and Gi.
As Gilman and collaborators had done, we also determined the molecular weight of Gs and Gi. The proteins had been obtained in a buffer containing the non-ionic detergent Lubrol PX and determination of the molecular weight of the protein component of the detergent–protein complex was a complex process requiring careful determination of sedimentation coefficients in normal and deuterated water (H2O and D2O) and several assumptions. G/F was reported by Gilman’s laboratory to have a protein mass of 70 kDa, which fell to 50 kDa upon activation with either a non-hydrolyzable GTP analogue or NaF, a reflection of the dissociation of α β into α* plus β (Sternweis et al., 1981 [69]). Our measurements, by the same techniques and with the same assumptions, yielded a protein mass of 95.5 kDa (Codina et al., 1984 [70]). Thinking that perhaps Gilman’s group had lost something during the purification, we looked carefully at our Coomassie blue stained gels and noted that we had something running with the dye front. This turned out to be the γ subunit, present in the Ns (Gs) and Ni (Gi) preparations, as well as in what we had thought to be free β (Hildebrandt et al., 1984 [71], Fig. 16A). With the discovery of the γ subunits we drew the model of hormone-stimulated adenylyl cyclase shown in Fig. 16B (Hildebrandt et al., 1984 [71]). Eventually, Gilman’s group also found γ at the dye front of their gels. The differing Mr estimates were simply a reflection of the wide error intrinsic to the methodology used. Fig. 17 presents a dynamic view of G proteins incorporating the receptor-activated GDP–GTP exchange reaction, the subunit dissociation reaction, the GTPase activity and the re-assembly of the trimer upon GTP hydrolysis, ready for reactivation by the receptor. With H denoting hormone or agonist, the reaction scheme incorporates my bias as to which the likely reaction products may be when GTP binds to the HR::αβγ. Alternative dissociation–reassociation paths were discussed earlier (Birnbaumer, 1993 [72]) and are still under study.
Fig. 16.
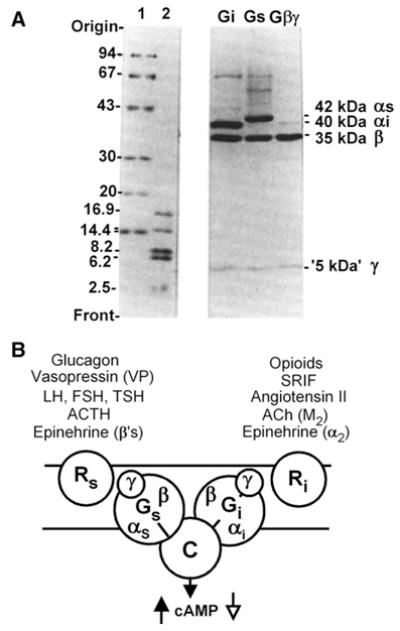
A, SDS-PAGE analysis of human erythrocyte Gs and Gi proteins showing αβγ subunit composition. Lanes 1 and 2, molecular weight standards B, 1984 model of adenylyl cyclase susceptible to both stimulatory and inhibitory inputs mediated by respective stimulatory (Gs) and inhibitory (Gi) regulatory components. Each G protein is shown as a heterotrimer and under the control of a distinct set of receptors responsible for distinct hormonal specificities exhibited by adenylyl cyclases from different tissues and cells (Adapted from Hildebrandt et al., 1984 [67]).
Fig. 17.
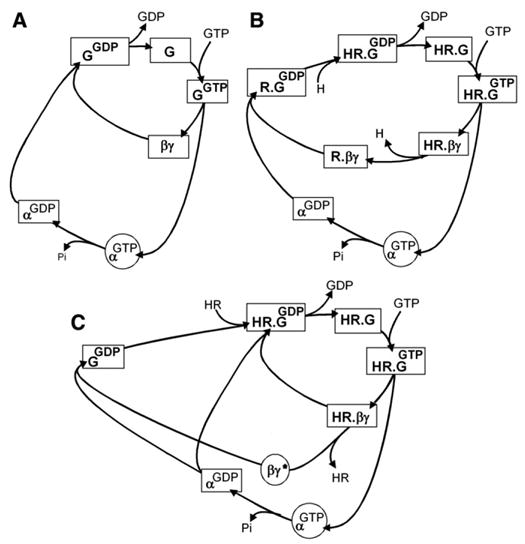
The double regulatory cycle of a G protein: activation involves GTP binding and subunit dissociation; deactivation involves GTP hydrolysis and subunit reassociation. (A) In the absence of receptor — at steady state most of the G protein is in the “resting” GDP state. (B and C) Under control of receptor — in the presence of hormone, at steady state, the slow step is the hydrolysis of GTP and α.GTP accumulates. (B) If the HR complex does not dissociate from βγ-signaling is exclusively though the α.GTP complex; (C) If the HR complex dissociates from βγ before the GTPase hydrolyzes GTP-signaling will be through both, α. GTP and the βγ dimer. (From Birnbaumer, 1993 [72]).
It was while we were doing these analyses of our purified proteins, that John Hildebrandt in my laboratory raised the possibility that Gs and Gi might be structurally related to the light activated GTPase. These thoughts are discussed in his paper (Hildebrandt et al., 1984 [71]) and join those of Bitensky, who was on record since 1978 for having at a Summer ICN-UCLA symposium, pointed to similarities between the visual-phosphodiesterase and the hormone-stimulated adenylyl cyclase signal transduction systems (Shinozawa et al., 1979 [73]).
Molecular cloning, of course, confirmed the structural similarities among not only the three heterotrimeric G proteins identified in this article, but among 16 such G proteins with 16 distinct α subunits, all sharing a set of β γ dimers encoded in 5 β and 12 γ subunit genes, and all activated by a much larger family of seven transmembrane receptors, now referred to generically as GPCRs, G protein-coupled receptors, each catalyzing the activation of its cognate set of G proteins by promoting the exchange of resident GDP with the activating ligand, GTP. The many genes and functions of heterotrimeric G proteins have recently been reviewed (Birnbaumer, 2004 [74]; Wettschureck and Offermanns, 2005 [75]).
Acknowledgments
I am deeply indebted to Mariel Birnbaumer, Joel Abramo-witz and members from my present laboratory for their critical reading and editorial assistance during the preparation of the manuscript. Supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
1
It should be noted that if instead of studying adenylyl cyclase I would have been looking into the mechanism of action of growth factors acting through receptor tyrosine kinases — e.g. epidermal growth factor (EGF) or platelet derived growth factor (PDGF) or at guanylyl cyclases — e.g. atrial natriuretic peptide- (ANP) or guanylin-stimulated cGMP formation — Sutherland’s view would have been on target: each receptor molecule is also the effector enzyme.
2
Even though we had discovered a requirement for GTP in hormonal stimulation of adenylyl cyclase by following up on the finding that GTP affected binding of 125I-glucagon to liver membranes, and the role of GTP was becoming understandable, binding of 125I-glucagon to liver membranes in the presence of GTP was still too slow to be responsible for the stimulatory effect seen with glucagon: the cyclase reached steady state activation within seconds but 125I-glucagon binding continued to increase for minutes on end (Fig. 6), without visible change in activity (Fig. 7). Mistakenly, I assumed that we were looking at two or more types of binding sites, of which some — a few-coupled to adenylyl cyclase, but the majority played no functional role, being acceptor sites instead of receptors (Birnbaumer et al., 1974 [76]). The conundrum was solved 12 years later, with the characterization of two iodinated forms of glucagon: one [I-Tyr10]glucagon, being biologically active and binding with a 5- to 8-fold higher affinity and full cyclase stimulating activity than the parent un-iodinated glucagon, and the other [I-Tyr13]glucagon, that bound with lower affinity and did not activate adenylyl cyclase (Rojas et al., 1983 [77]). Instead of dealing with a heterogeneous set of binding sites, we had been dealing with a heterogeneous set of iodinated glucagons. In the presence of GTP, the active [125I-Tyr10]glucagon bound rapidly and dissociated accordingly, while the [125I-Tyr13]glucagon bound slowly. This product had been the bulk of the iodinated material we had been offering to the membranes and thus led to the bulk of the observed binding.
3
We had purified Gs, then Ns, without using fluoride as a stabilizing agent. Gilman’s group stabilized Gs, then G/F, with aluminum fluoride and Mg (AMF) in their buffers. Without knowing, AMF had caused Gi, but not Gs, to dissociate at cold room temperatures into αi[Al4F−]-GDP plus βγ. This caused the Dallas group to “loose” Gi during their initial purification of Gs. Our Gi stayed as a trimer. By chance, we had co-purified Gi with Gs — which have very similar biochemical fractionation behaviors.
References
- 1.Butcher RW, Sneyd JGT, Park CR, Sutherland EW. Effect of insulin on adenosine 3′,5′-monophosphate in rat epididymal fat pads. J Biol Chem. 1966;241:1651–1653. [PubMed] [Google Scholar]
- 2.Rodbell M, Jones AB, Chiappe de Cingolani GE, Birnbaumer L. Actions of insulin and catabolic hormones on the plasma membrane of the fat cell. Recent Prog Horm Res. 1968;24:215–254. doi: 10.1016/b978-1-4831-9827-9.50011-3. [DOI] [PubMed] [Google Scholar]
- 3.Robison GA, Butcher RW, Sutherland EW. Adenyl cyclase as an adrenergic receptor. Ann N Y Acad Sci. 1967;139:703–723. doi: 10.1111/j.1749-6632.1967.tb41239.x. [DOI] [PubMed] [Google Scholar]
- 4.Birnbaumer L, Pohl SL, Rodbell M. Adenyl cyclase in fat cells. I. Properties and the effects of adrenocorticotropin and fluoride. J Biol Chem. 1969;244:3468–3476. [PubMed] [Google Scholar]
- 5.Birnbaumer L, Rodbell M. Adenyl cyclase in fat cells: II. Hormone receptors. J Biol Chem. 1969;244:3477–3482. [PubMed] [Google Scholar]
- 6.Rodbell M, Birnbaumer L, Pohl SL. Adenyl cyclase in fat cells: III. Stimulation by secretin and the effects of trypsin on the receptors for lipolytic hormones. J Biol Chem. 1970a;245:718–722. [PubMed] [Google Scholar]
- 7.Rodbell M, Birnbaumer L, Pohl SL, Krans HMJ. Hormone receptors and adenyl cyclase activity in mammalian cells. In: Rodbell M, Condliffe P, editors. Colloquium on the Role of Adenyl Cyclase and Cyclic AMP in Biology. Fogarty International Center, U.S. Government Printing Office; Washington, D.C.: 1970b. pp. 59–76. [Google Scholar]
- 8.Berthet C, Rall TWJ, Sutherland EW. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem. 1957;224:463–475. [PubMed] [Google Scholar]
- 9.Pohl M, Birnbaumer L, Rodbell M. Glucagon-sensitive adenyl cyclase in plasma membranes of hepatic parenchymal cells. Science. 1969;164:566–567. doi: 10.1126/science.164.3879.566. [DOI] [PubMed] [Google Scholar]
- 10.Pohl SL, Birnbaumer L, Rodbell M. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. I. Properties. J Biol Chem. 1971;246:1849–1856. [PubMed] [Google Scholar]
- 11.Birnbaumer L, Pohl SL, Rodbell M. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. II. Comparison between glucagon- and fluoride-stimulated activities. J Biol Chem. 1971;246:1857–1860. [PubMed] [Google Scholar]
- 12.Freychet P, Roth J, Neville DM., Jr Monoiodoinsulin: demonstration of its biological activity and binding to fat cells and liver membranes. Biochem Biophys Res Commun. 1971a;43:400–408. doi: 10.1016/0006-291x(71)90767-4. [DOI] [PubMed] [Google Scholar]
- 13.Freychet P, Roth J, Neville DM., Jr Insulin receptors in the liver: specific binding of 125i-insulin to the plasma membrane and its relation to insulin bioactivity. Proc Natl Acad Sci USA. 1971b;68:1833–1837. doi: 10.1073/pnas.68.8.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Kretser DM, Catt KJ, Paulsen CA. Studies on the in vitro testicular binding of iodinated luteinizing hormone in rats. Endocrinology. 1971;88:332–337. doi: 10.1210/endo-88-2-332. [DOI] [PubMed] [Google Scholar]
- 15.Catt KJ, Dufau ML, Tsuruhara T. Studies on a radioligand–receptor assay system for luteinizing hormone and chorionic gonadotropin. J Clin Endocrinol Metab. 1971;32:860–863. doi: 10.1210/jcem-32-6-860. [DOI] [PubMed] [Google Scholar]
- 16.Rodbell M, Krans HMJ, Pohl SL, Birnbaumer L. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. III. Binding of glucagon: method of assay and specificity. J Biol Chem. 1971a;246:1861–1871. [PubMed] [Google Scholar]
- 17.Lefkowitz RJ, Roth J, Pricer W, Pastan I. ACTH receptors in the adrenal: specific binding of ACTH-125I and its relation to adenyl cyclase. Proc Natl Acad Sci USA. 1970;65:745–752. doi: 10.1073/pnas.65.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodbell M, Krans HMJ, Pohl SL, Birnbaumer L. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. IV. Binding of glucagon: effect of guanyl nucleotides. J Biol Chem. 1971b;246:1872–1876. [PubMed] [Google Scholar]
- 19.Rodbell M, Birnbaumer L, Krans HMJ, Pohl SL. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role for guanyl nucleotides in glucagon action. J Biol Chem. 1971c;246:1877–1882. [PubMed] [Google Scholar]
- 20.Rodbell M, Birnbaumer L, Pohl SL, Krans HM. Properties of the adenyl cyclase systems in liver and adipose cells: the mode of action of hormones, Proceedings of the IVth Capri Conference on the Etiology of Diabetes Mellitus (March 1969) Acta Diabetol Lat. 1970;7(Suppl 1):9–63. [PubMed] [Google Scholar]
- 21.Jarett M, Reuter L, McKeel DW, Smith RM. Loss of adenyl cyclase hormone receptors during purification of fat cell plasma membranes. Endocrinology. 1971;89:1186–1190. doi: 10.1210/endo-89-5-1186. [DOI] [PubMed] [Google Scholar]
- 22.Harwood JP, Low H, Rodbell M. Stimulatory and inhibitory effects of guanyl nucleotides on fat cell adenylate cyclase. J Biol Chem. 1973;248:6239–6245. [PubMed] [Google Scholar]
- 23.Harwood JP, Rodbell M. Inhibition by fluoride ion of hormonal activation of fat cell adenylate cyclase. J Biol Chem. 1973;248:4901–4904. [PubMed] [Google Scholar]
- 24.Birnbaumer L. Hormone-sensitive adenylyl cyclases: useful models for studying hormone receptor functions in cell-free systems. Biochim Biophys Acta (Reviews on Biomembranes) 1973;300:129–158. doi: 10.1016/0304-4157(73)90002-6. [DOI] [PubMed] [Google Scholar]
- 25.Krishna G, Harwood JP, Barber AJ, Jamieson GA. Requirement for guanosine triphosphate in the prostaglandin activation of adenylate cyclase of platelet membranes. J Biol Chem. 1972;247:2253–2254. [PubMed] [Google Scholar]
- 26.Londos DC, Salomon Y, Lin MC, Harwood JP, Schramm M, Wolff J, Rodbell M. 5′-Guanylylimidodiphosphate, a potent activator of adenylate cyclase systems in eukaryotic cells. Proc Natl Acad Sci USA. 1974;71:3087–3090. doi: 10.1073/pnas.71.8.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schramm M, Rodbell M. A persistent active state of the adenylate cyclase system produced by the combined actions of isoproterenol and guanylyl imidodiphosphate in frog erythrocyte membranes. J Biol Chem. 1975;250:2232–2237. [PubMed] [Google Scholar]
- 28.Cassel D, Selinger Z. Catecholamine-stimulated GTPase activity in turkey erythrocyte membranes. Biochim Biophys Acta. 1976;252:538–551. doi: 10.1016/0005-2744(76)90206-0. [DOI] [PubMed] [Google Scholar]
- 29.Sharp GW, Hynie S. Stimulation of intestinal adenyl cyclase by cholera toxin. Nature. 1971;229:266–269. doi: 10.1038/229266a0. [DOI] [PubMed] [Google Scholar]
- 30.Vaughan M, Pierce NF, Greenough WB., III Stimulation of glycerol production in fat cells by cholera toxin. Nature. 1970;226:658–659. doi: 10.1038/226658a0. [DOI] [PubMed] [Google Scholar]
- 31.Schafer DE, Lust WD, Sircar B, Goldberg ND. Elevated concentration of adenosine 3′:5′-cyclic monophosphate in intestinal mucosa after treatment with cholera toxin. Proc Natl Acad Sci USA. 1970;67:851–856. doi: 10.1073/pnas.67.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassel D, Selinger Z. Mechanism of adenylate cyclase activation by cholera toxin: inhibition of GTP hydrolysis oat the regulatory site. Proc Natl Acad Sci USA. 1977;74:3307–3311. doi: 10.1073/pnas.74.8.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassel D, Selinger Z. Mechanism of adenylate cyclase activation through the beta-adrenergic receptor: catecholamine-induced displacement of bound GDPby GTP. Proc Natl Acad Sci USA. 1978;75:4155–4159. doi: 10.1073/pnas.75.9.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandt DR, Asano T, Pedersen SE, Ross EM. Reconstitution of catecholamine-stimulated guanosinetriphosphatase activity. Biochemistry. 1983;22:4357–4362. doi: 10.1021/bi00288a002. [DOI] [PubMed] [Google Scholar]
- 35.Pfeuffer T, Helmreich EJM. Activation of pigeon erythrocyte membrane adenylate cyclase by guanylnucleotide analogs and separation of nucleotide binding protein. J Biol Chem. 1975;250:867–876. [PubMed] [Google Scholar]
- 36.Pfeuffer T, Eckstein F. Topology of the GTP-binding site of adenylyl cyclase from pigeon erythrocytes. FEBS Lett. 1976;67:354–358. doi: 10.1016/0014-5793(76)80563-7. [DOI] [PubMed] [Google Scholar]
- 37.Pfeuffer T. GTP-Binding proteins in membranes and the control of adenylate cyclase activity. J Biol Chem. 1977;252:7224–7234. [PubMed] [Google Scholar]
- 38.Ross EM, Gilman AG. Resolution of some components of adenylate cyclase necessary for catalytic activity. J Biol Chem. 1977;252:6966–6969. [PubMed] [Google Scholar]
- 39.Bourne HR, Coffino P, Tomkins GM. Selection of a variant lymphoma cell deficient in adenylate cyclase. Science. 1975;187:750–752. doi: 10.1126/science.163487. [DOI] [PubMed] [Google Scholar]
- 40.Northup JK, Sternweis PC, Smigel MD, Schleifer LS, Ross EM, Gilman AG. Purification of the regulatory component of adenylate cyclase. Proc Natl Acad Sci USA. 1980;77:6516–6520. doi: 10.1073/pnas.77.11.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gill DM. Involvement of nicotinamide adenine dinucleotide in the action of cholera toxin in vitro. Proc Natl Acad Sci USA. 1975;72:2064–2068. doi: 10.1073/pnas.72.6.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassel D, Pfeuffer T. Mechanism of cholera toxin action: covalent modification of the guanyl nucleotide-binding protein of the adenylate cyclase system. Proc Natl Acad Sci USA. 1978;75:2669–2673. doi: 10.1073/pnas.75.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gill DM, Meren R. ADP-Ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase. Proc Natl Acad Sci USA. 1978;75:3050–3054. doi: 10.1073/pnas.75.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Northup JK, Sternweis PC, Gilman AG. The subunits of the stimulatory regulatory component of adenylate cyclase. Resolution, activity and properties of the 35,000 dalton (beta) subunit. J Biol Chem. 1983a;258:11361–11368. [PubMed] [Google Scholar]
- 45.Northup JK, Smigel MD, Sternweis PC, Gilman AG. The subunits of the stimulatory regulatory component of adenylate cyclase. Resolution of the activated 45,000-dalton (alpha) subunit. J Biol Chem. 1983b;258:11369–11376. [PubMed] [Google Scholar]
- 46.Porte D., Jr A receptor mechanism for the inhibition of insulin release by epinephrine in man. J Clin Invest. 1967;46:86–94. doi: 10.1172/JCI105514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanura K, Nigimori M, Murai S, Yajima M, Ito K, Katada T, Ui M, Isahi S. Subunit structure of islet activating protein, pertussis toxin, in conformity with the A–B model. Biochemistry. 1982;21:5516–5522. doi: 10.1021/bi00265a021. [DOI] [PubMed] [Google Scholar]
- 48.Katada T, Ui M. Slow interaction of islet-activating protein with pancreatic islets during primary culture to cause reversal of alpha-adrenergic inhibition of insulin secretion. J Biol Chem. 1980;255:9580–9588. [PubMed] [Google Scholar]
- 49.Katada T, Ui M. Islet-activating protein; a modifier of receptor-mediated regulation of rat islet adenylate cyclase. J Biol Chem. 1981;256:8310–8317. [PubMed] [Google Scholar]
- 50.Katada T, Ui M. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc Natl Acad Sci USA. 1982;79:3129–3133. doi: 10.1073/pnas.79.10.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hildebrandt JD, Sekura RD, Codina J, Iyengar R, Manclark CR, Birnbaumer L. Stimulation and inhibition of adenylyl cyclases is mediated by distinct proteins. Nature. 1983;302:706–709. doi: 10.1038/302706a0. [DOI] [PubMed] [Google Scholar]
- 52.Codina JD, Hildebrandt J, Iyengar R, Birnbaumer L, Sekura RD, Manclark CR. Pertussis toxin substrate, the putative Ni of adenylyl cyclases, is an alpha/beta heterodimer regulated by guanine nucleotide and magnesium. Proc Natl Acad Sci USA. 1983;80:4276–4280. doi: 10.1073/pnas.80.14.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bokoch GM, Katada T, Northup JK, Hewlett EL, Gilman AG. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J Biol Chem. 1983;258:2071–2075. [PubMed] [Google Scholar]
- 54.Bitensky MW, Gorman RE, Miller WH. Adenyl cyclase as a link between photon capture and changes in membrane permeability of frog photoreceptors. Proc Natl Acad Sci USA. 1971;68:561–562. doi: 10.1073/pnas.68.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller WH, Gorman RE, Bitensky MW. Cyclic adenosine monophosphate: function in photoreceptors. Science. 1971;174:295–297. doi: 10.1126/science.174.4006.295. [DOI] [PubMed] [Google Scholar]
- 56.Bitensky MW, Gorman RE, Miller WH. Digitonin effects on photoreceptor adenylate cyclase. Science. 1972;175:1363–1364. doi: 10.1126/science.175.4028.1363. [DOI] [PubMed] [Google Scholar]
- 57.Miki N, Keirns JJ, Marcus FR, Freeman J, Bitensky MW. Regulation of cyclic nucleotide concentrations in photoreceptors: an ATP-dependent stimulation of cyclic nucleotide phosphodiesterase by light. Proc Natl Acad Sci USA. 1973;70:3820–3824. doi: 10.1073/pnas.70.12.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miki N, Baraban JM, Keirns JJ, Boyce JJ, Bitensky MW. Purification and properties of the light-activated cyclic nucleotide phosphodiesterase of rod outer segments. J Biol Chem. 1975;250:6320–6327. [PubMed] [Google Scholar]
- 59.Keirns JJ, Miki N, Bitensky MW, Keirns M. A link between rhodopsin and disc membrane cyclic nucleotide phosphodiesterase. Action spectrum and sensitivity to illumination. Biochemistry. 1975;14:2760–2766. doi: 10.1021/bi00683a032. [DOI] [PubMed] [Google Scholar]
- 60.Wheeler GL, Matuto Y, Bitensky MW. Light-activated GTPase in vertebrate photoreceptors. Nature. 1977;269:822–824. doi: 10.1038/269822a0. [DOI] [PubMed] [Google Scholar]
- 61.Wheeler GL, Bitensky MW. A light-activated GTPase in vertebrate photoreceptors: regulation of light-activated cyclic GMP phosphodiesterase. Proc Natl Acad Sci USA. 1977;74:4238–4242. doi: 10.1073/pnas.74.10.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Godchaux W, III, Zimmerman WF. Membrane-dependent guanine nucleotide binding and GTPase activities of soluble protein from bovine rod cell outer segments. J Biol Chem. 1979;254:7874–7884. [PubMed] [Google Scholar]
- 63.Kuhn H. Light-induced, reversible binding of proteins to bovine photoreceptor membranes. Influence of nucleotides. Neurochem Int. 1980;1:269–285. doi: 10.1016/0197-0186(80)90066-2. [DOI] [PubMed] [Google Scholar]
- 64.Baehr W, Morita EA, Swanson RJ, Applebury ML. Characterization of bovine rod outer segment G-protein. J Biol Chem. 1982;257:6452–6460. [PubMed] [Google Scholar]
- 65.Shinozawa T, Uchida S, Martin E, Cafiso D, Hubbell W, Bitensky M. Additional component required for activity and reconstitution of light-activated vertebrate photoreceptor GTPase. Proc Natl Acad Sci USA. 1980;77:1408–1411. doi: 10.1073/pnas.77.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shinozawa T, Bitensky MW. Purification and characteristics of photoreceptor light-activated guanosinetriphosphatase. Biochemistry. 1981;20:7068–7074. doi: 10.1021/bi00528a003. [DOI] [PubMed] [Google Scholar]
- 67.Fung BK, Hurley JB, Stryer L. Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci USA. 1981;78:152–156. doi: 10.1073/pnas.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deterre P, Bigay J, Forquet F, Robert M, Chabre M. cGMP phosphodiesterase of retinal rods is regulated by two inhibitory subunits. Proc Natl Acad Sci USA. 1988;85:2424–2428. doi: 10.1073/pnas.85.8.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sternweis PC, Northup JK, Smigel MD, Gilman AG. The regulatory component of adenylate cyclase. Purification and Properties. J Biol Chem. 1981;256:11517–11527. [PubMed] [Google Scholar]
- 70.Codina J, Hildebrandt JD, Sekura RD, Birnbaumer M, Bryan J, Manclark CR, Iyengar R, Birnbaumer L. Ns and Ni, the stimulatory and inhibitory regulatory components of adenylyl cyclases. Purification of the human erythrocyte proteins without the use of activating regulatory ligands. J Biol Chem. 1984;259:5871–5886. [PubMed] [Google Scholar]
- 71.Hildebrandt JD, Codina J, Risinger R, Birnbaumer L. Identification of a gamma subunit associated with the adenylyl cyclase regulatory proteins Ns and Ni. J Biol Chem. 1984;259:2039–2042. [PubMed] [Google Scholar]
- 72.Birnbaumer L. A new look at receptor mediated activation of a G protein. In: Dickey B, Birnbaumer L, editors. GTPases in Biology, Handbook of Experimental Pharmacology. 108/I. Springer Verlag; Heidelberg: 1993. pp. 31–38. Chapter 3. [Google Scholar]
- 73.Shinozawa T, Sen I, Wheeler G, Bitensky MW. Predictive value of the analogy between hormone-sensitove adenylate cyclase and light-sensiitve photoreceptor cyclic GMP phosphodiesterase: a specific role for a light-sensitive GTPase as a component in the activation sequence. J Supramol Struct. 1979;10:185–190. doi: 10.1002/jss.400100208. [DOI] [PubMed] [Google Scholar]
- 74.Birnbaumer L. Signal transduction by G proteins: basic principles, molecular diversity and structural basis of their action. In: Bradshaw RA, Dennis ED, Hamm HE, editors. Handbook of Cell Signaling. Vol. 2. Academic Press; 2004. pp. 557–569. [Google Scholar]
- 75.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 76.Birnbaumer L, Pohl SL, Kaumann AJ. Receptors and acceptors: a necessary distinction in hormone binding studies. In: Greengard P, Robison GA, editors. Advances in Cyclic Nucleotide Research. Vol. 4. Raven Press; New York: 1974. pp. 239–281. [PubMed] [Google Scholar]
- 77.Rojas FJ, Swartz RL, Iyengar R, Garber AJ, Birnbaumer L. Monoiodoglucagon: synthesis, purification by high pressure liquid chromatography and characteristics as a receptor probe. Endocrinology. 1983;113:711–719. doi: 10.1210/endo-113-2-711. [DOI] [PubMed] [Google Scholar]
- 78.Birnbaumer L, Pohl SL, Rodbell M, Sundby F. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. Vii. Hormonal stimulation: reversibility and dependence on concentration of free hormone. J Biol Chem. 1972;247:2038–2043. [PubMed] [Google Scholar]