Concatenated Metallothionein as a Clonable Gold Label for Electron Microscopy (original) (raw)
. Author manuscript; available in PMC: 2008 Oct 1.
Published in final edited form as: J Struct Biol. 2007 Jul 10;160(1):70–82. doi: 10.1016/j.jsb.2007.06.010
Abstract
Localization of proteins in cells or complexes using electron microscopy has mainly relied upon the use of heavy metal clusters, which can be difficult to direct to sites of interest. For this reason, we would like to develop a clonable tag analogous to the clonable fluorescent tags common to light microscopy. Instead of fluorescing, such a tag would initiate formation of a heavy metal cluster. To test the feasibility of such a tag, we exploited the metal-binding protein, metallothionein (MT). We created a chimeric protein by fusing one or two copies of the MT gene to the gene for maltose binding protein. These chimeric proteins bound many gold atoms, with a conservative value of 16 gold atoms per copy of metallothionein. Visualization of gold-labeled fusion proteins by scanning electron microscopy required one copy of metallothionein while transmission electron microscopy required two copies. Images of frozen-hydrated samples of simple complexes made with anti-MBP antibodies hint at the usefulness of this method.
Keywords: metallothionein, mass spectrometry, transmission electron microscopy, nanocluster, gold tag, protein label
Introduction
Electron cryo-microscopy of cells, organelles and macromolecular assemblies is one of the key frontiers in structural cell biology, yet localization of component proteins in three-dimensional (3D) reconstructions continues to be a problem. For cells and tissues, one system that shows promise is the genetic addition of a tetracysteine motif to a protein of interest. This motif binds fluorescent biarsenical compounds that can be photo-reacted within chemically fixed samples to provide densities suitable for localization (Adams et al. 2002). However, methods requiring such fixation suffer from the possibility that such treatments distort protein structure. Another straightforward way to localize a protein of interest is to perform correlation-based matching to 3D maps of isolated components determined by x-ray crystallography, NMR, or even electron cryo-microscopy (Nickell et al. 2006). This method has worked for large complexes; for example, the proteasome was localized within electron tomograms of phantom cells (Bohm et al. 2000), and the 70S ribosome has been identified in Spiroplasma melliferum (Ortiz et al. 2006). However, small proteins are more difficult to localize. For a small protein in a multiprotein complex, correlation-based methods may not always provide a definitive answer.
With macromolecular complexes, an alternative localization method has been to identify proteins of interest through genetic modifications. For higher resolution structures, the reconstruction of a complex in which the protein of interest has been replaced by a chimera of the protein of interest engineered with a tag protein, may result in an additional lobe of density that marks the position of the protein of interest (Wendt et al. 2002). Another alternative has been to delete a domain in the protein of interest, which can result in a missing feature in the reconstruction (Thomas et al. 2001). For more difficult situations, where the structures are larger and are less well resolved, highly visible heavy metal clusters can be used as effective labels. Such a label can be indirect, whereby a metal cluster is attached to a protein of interest by an antibody or other known interacting protein, or a label can be direct, where the metal cluster is attached directly to the protein of interest. Whether indirect or direct, labelling of the protein may be sparse because of inaccessibility of the tag to its target or because of low affinity of the tag for its target. Moreover, other sites may be labeled because of adventitious binding of the tag to other proteins (Jahn 1999; Hainfeld and Powell 2000). Together, these difficulties point out the need for more efficient, specific methods of tagging a protein of interest. In light microscopy, these labelling difficulties have been solved by the discovery of fluorescent proteins, e.g., green fluorescent protein, which can be added to the protein of interest to form of a chimera. The creation of a clonable label could provide the same advantages for electron microscopy provided that the label had high visibility in the electron microscope.
Ideally, we want a small protein that would initiate formation of a heavy metal cluster from a heavy metal salt or organometallic compound. We selected the cysteine-rich protein, metallothionein (MT) as a possible label. It has only ~60 amino acids, a third of which are cysteine, and it can bind heavy metal atoms with high affinity. Among these, gold is one of the most tightly bound (Nielson and Winge 1983; Nielson et al. 1985). Aurothiomalate, a gold(I)-containing, anti-arthritic compound, can deliver ~20 gold atom bound to a single MT protein (Schmitz et al. 1980; Laib et al. 1985). In addition, silver(I) and mercury(II) can also be bound with similar stoichiometries (Lu et al. 1993) (Palacios et al. 2003). Extended X-ray absorption fine structure (EXAFS) studies on the mercury complex suggested that metal atoms were bridged between two sulphurs with no additional ligand and that the protein may have refolded into a single domain (Jiang et al. 1994). Gold appears to do the same, and indeed we found that up to 40 gold atoms may at times be bound to a single MT subunit (Mercogliano and DeRosier 2006).
In our previous work, we have suggested that Zn7-MT was able to use its 20 cysteines to bind gold by a mechanism like that used to synthesize gold clusters such as the popular Nanogold® (Mercogliano and DeRosier 2006). These findings encouraged us to test concatenated MT as a clonable electron microscopy label. Here, we show that simple fusion proteins composed of maltose binding protein (MBP) and MT can bind gold in proportion to the number of copies of MT with 12 to 20 gold atoms per MT. The gold-labeled chimera of MBP with a single concatenated MT (MBP-MT) was visible by scanning transmission electron microscopy (STEM). The gold-labeled chimera of MBP with two copies of MT (MBP-MT2) was visible by conventional transmission electron microscopy (TEM). Finally, electron cryo-micrographs of antibody complexes with MBP-MT2 provide insights into the visibility of the gold label in frozen-hydrated preparations.
Material and Methods
Creation of MBP fusion proteins with MT
Plasmids for expressing MBP fusion proteins were created by cloning copies of MT into vector pMAL-c2x from New England Biolabs (Beverly, MA). The MT gene was amplified from pET-3d-MT (a gift from the Winge laboratory, University of Utah), which contains the MT gene within the pET-3d vector (Novagen, Madison, WI).
One fusion product contained MT directionally cloned onto the C-terminus of MBP and was designated pMAL-c2x-MT. The MT gene was amplified with the forward primer (5′-CTCGGGATCGAGGGAAGGATTTCAAGATATACCATGGACCCC-3′), which codes for the MBP linker region containing an XmnI and NcoI site fused in frame to the MT gene, and the reverse primer (5′-GACTCTAGAGGATCCTAGGCACAGCACGTG-3′), which contains the MT gene stop codon and a subsequent BamHI site. Both the PCR product and pMAL-c2x vector were digested with XmnI and BamHI and were later ligated together to generate the final plasmid.
The second fusion product contained two copies of the MT gene fused in frame and was designated pMAL-c2x-MT2. In this construct, the only translational modification was the change of an alanine to an aspartic acid at the junction between the two MT genes. The second copy of the MT gene was inserted into the pMAL-c2x-MT plasmid before the existing MT copy. The inserted MT gene was amplified from pET-3d-MT using the same forward primer as used for the pMAL-c2x-MT plasmid (above), and the reverse primer (5′-GTGACCACATGTCACAGCACGTGCACTTGTCC -3′), which contains an AflIII site at the MT-MT junction. The pMAL-c2x-MT plasmid was prepared by digesting with XmnI and NcoI, and the PCR product was constructed with XmnI and AflIII. Since NcoI and AflIII produced equivalent DNA ends, the second copy of MT was inserted with removal of both restriction sites at the ligation junction.
The products of the ligation reactions for both constructs were transformed into Novablue (Novagen) E. coli cells. Properly fused constructs were isolated by screening for over-expressed proteins of the expected molecular weight. Isolated colonies grown in LB (5 gr yeast extract, 10 gr tryptone, and 5 gr sodium chloride in 1 L) with selection were checked for expression after induction with 1mM isopropyl-B-D-thiogalactopyranoside (IPTG) for 2hr. Clarified cell extracts from individual colonies were run on SDS polyacrylamide gels to check for the molecular weight of the induced protein. Colonies showing over-expressed protein of the expected molecular weight were grown a second time followed by isolation of the newly formed plasmid. Finally, the DNA was checked by restriction analysis and was subsequently sequenced.
Expression of MBP Fusion Proteins
Sequence verified plasmids were transformed into TB1 E.coli cells. Starter cultures from single colonies were grown in LB media with ampicillin selection overnight at 30°C with aeration. The next morning, 1L growth cultures were inoculated with 5ml of overnight culture and grown with selection and with 1% glucose until an OD600 of 0.5 was reached. Protein production was induced with 0.2mM IPTG. After 0.5hr, zinc sulfate was added to 0.2mM to fill metal binding sites within the MT domains. After 2hr of induction, cells were pelleted at 6000 x g. Cell pellets were placed in 50ml conical tubes, flash frozen in liquid nitrogen, and stored at −70°C until the day of purification.
Purification of MBP Fusion Proteins
Fusion proteins were purified with slight modification to the New England Biolabs standard MBP purification procedure. Briefly, cells were defrosted the day of purification, and all steps of the procedure were performed at 4°C. Cells were resuspended with the addition 25ml of Wash buffer (20mM Trizma Base pH 7.5, 150mM sodium chloride, and 0.1mM β-mercaptoethanol (all from Sigma Chemical Corp, St. Louis, MO)). Cell suspensions were lysed by 8 sonication cycles of 30sec pulses and 1min rest periods on ice with a Branson 2000 sonicator. Lysis was monitored with Total Protein Concentration system (Biorad, Hercules, CA).
Once lysed, cell suspensions were spun at 9000 × g for 20min. The resulting supernatants were diluted in Wash buffer to 100ml and were then loaded onto a pre-equilibrated 5ml amylose column (New England Biolabs). After loading, the column was washed with 10 column volumes of Wash buffer. Bound protein was eluted in 0.5ml fractions in Wash buffer supplemented with 10mM maltose (Sigma Chemical Corp) but without β-mercaptoethanol or other reducing agents. Protein concentrations of eluted fractions were quantified with Total Protein Concentration system. Typically, eluted peak fractions were found to have concentrations of about 5mg/ml. Aliquots were flash frozen in liquid nitrogen in 100μl volumes and stored at −70°C.
Gold Incubation and Preparation of MBP Fusion Proteins
Gold incubation experiments with chimeric MBP fusion protein samples were carried out in 100μl final volume for 3hr at 37°C. For the chimera containing a single MT, the final concentrations during incubations were 50μM protein, 10mM disodium aurothiomalate, and 25mM Tris-HCl pH 7.5. Similarly, for incubations of the dual MT chimera, the concentrations were 50μM protein, 20mM disodium aurothiomalate, and 25mM Tris-HCl pH 7.5. These concentrations provide a 20 to 1 ratio of aurothiomalate to cysteine. Control samples of identical volume and concentration were prepared in the same way but without the addition of the aurothiomalate. After incubation, samples were desalted over a Superdex10/30HR column (Pharmacia, Piscataway, NJ) at 0.5ml/min on an Akta FPLC (Pharmacia) pre-equilibrated with 100mM ammonium acetate buffer pH 6.0. Sample elution was monitored with the UV detector set at a wavelength of 280nm, and 0.5ml fractions were collected.
MALDI Mass Spectrometry of Fusion Samples
Desalted samples with and without bound gold were analyzed by MALDI mass spectrometry. All MALDI samples were run at the Brandeis University Biochemistry Core Facility on a PerSeptive Biosystems Voyager (Framingham, MA). Samples were diluted in one of two matrixes; either a 5mg/ml sinapinic acid (SA) (D7927, Sigma Chemical Corporation) solution or a 10mg/ml 6-azo-thiothymidine (ATT) (27,551-4, Aldrich Chemical Company) in a water:acetonitrile (50%:50% vol:vol). Samples were diluted 10 or 20 fold in matrix solution and spotted onto the MALDI sample plate with 2ul/well to obtain a strong signal with minimal amounts of protein. Droplets were dried and then placed into the mass spectrometer. Spectra were collected with a 25,000V acceleration voltage in positive ion mode usually using a mass/charge ratio range of 20,000 to 100,000. Samples that did not yield strong signal were concentrated in a Savant vacuum concentrator (Holbrook, New York) and retested in the MALDI mass spectrometer.
STEM of Fusion Protein Samples
Some of the desalted MBP fusion samples were sent for STEM imaging at Brookhaven National Laboratories. Samples were sent flash frozen in liquid nitrogen and shipped on dry ice. Samples were defrosted, applied to grids containing a thin carbon foil, and mixed with a small amount of tobacco mosaic virus, a mass standard. Grids were rinsed with ammonium acetate buffer, and excess buffer was wicked away. Finally, the grids were flash frozen and slowly freeze-dried overnight to remove remaining buffer. Grids were imaged at 40keV within the STEM using both the high angle and low angle dark field annular detectors producing images containing 512×512 pixels.
Transmission Electron Microscopy of Fusion Samples
Desalted samples of MBP fusion proteins in ammonium acetate buffer pH 6.0 were saved for viewing in the transmission electron microscope. Quantifoil (Jena, Germany) grids with 1μm diameter holes were used to support thin (<20 nm) continuous carbon foils. Carbon foils were prepared by depositing carbon onto freshly cleaved smooth mica in an Edwards (West Sussex, UK) carbon evaporator. Thin foils were then floated on water, and pieces of the foil were picked up on to the Quantifoil grids and dried 24hr before proceeding. Grids were prepared by glow discharging followed by application of 3ul of sample for 30sec. The grids were rinsed twice with ammonium acetate buffer, carefully blotted with Whatman (Kent, UK) filter paper to remove excess buffer, and allowed to fully dry. Low resolution TEM images were collected on a CM12 (Philips-FEI, Eindhoven, The Netherlands) at 120 keV with a nominal microscope magnification of 160,000 times and with a defocus of ~700 nm on a Gatan Slow Scan CCD (Pleasanton, CA). High-resolution TEM images were collected on a FEI F30 (Philips-FEI, Eindhoven, The Netherlands) at 300keV at nominal microscope magnification of 230,000 times under low dose conditions (<2000 e/nm2) with a defocus between 0.5μm and 1.5 μm to a Tem-Cam F2224HD CCD (TVIPS GmbH, Gauting, Germany).
Preparation of MBP-MT2 Antibody Complexes
MBP monoclonal antibodies (1 mg/ml in 50% glycerol) were purchased form New England Biolabs (Bedford, MA). Prior to incubation with MBP-MT2 fusion protein, antibodies were buffer exchanged via cycles of dilution and reconcentration using a 10 kDa MWCO microconcentration device (Pall, East Hills, NY). Briefly, 100μl of antibodies were diluted in TBS buffer (25 mM Tris-Cl, 200mM sodium chloride [pH7.5]) to a volume of 500μl and reconcentrated to 50μl. This cycle was repeated 2 more times.
Prior to making complexes, monomeric, gold-incubated MBP-MT2 protein was prepared as above. Monomeric fractions were combined and concentrated to 20μl within a Speedvac (Savant, Waltham, MA) vacuum concentrator giving a concentration of about 4mg/ml. Antibody complexes were formed by incubating 25μl of concentrated antibody with 20μl of concentrated MBP-MT2 protein and 10μl of TBS buffer at room temperature for 1hr. Samples were then separated on a Pharmacia 3.2/30 Superose 12 column using a Pharmacia Akta FPLC pre-equilibrated with 100mM ammonium acetate buffer pH 6.0 and collected in 100μl fractions.
Imaging of MBP-MT2 Antibody Complexes
Samples containing complex eluted from the sizing column and control samples of antibody alone were visualized in the TEM in negative stain. For this, 400 mesh TEM grids supporting carbon foils were glow discharged in air, and 3μl of sample was applied to the grid for 30 sec. Grids were stained with several drops of filtered 2% uranyl acetate stain, excess stain was wicked away with Whatman (Kent, UK) #1 filter paper, and grids were allowed to dry. Samples were visualized in a Morgagni TEM (FEI, Eindhoven, The Netherlands) using an 80 keV acceleration voltage, and images were collected with a 2k × 2k CCD camera (Hamamatsu, Japan).
Antibody complex was prepared for electron cryo-microscopy on Quantifoil® R 1.2/1.3 (Jena, Germany) holey grids. Three 5μL drops of sample were placed on a negatively glow discharged grid and blotted with Whatman #1 filter paper between application of drops. After the final blot, the sample was plunged into liquid-nitrogen-cooled liquid ethane to vitrify the sample. For electron cryo-microscopy, a grid was placed into a pre-cooled Gatan (Pleasanton, CA) 626 single tilt cyro-holder. The grid was then transferred into a FEI F30 (Philips-FEI, Eindhoven, The Netherlands) at 200keV, and images were collected with a nominal microscope magnification of 78,000 times under low dose conditions (<2000 e/nm2) with a defocus between 0.75 μm and 1.5 μm defocus on a Tem-Cam F2224HD CCD (TVIPS GmbH, Gauting, Germany) or on SO-163 Kodak (Rochester, NY) film.
Results
Cloning and purification MBP-metallothionein fusion proteins
To test if concatenation of MT gene products could serve as a clonable TEM label, two plasmids were constructed. One and two copies of the mouse MT-1 gene were cloned onto the gene for MBP to create a chimeric protein (Figure 1, top left). The inserted DNA preserved the methionine start codon of MT and added the protein sequence ISRVT to the pre-existing MBP-linker region. The expressed fusion protein with a single MT copy was designated MPB-MT. A second copy of the MT gene was added ahead of the first MT copy (Figure 1, lower left) in the pMAL-c2x-MT plasmid. The two MT genes in pMAL-c2x-MT2 are connected with no extra residues, but the final alanine in the added MT gene (first MT copy in the fusion protein) was mutated to an aspartic acid. The absence of a linker between the MT genes was chosen to maximize the ability of the MBP-MT2 to concentrate gold atoms and subsequently act as a TEM label.
Figure 1.
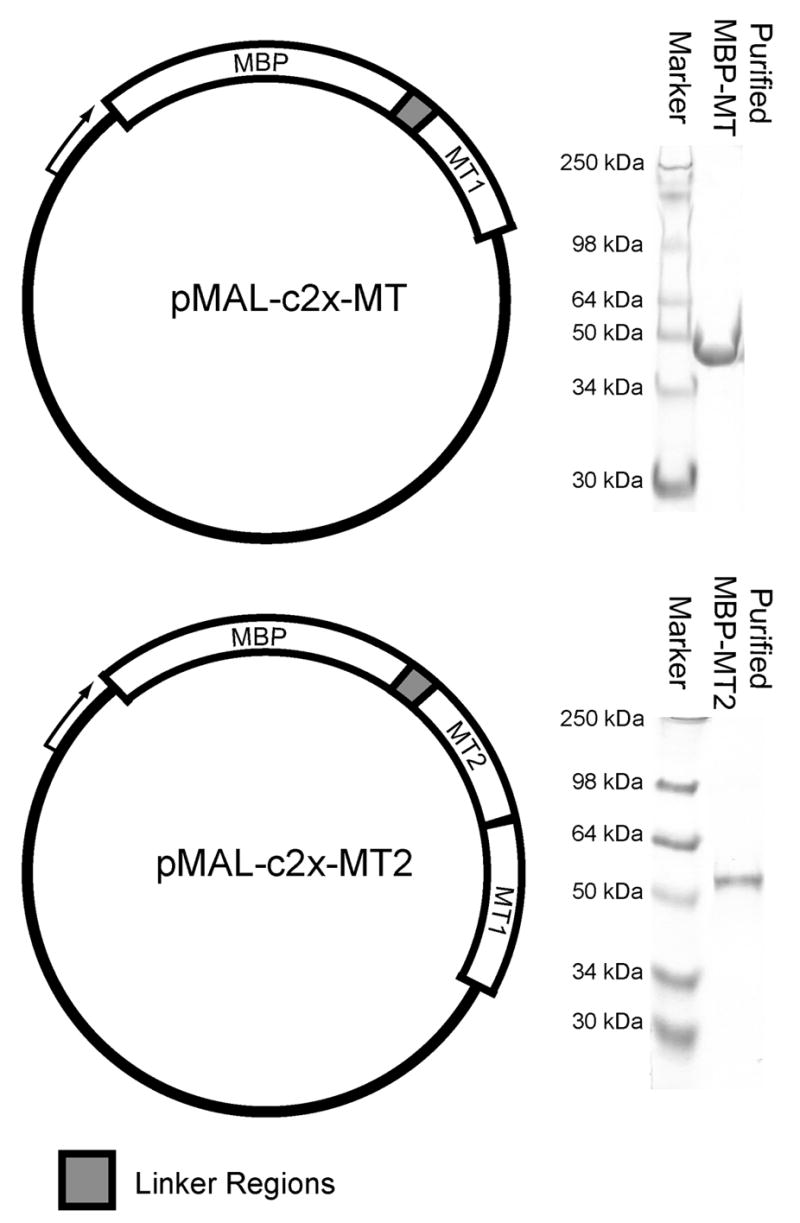
Cloning and purification of MBP fusion proteins with MT. (top, left) MBP was cloned as a fusion protein to a single copy of mouse MT-1 as shown by the plasmid map describing the layout of the pMAL-c2x-MT plasmid. (top, right) After expression and purification, SDS-PAGE showed highly purified MBP-MT protein. (bottom, left) The plasmid, pMAL-c2x-MT2, able to express MBP fused to concatenated MT was created by inserting a second copy of the gene in front of the existing copy. (bottom right) This plasmid was used to express and purify MBP-MT2 as shown by SDS-PAGE.
Purification of both MBP-MT and MBP-MT2 was performed using a modified New England Biolabs protocol. The first modification was the removal of EDTA or other chelators from all buffers. This was to prevent removal of the protective metal (Zn) atoms from the MT metal binding sites. The second modification was the use of β-mercaptoethanol at a concentration of 0.1 mM, which is ten fold less than the standard procedure. In purification procedures with metallothionein, it is common to use β-mercaptoethanol over stronger dithiol-type reducing agents due to the chelating ability of dithiols (Suzuki 1991). The lower β-mercaptoethanol concentration rather than a non-thiol based reducing agent such as Tris-carboxyethylphosphine (TCEP) was chosen to avoid interference in subsequent reactions of the purified proteins with gold compounds. Consequently, the fusion proteins were eluted from the amylose columns with a buffer containing 10 mM maltose but no reducing agent. Typically, peak elution fractions contained 5mg/ml of fusion protein with no obvious contaminating proteins (Figure 1, right).
Gold binding by MBP-MT and MBP-MT2
To fill metal binding sites in MT with gold, fusion protein samples were incubated with aurothiomalate. Samples were incubated for 3hr at 37°C with a molar excess of gold to cysteine by a 20 to 1 ratio. After incubations, samples were passed over a Superdex 10/30 HR column to separate free gold from gold-bound protein. Example elution profiles from size exclusion chromatography of MBP-MT and of MBP-MT2 can be seen in Figure 2A and 2C, respectively. Control samples containing aurothiomalate only (blue traces) elute as a single peak at about 19 ml after injection. Control samples of MBP-MT and MBP-MT2 alone (red) show a series of increasing peaks ranging from about 10 ml to 16 ml volume after sample injection. These peaks correspond to different oligomeric states of the protein. The last eluted, largest peak (red arrows) of each trace is the monomeric protein. This was confirmed by MALDI mass spectrometry of MBP-MT and MBP-MT2 (Figure 2B top and Figure 2D top, respectively). MALDI mass spectrometry of higher oligomeric state of these protein showed corresponding mass shifts (not shown), but signal for these peaks was weak because they are less populated and their higher masses make them less detectable by this method. It is important to this work and to the prospect of using MT fusion proteins as TEM labels that these experiments show oligomerization is not the result of incubation with our source of gold atoms. We believe this is the case since control samples without gold produce chromatographic profiles similar to those incubated with gold, and as such oligomerization was previously present (Figure 2A and 2C). Furthermore, because SDS-PAGE gels run with reducing agents show monomeric protein, the oligomerization is likely caused by MT cysteine oxidation (Figure 1, right).
Figure 2.

Gold Binding by MBP fusion proteins. (A and C) Chromatography on a Superdex 10/30HR column shows distinguishable peaks associated with gold-bound MBP-MT and MBP-MT2 fusion proteins. Proteins treated with aurothiomalate (green) or untreated protein (red) are cleanly separated from aurothiomalate run separately (blue). Purified proteins (red) show complete separation from unbound gold (blue) and later elution as compared to gold-bound peaks (green). Note that aurothiomalate incubated protein is shifted to earlier elution compared to untreated protein samples. The unreacted aurothiomalate appears as a peak with a slightly faster shoulder corresponding to released thiomalate, which are probably thiomalate dimers; All profiles were collected at 280nm and scaling factors (in parentheses) were used to compare results because of a gold-associated absorbance. (B and D) Monomeric peaks of wild type and gold-bound protein (red and green arrows) were further analyzed by MALDI mass spectrometry. In each case, gold-incubated proteins (bottom spectra) are shifted to higher mass-to-charge values than the wild-type proteins (top spectra). For each peak shown, the mass-to-charge value corresponding to the maximum observed amplitude is listed as well as the charge state of the peak (in parentheses). (E) Absorbance vs. wavelength spectra for aurothiomalate (blue), a 5x scaled MBP-MT2 (red), and gold-incubated MBP-MT2 (green) samples. Upon gold incubation a shoulder above 300nm emerges
Fusion protein samples incubated with aurothiomalate show altered chromatographic profiles. Peaks attributed to gold-bound MBP-MT and MBP-MT2 (Figure 2A and 2C, red) show similarly shaped elution profiles to apo-proteins (green). An additional peak eluted at about 19 ml is comparable to the aurothiomalate control (blue). The extra shoulder on this peak likely represents the production of free thiomalic acid upon transfer of gold atoms to the fusion protein. The thiomalic acid in this shoulder either may bind to unreacted aurothiomalate in a bridging ligation to the gold atom or may react with itself to form a disulphide. In either case, the unreacted reagents and newly formed product are distinguishable from the gold-bound protein and thus show that this column provides proper separation. All gold-bound proteins elute more quickly from the Superdex 10/30 column than their apo-protein counterparts. This quicker mobility supports the idea that there is a increase in the hydrodynamic radius of the protein upon gold incubation and may also indicate a structural change that accompanies gold binding.
To measure the number of bound gold atoms, gold-labeled monomeric MBP-MT and MBP-MT2 samples (see peaks marked by green arrows in Fig. 2) were subjected to MALDI mass spectrometry. Even given the low pH matrices used to prepare samples for MALDI, tightly bound metals, such as gold, should remain bound for detection by this method. Apo-MBP-MT is expected to have a mass of 48682 amu. When measured with MALDI mass spectrometry, a peak mass of 48771 amu was recorded (Figure 2A top). We believe that this mass difference may indicate that metal atoms acquired during expression and purification may still be associated with the protein. When monomeric gold-bound MBP-MT was analyzed, the +1 charged mass peak shifted to 51204 amu with an extended distribution toward higher mass values. This increase in peak mass and in distribution is comparable to previous MALDI results performed on gold-incubated MT (Mercogliano and DeRosier 2006). The difference in masses at maximal peak amplitude corresponds to the addition of about 12 gold atoms bound to the MT portion of the fusion protein.
Similarly, MBP-MT2 was measured in its apo-protein and gold-bound states with MALDI mass spectrometry. The measured and expected values of apo-MBP-MT2 agree perfectly at 55146 amu, though a shoulder with increased mass may indicate some bound metal. Upon incubation with gold, the mass peak shifts from 55146 amu to 62641 amu (Figure 2D). Assuming that gold binds to the fused MT portions of the protein as single atoms with no carrier ligands as was shown previously (Mercogliano and DeRosier 2006), this shift signifies around 38 gold atoms. In addition, the extended distribution of mass values recorded up to about 68000 amu would suggest some MBP-MT2 proteins may have acquired up to 65 gold atoms. These higher values are similar to those found in gold clusters already synthesized as TEM labels.
Spectrophotometric measurements of gold-incubated samples may give clues to the nature of the bonds forming during our gold incubations. Figure 2E shows UV-Vis spectra for aurothiomalate (blue), a 5 times scaled MBP-MT2 sample (red), and a sample of MBP-MT2 incubated with aurothiomalate (green). While all three samples show strong absorption below 280nm, only the incubated mixture of MBP-MT2 with aurothiomalate demonstrates absorption above 300nm. The reaction with aurothiomalate causes a strong absorption shoulder from about 300nm to 350nm with a gradual fall off. Although these spectra are difficult to interpret without structural information, such extended absorption shoulders above 300nm have been observed with synthesized gold clusters (Gutierrez et al. 1999; Schaaff and Whetten 2000) and may suggest similar bond formation here.
Visualization of gold-bound MBP-MT and MBP-MT2
Samples characterized by chromatography and mass spectrometry were visualized with TEM and STEM. TEM had the advantage that grids could be prepared as they came off the separation column. For TEM, samples were viewed on thin carbon foils to reduce the background of thicker carbon foils. STEM imaging was also attempted since it has the advantage of greater contrast since only electrons scattered by the sample are collected to make an image. This allows for even very small clusters of only a few metal atoms to be visualized.
Samples (Figure 2A, green arrow) of monomeric, gold-incubated MBP-MT displayed few discernable gold clusters when visualized by TEM (Figure 3A, arrows). For comparison, we examined grids with Nanogold® clusters. These clusters are directly visible with apparent diameters of about 1.4nm as expected (Figure 4G), though some variation in size/visibility is apparent (Figure 3G, compare individual clusters within hexamers). Unlike their TEM equivalents, STEM images of gold-incubated MBP-MT samples show many examples of small clusters (Figure 3B). We believe that similar numbers of small clusters are also present in the TEM sample (Figure 3A), yet their identification is unreliable due to their small size, which is similar to the grain in carbon foil background. It is noteworthy that fusions with a single copy of MT may be useful as a STEM label.
Figure 3.

Visual comparison of gold-incubated MT fusion proteins. To test the visibility of gold-incubated MBP fusion protein with MT, samples were spotted on to thin carbon foils and visualized by TEM (left panel) and STEM (right panels). Gold clusters can be seen as the very dark spots in these images. Example clusters for each case are denoted by arrows or at the center of open symbols for each sample. Additional clusters can also be seen, especially in images from MBP-MT2 and the Nanogold®. Gold incubated samples of monomeric MBP-MT (A and B) are not as visible as gold incubated monomeric MBP-MT2 (C and D). To insure that MBP-MT2 sample (C and D) were monomeric, the chromatography peak associated with trimer was also visualized. As expected, groups of two of three clusters were observed (E and F). As a control, 1.4nm Nanogold® clusters were also imaged (G and H).
Figure 4.

Visualization and analysis of MBP-MT2 gold clusters. (A) For analysis, images (left) were taken at about 500 nm, 750 nm, and 1.0 μm defocus. From these images, rotational power spectra were calculated and plotted to determine the locations of the zeros within each contrast transfer function (CTF) and according the exact defocus of each image. Scale Bar = 4 nm (B) HRTEM corrected image and particle size analysis of MBP-MT2 gold clusters. The enhanced, corrected image (left) was calculated after CTF correcting, aligning, and averaging the three defocused images. To determine the area associated with each particle, a binary image was produced (center). The intensity threshold value used to produce this binary image was determined by choosing a value that best approximated the area of the particles without selection of pixels within the background carbon. An image produced (right) by subtracting the corrected image (left) from the binary image (center) shows that the best threshold value produces an underestimate of particle size by 1 to 2 pixels along the edge of each particle as witnessed by the white glow around the black particles. Scale bar = 4 nm (left) and 2 nm (right) (C) Particle analysis from the underestimated binary images produces a skewed distribution (circles, dashed line) with an average diameter with a peak at about 1.05 nm. A cumulative distribution function produced from this distribution shows a 50% point at about 1.18 nm. Correcting the underestimate by 2 to 4 pixels, we suggest a 50% point of between 1.30 nm to 1.43 nm.
Samples of gold-incubated MBP-MT2 demonstrated more visible clusters. Notably, MBP-MT2 samples prepared for TEM appeared uniform and showed sizes equivalent, if not slightly larger than, the 1.4nm Nanogold® control sample (Figure 3G). This is puzzling since our clusters should have fewer gold atoms than a Nanogold® cluster (see Discussion). However, STEM images of MBP-MT2 samples, though more visible than their single MT counterparts, show groupings of small gold clusters, rather than single clusters (Figure 3D). Assuming these groupings form from monomeric, gold-bound MBP-MT2 proteins, we suggest that the concatenation of MT subunits does not result in a single cluster but instead in a tight grouping of smaller clusters. Note that in TEM these could easily appear as a single cluster. Although MBP-MT2 gold clusters appear to be an equivalent size to Nanogold® clusters, the Nanogold® clusters appear more uniform in density. However, part of this size effect may result from the imaging condition, such as the amount of defocus, used in these experiments. Nevertheless, gold-incubated MBP-MT2 samples form identifiable densities in TEM and STEM.
Although gold-incubated MBP-MT2 samples showed regular, easily visible clusters in TEM and STEM, it was still unclear if these clusters resulted from single proteins or aggregates of protein formed during grid preparation. To answer this question, samples prepared from fractions of oligomerized gold-incubated MBP-MT2 (Figure 2C) were also imaged. These fractions contain oligomers with 2 to 3 copies of MBP-MT2. TEM and STEM images of these samples can be seen in Figure 3E and 3F, respectively. Interestingly, TEM images of dilute, well-separated gold-incubated MBP-MT2 oligomers show a group of 2 to 3 small gold clusters. This leads us to conclude that each cluster likely resulted from a one of the MBP-MT2 copies in the oligomer. Similarly, STEM images (Figure 3F) show groupings with about 2 to 3 times the number of clusters present in the monomeric MBP-MT2 (Figure 3D). Some of the observable differences between TEM and STEM results may arise from sample preparation. Notably, TEM samples were prepared soon after column elution, while STEM samples were flash frozen, defrosted, and then freeze-dried. The extra STEM preparation steps may alter gold-bound complexes. None the less, these results show that 2 copies of MT should be able to form a TEM label.
Visual characterization of MBP-MT2
To better visualize and characterize clusters, gold-incubated MBP-MT2 samples were imaged using a procedure similar to the image restoration method reported by Typke et al. (1992). This method essentially relies on taking multiple images of the same object at different defocus value, phase correcting the CTFs [Contrast Transfer Function] of these images, aligning the corrected images, and averaging the images together. The final average provides an image with improved quality.. Figure 4A (left 3 images) shows three images taken at a nominal microscope magnification of 230,000X at three different defocus values of an area a TEM grid carbon foil support containing gold-incubated MBP-MT2. This magnification corresponds to a pixel spacing of 0.062 nm on the CCD camera. The quality of these images, exact defocus values, and CTF can be derived from the rotationally averaged power spectra that are plotted in Figure 4A (right). These plots show clear modulation out to at least 0.25 reciprocal angstroms (0.4 nm resolution). Defocus values and phase flipping, using a wiener filter with a delta of the gauss curves around the CTF zeros of 0.2, were performed with IMAGIC–5 (van Heel et al. 1996). These images were then averaged together as shown in Figure 4B (left) to provide a more reliable image than the original three images. Interestingly, this method shows that gold particles vary in shape and size. Nearly all gold-incubated MBP-MT2 clusters are non-spherical unlike the defined shape report for Nanogold® (Safer et al. 1982; Jahn 1999). This non-uniform shape of MBP-MT2 gold clusters perhaps suggests that MT may accumulate gold atoms via the four individual domains in the MT2 construct.
Particle size analysis on the enhanced images allows for an estimation of the average diameter of our MBP-MT2 gold clusters. Analysis was performed using an automated area analysis program in IMAGEJ (Abramoff et al. 2004). Briefly, the method entails setting a threshold pixel intensity value for particles in the image to create a binary image on which area calculations can be performed. The threshold value was set by visual inspection to a value that best filled identifiable gold clusters. One drawback to this method is that weaker gold cluster pixel values, such as those near the edges of particles, have similar intensities to extreme values associated with the carbon support film, yet these carbon-associated pixels do not group into clusters but rather formed diffuse speckling throughout the image. Therefore, a minimum particle size cut-off corresponding to an average diameter 0.7 nm was set within the program parameters to discriminate against the pixels corresponding to dense carbon. As output, the program generates a binary image with the detected particles shown in black within Figure 4B (center). From this image, the projected area of each particle was determined. From each area, A, the average diameter, d̄, of particles could be calculated:
This distribution of average diameters is shown in Figure 4C (circles, dashed line). The curve has a peak at about a diameter of 1.05 nm with an extended tail towards larger diameters. This curve is reminiscent of the extended distributions of the mass spectrometry data (Figures 2B and 2D). The cumulative distribution plot derived from this data shows that the 50% point of the data is at about 1.18 nm. Given Nanogold® has about 70 gold atoms and an apparent diameter of 1.4 nm (Safer et al. 1982; Jahn 1999), this determined particle diameter value is more in line with the size expected for particles with 38 to 65 gold atoms as determined by mass spectrometry. However, this area determination method likely underestimates the edges of the gold clusters as described above.
To evaluate the possible error associated with the particle analysis method, the CTF-corrected image (Figure 4B, left) was subtracted from the threshold binary image (Figure 4B, center). In this way, dark areas in the corrected image that do not overlap with dark areas in the binary image will create light areas in the subtracted image. This effect of non-overlap can be seen as the bright pixels along the edges of clusters in Figure 4B (right), and thus signifies that our method will underestimate the edges of on area by 1 to 2 pixels. Hence, we suggest that the 50% point of the cumulative distribution function is more likely between 1.30 nm to 1.43 nm. This implies that nearly half of the particles appear slightly larger in diameter than Nanogold® in projection images.
Antibody complex: a test case
To evaluate concatenated protein with 2 copies of MT as a component of a simple complex, MBP antibodies (MBPAb) were saturated with gold-incubated MBP-MT2, and the complex was purified by size exclusion chromatography. The results of the separation can be seen in Figure 5A. Controls of antibody alone (closed triangles) and gold-incubated MBP-MT2 alone (open squares) are well separated from samples of complex (circles), which have a peak at about 2.0 ml. Complexes made with only a small excess of gold-incubated MBP-MT2 (open circles) may not have saturated antigen sites. We therefore used complexes produced with a larger excess of antigen (closed circles). To make certain that these samples contained fully saturated complex, fractions collected from the antibody alone sample (Figure 5A, closed triangles) were compared by TEM to the first elution peak from complex-containing samples (Figure 5A, closed circles).
Figure 5.

Preparation of gold-labeled MBP-MT antibody complex. (A) Antibody complexes composed of anti-MBP antibody and gold-incubated MBP-MT2 were prepared on a Superose 12 column. Incubation with low (open circles) and high (closed circles) concentrations of MBP-MT2 resulted in a peak at about 2ml. This was clearly separated from gold-incubated MBP (open squares) and an anti-MBP peak (closed triangles). To guarantee the saturation of antigen binding sites, only the first elution peak from incubations with excess MBP-MT2 (closed circles) was used for EM. For the antibody and antibody images shown, the gallery of images in the left three columns shows typical views of stained complex. The right two columns show an enlarged image of the first image of each set (4th column) and a contour map with the location of the protein shaded in black. (B) Antibodies without MBP-MT2 show the signature similarly sized triple-armed appearance in negative stain. (C) This appearance is in contrast the two large and on small domain appearance of antibody complex. This is best seen in the enlarged image that shows two arms that are significantly larger than the third. All scale bars = 10 nm.
Images of MBP antibody and antibody complexes in negative stain both show three-armed structures. With the antibody alone (Figure 5B), all three arms appear similar in size whereas images of antibody complex with MBP-MT2 (Figure 5C) show two arms larger than the third, due presumably to the two MBP-MT2 molecules. Unfortunately, the uranyl acetate negative stain provides contrast that is similar to that expected from the gold clusters, and the gold is not seen. Attempts to visualize antibody complexes with less dense negative stains proved unsuccessful. The poorer staining quality and lesser contrast associated with these less dense stains made identifying antibody complexes difficult. Nevertheless, these samples in uranyl acetate confirm that antibody complex was formed.
In an attempt to visualize the gold clusters in this simple complex, grids of these samples were prepared for cryo-EM. Figure 6A shows field of gold-labeled MBP-MT2 antibody complex near the edge of a holey carbon film taken at 1 um defocus at 200kV. Groups of nanometer-sized densities separated by medium or lower density material, often in groups, can be seen. Given this low defocus used, we believe that the darker area in the image represent gold clusters while the moderate intensity regions may signify regions of protein. To test the hypothesis that these clusters are gold, the image was CTF corrected, and areas of the image were examined for pixel intensity values in these regions. As controls, areas of ice and the carbon support film were also analyzed as diagrammed in Figure 6B. The results of this analysis are shown in Figure 6C. The areas presumed to be gold (solid line) have pixel intensity values lower than the relatively thick carbon support (thin dashed line) and much lower than the surrounding ice values (medium dashes line). Although this is not proof that these regions are gold cluster, their sizes and relative pixel intensity values is consistent with this possibility. Additionally, some examples of cryo-EM imaged antibody complex in characteristic views can be seen in Figure 6D.
Figure 6.

Potential use of gold-labeled MBP-MT2 (A) Cryo-EM images of antibody complexes frozen within the holes of carbon films show the potential usefulness of this method. The low (about −1 μm) defocus image shows diffuse spots that likely result from gold particles. The spots appear to be in clusters suggesting some degree of aggregation of antibody complex. The schematic image (B) is shown as an aid to interpret the image shown in (A). To help verify the identity of the gold, a CTF-corrected image was sampled and analysed for the intensity values found within the image. The solid areas in the schematic image show the locations of pixel intensity sampling used for this analysis. All presumed gold areas, which were masked out with surrounding ice areas, were totalled as one area. (C) The graph shows the distribution of pixel intensities determined by the pixel intensity analysis. The presumed gold areas show intensity values darker than the thick carbon film used as a support and thus suggest their identity as gold. (D) With a combination of complex flexibility, variation in orientation, and some degree of aggregation, EM images from cryo-prepared antibody complex only occasionally show the characteristic views seen in negative stain. Areas of darker density (arrows) can sometimes be seen at the ends of the longer arms of the density; thee are presumably due to the gold clusters and thus hint at the potential of this method.
Discussion
Gold binding by the concatenated MT proteins
Fusion proteins with concatenated MT are able to bind gold as was shown in previous studies. In this work, incubated MBP-MT samples acquired about 13 gold atoms while MBP-MT2 samples, with its two copies of MT, acquired about 38 gold atoms, or about 19 per MT, as determined at their mass spectrometry peak amplitudes. From these experiments we get an value of 16 gold atoms per copy of MT, which are in agreement with past studies that showed a range of metal binding stoichiometries with 12 to 20 gold atoms bound the a single MT copy (Laib et al. 1985; Shaw III et al. 1990). Also in this work, distributions associated with these peaks are skewed toward values greater than their peak amplitudes. These distributions indicate a range of gold binding with their upper end fall off values corresponding to a gold to cysteine ratio of 1 to 1 or greater. Similar ratios have been seen not just for gold but also for silver and mercury (Lu et al. 1993; Li and Otvos 1996). Given the skewed shape of the distributions, the values we have witnessed previously for unfused-MT (Mercogliano and DeRosier, 2006), and the well-established fact that a single copy of MT will accommodate 20 gold atoms (Laib et al. 1985), we believe that the average value of 16 gold atoms/MT found here is a conservative value. When considering gold clusters currently available as TEM labels, gold-bound MT is slightly better than undecagold, which can only be visualized through averaging, but has about 3 to 4 times fewer gold atoms than Nanogold® sized clusters, which have about 70 gold atoms and can be seen directly (Safer et al. 1982; Jahn 1999). Thus it should take 3 to 4 copies of MT to form similarly visible clusters.
Visualization of MT fusion proteins
In our results, MBP fusion proteins with only 2 copies of MT appear almost as visible as Nanogold®. Many gold clusters formed with MBP-MT2 appeared as 1.4 nm or slightly larger clusters in TEM, although at times they appear less intense than their Nanogold® counterparts. STEM imaging hints at the possible reason for this observation. The MT-formed clusters in STEM images appear more diffuse as if the gold atoms are not packed in a single, tight gold cluster. With the lower magnification TEM images where defocus blurs out the individual atoms, the clusters appear as a single mass. Conversely, higher magnification TEM images show clusters with more irregular shapes. This again suggests that gold atoms may be sequestered into smaller domains within the MT subunits. Since the mass spectrometry data demonstrates that a proportion of the clusters have gold atoms bound at a ratio greater than one gold atom per cysteine, this would suggest some gold atoms have been reduced to gold(0). Thus, it was hoped that a crystal lattice might form in such clusters. However, no crystalline material was observed. This is not surprising since if such a lattice is present in the samples, it would be small, consisting of only a few atoms, and also rare since only a proportion of clusters contain these reduced atoms. Furthermore, since imaging suggests binding may be localized within the MT domains, such reduced gold atoms may be correspondingly distributed. The difference witnessed between the two imaging techniques may be accounted for by the differences in the preparations of the samples for viewing by the two methods (see Materials and Methods). These differences may have altered the sample and thereby account for the difference in appearance between TEM and STEM. Nevertheless, 2 copies of MT are sufficient to be directly visible in TEM images.
Concatenated MT as a labeling method
Visualization of concatenated MT-containing fusion proteins in biological complexes has proven difficult. We failed in two attempts to produce functional MT-fusions to other proteins (RecA and kinesin). It is unclear whether these difficulties result from the general difficulty of making functional protein chimeras or from a specific problem with MT fusions perhaps arising from the large number of cysteines. Chromatography of MT-fused MBP protein (Figure 2A and 2C) did show oligomerization, which might contribute to the difficulties. Oligomerization most likely is a consequence of the large number of cysteines in MT. If this is the case, preparation of MT with strong binding, oxygen insensitive metals such as gold or cadmium, as well as more rigorous purification steps may provide more stable material.
We were able to retain maltose binding activity in the MBP-MT chimera. We made a complex of this using an anti-MBP antibody. The characteristic appearance and ease of formation of these complexes (Figure 5C and 6D) leads us to believe that the incubations used to fill gold binding sited in MT have not adversely affected the MBP. At times, cryo-EM images hint at more strongly scattering densities associated with the extended arms of the antigen (see arrow in Figure 6). These are consistent with the size and expected location of gold clusters in the complex. Unfortunately, the small size and flexibility of the complex make detection of identifiable views and image averaging impractical. Our results show the potential of this method but more work is still needed to bring it to fruition.
Acknowledgments
We would like the thank Martha Simon and Joseph Wall of the Brookhaven National Laboratory for STEM imaging. This work was supported by National Institute of General Medical Sciences grants R01-GM26357, R01-GM35433, and P01-GM62580.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- Bohm J, Frangakis AS, Hegerl R, Nickell S, Typke D, Baumeister W. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proc Natl Acad Sci U S A. 2000;97:14245–14250. doi: 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez E, Powell RD, Furuya FR, Hainfeld JF, Schaaff TG, Shafigullin M, Stephens PW, Whetten RL. Greengold, a giant cluster compound of unusual electronic structure. Eur Phys J D. 1999;9:647–651. [Google Scholar]
- Hainfeld JF, Powell RD. New frontiers in gold labeling. J Histochem Cytochem. 2000;48:471–480. doi: 10.1177/002215540004800404. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- Jahn W. Review: chemical aspects of the use of gold clusters in structural biology. J Struct Biol. 1999;127:106–112. doi: 10.1006/jsbi.1999.4123. [DOI] [PubMed] [Google Scholar]
- Jiang DT, Heald SM, Sham TK, Stillman MJ. Structures of the Cadmium, Mercury, and Zinc Thiolate Clusters in Metallothionein. J Am Chem Soc. 1994;116:11004–11013. [Google Scholar]
- Laib JE, Shaw CF, 3rd, Petering DH, Eidsness MK, Elder RC, Garvey JS. Formation and characterization of aurothioneins: Au,Zn,Cd-thionein, Au,Cd-thionein, and (thiomalato-Au)chi-thionein. Biochemistry. 1985;24:1977–1986. doi: 10.1021/bi00329a027. [DOI] [PubMed] [Google Scholar]
- Li H, Otvos JD. HPLC characterization of Ag+ and Cu+ metal exchange reactions with Zn- and Cd-metallothioneins. Biochemistry. 1996;35:13937–13945. doi: 10.1021/bi961402f. [DOI] [PubMed] [Google Scholar]
- Lu W, Zelazowski A, Stillman MJ. Mercury Binding to metallothionein: Formation of the Hg18-MT Species. Inorg Chem. 1993;32:919–926. [Google Scholar]
- Mercogliano CP, DeRosier DJ. Gold nanocluster formation using metallothionein: mass spectrometry and electron microscopy. J Mol Biol. 2006;355:211–223. doi: 10.1016/j.jmb.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Nickell S, Kofler C, Leis AP, Baumeister W. A visual approach to proteomics. Nat Rev Mol Cell Biol. 2006;7:225–230. doi: 10.1038/nrm1861. [DOI] [PubMed] [Google Scholar]
- Nielson KB, Atkin CL, Winge DR. Distinct metal-binding configurations in metallothionein. J Biol Chem. 1985;260:5342–5350. [PubMed] [Google Scholar]
- Nielson KB, Winge DR. Order of metal binding in metallothionein. J Biol Chem. 1983;258:13063–13069. [PubMed] [Google Scholar]
- Ortiz JO, Förster F, Kürner J, Linaroudis AA, Baumeister W. Mapping 70S ribosomes in intact cells by cryoelectron tomography and pattern recognition. J Struct Biol. 156:334–341. doi: 10.1016/j.jsb.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Palacios O, Polec-Pawlak K, Lobinski R, Capdevila M, Gonzalez-Duarte P. Is Ag(I) an adequate probe for Cu(I) in structural copper-metallothionein studies? The binding features of Ag(I) to mammalian metallothionein 1. J Biol Inorg Chem. 2003;8:831–842. doi: 10.1007/s00775-003-0481-4. [DOI] [PubMed] [Google Scholar]
- Safer D, Hainfeld J, Wall JS, Reardon JE. Biospecific labeling with undecagold: visualization of the biotin-binding site on avidin. Science. 1982;218:290–291. doi: 10.1126/science.7123234. [DOI] [PubMed] [Google Scholar]
- Schaaff TG, Whetten RL. Giant Gold-Glutathione Cluster Compounds: Intense Optical Activity in Metal-Based Transitions. J Phys Chem B. 2000;104:2630–2641. [Google Scholar]
- Schmitz G, Minkel DT, Gingrich D, Shaw CF., 3rd The binding of Gold(I) to metallothionein. J Inorg Biochem. 1980;12:293–306. doi: 10.1016/s0162-0134(00)80270-6. [DOI] [PubMed] [Google Scholar]
- Shaw CF, III, Laib JE, Savas MM, Petering DH. Biphasic Kinetics of Aurothionein Formation from Gold Sodium Thiomalate. Inorg Chem. 1990;29:403–408. [Google Scholar]
- Suzuki K. Methods in Enzymology. Academic Press; 1991. Purification of Vertebrate Metallothioneins; pp. 252–263. [DOI] [PubMed] [Google Scholar]
- Thomas D, Morgan DG, DeRosier DJ. Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J Bacteriol. 2001;183:6404–6412. doi: 10.1128/JB.183.21.6404-6412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt TG, Volkmann N, Skiniotis G, Goldie KN, Muller J, Mandelkow E, Hoenger A. Microscopic evidence for a minus-end-directed power stroke in the kinesin motor ncd. Embo J. 2002;21:5969–5978. doi: 10.1093/emboj/cdf622. [DOI] [PMC free article] [PubMed] [Google Scholar]