Differential body weight and feeding responses to high fat diet in rats and mice lacking cholecystokinin 1 receptors (original) (raw)
. Author manuscript; available in PMC: 2008 Jul 1.
Published in final edited form as: Am J Physiol Regul Integr Comp Physiol. 2007 Apr 4;293(1):R55–R63. doi: 10.1152/ajpregu.00002.2007
Abstract
Prior data have demonstrated differential roles for cholecystokinin (CCK) 1 receptors in maintaining energy balance in rats and mice. CCK1 receptor deficiency results in hyperphagia and obesity of Otsuka Long-Evans Tokushima Fatty (OLETF) rats, but not in mice. To ascertain the role of CCK1 receptors in high fat diet (HFD)-induced obesity, we compared alterations in food intake, body weight, fat mass, plasma glucose and leptin levels, and patterns of hypothalamic gene expression in OLETF rats and mice lacking CCK1 receptors in response to a 10-week exposure to HFD. As compared with Long-Evans Tokushima Otsuka (LETO) control rats, OLETF rats on HFD had sustained overconsumption over the 10-week period. High fat feeding resulted in greater increases in body weight and plasma leptin levels in OLETF than in LETO rats. In situ hybridization determinations revealed that while HFD reduced neuropeptide Y (NPY) mRNA expression in both the arcuate nucleus (Arc) and the DMH of LETO rats, HFD resulted in decreased NPY expression in the Arc but not in the DMH of OLETF rats. In contrast to these results in OLETF rats, HFD increased food intake and induced obesity to an equal degree in both wild type and CCK1 receptor−/− mice. NPY gene expression was decreased in the Arc in response to HFD, but was not detectable in the DMH in both wild type and CCK1 receptor−/− mice. Together, these data provide further evidence for differential roles of CCK1 receptors in the controls of food intake and body weight in rats and mice.
Keywords: Neuropeptide Y (NPY), proopiomelanocortin (POMC), dorsomedial hypothalamic nucleus, in situ hybridization
Peripheral cholecystokinin (CCK) acts as a satiety signal to limit meal size. Intraperitoneal injection of CCK decreases food intake and produces the earlier appearance of a behavioral satiety sequence (2). The actions of peripheral CCK in satiety are mediated through interactions with CCK1 receptors (previously named as CCK-A receptors). CCK1 receptor antagonists dose-dependently and competitively block the feeding inhibitory effect of exogenous CCK, whereas administration of a dose range of CCK-B receptor antagonist is ineffective (11, 21, 22, 27). Consistent with exogenous CCK’s actions, endogenous CCK is released from the duodenum and jejunum in response to the intra-luminal presence of nutrient-digestive products. Intraintestinal nutrient infusions reduce subsequent food intake, and such suppression is attenuated or blocked by CCK1 receptor antagonists (33). Furthermore, administration of CCK1 receptor antagonists increase food intake in a variety of test situations (22, 26).
CCK1 receptor disruption results in a deficit in CCK satiety actions in both rats and mice. The Otsuka Long-Evans Tokushima fatty (OLETF) rat is an animal model with a congenital CCK1 receptor deficiency, resulting from a 6847-base pair deletion spanning the promoter region and the first and second exons of the CCK1 receptor gene (31). OLETF rats develop disordered feeding behavior, characterized by increases in meal size, and they do not have a feeding inhibitory response to peripheral CCK administration (23). Similar to OLETF rats, CCK1 receptor−/− mice have no feeding response to peripheral exogenous CCK (19) and consume larger meals (7). Although both rats and mice lacking CCK1 receptors have a deficit in peripheral CCK satiety, important aspects of their phenotypes differ. CCK1 receptor disruption results in hyperphagia and obesity in OLETF rats, but does not alter overall daily food intake and body weight in CCK1 receptor−/− mice. We have suggested that the hyperphagia and obesity in OLETF rats is the outcome of both a deficit in peripheral CCK satiety, leading to increased meal size (23), and a deficit in central CCK signaling, resulting in dysregulation of neuropeptide Y (NPY) gene expression in the dorsomedial hypothalamus (DMH) and an overall dysfunction in energy balance control (4). In support of the view that CCK regulates DMH NPY signaling to control food intake, DMH NPY neurons contain CCK1 receptors and parenchymal picomole administration of CCK into the DMH inhibits food intake and down-regulates NPY gene expression in the DMH in intact rats (7). In contrast, intact mice do not have CCK1 receptors in the DMH and NPY gene expression is not altered in the DMH of CCK1 receptor−/− mice (7). Thus, mice lacking CCK1 receptors do not have a deficit in the regulation of NPY gene expression in the DMH, and appear to be able to compensate for the absence of CCK satiety signaling. They do not become hyperphagic and obese.
We have demonstrated that OLETF rats have deficits in their ability to alter their food intake to increases in dietary fat content (28), findings that are consistent with a feedback role of CCK in mediating the effects of dietary fats. Such deficits result in insensitivity to lipid preloads and increased rates of body weight gain when exposed to a high fat diet. It is unclear from these experiments whether such deficits result from an absence of peripheral CCK satiety signaling, an absence of central CCK signaling or both. Since CCK1 receptor−/− mice have a similar absence of peripheral CCK satiety signaling but appear to have intact controls of overall food intake and body weight, a comparison of OLETF rats and CCK1 receptor−/− mice in response to high fat diet would address this issue. In the present experiments, we sought to determine whether CCK1 receptor deficiency has an impact on high fat diet-induced obesity of rats and mice. We compared alterations in food intake, body weight, fat mass, plasma glucose and leptin levels, and patterns of hypothalamic gene expression for NPY, proopiomelanocortin (POMC) and corticotrophin-releasing factor (CRF) in OLETF rats and CCK1 receptor−/− mice with access to a high fat diet for 10 weeks. Overall, the present experiments demonstrate differential feeding and body weight responses to high fat diet in rats and mice lacking CCK1 receptors.
METHODS
Animals and high fat diets
Male OLETF and age matched male LETO rats were obtained as a generous gift of the Tokushima Research Institute, Otsuka Pharmaceutical, Tokushima, Japan. Animals were 5 week old when they arrived in our laboratory. On arrival, there were no significant differences in body weight between two strains weighing 101–135g in OLETF rats and 99–118g in LETO rats (_P_>0.05). All animals were individually housed in hanging wire mesh cages and maintained on a 12:12-h light-dark cycle (lights on at 6:00 AM) in a temperature-controlled colony room (22–23°C) and, for the initial 2 weeks in the laboratory, were maintained with ad libitum access to a standard chow diet. A strain of CCK1 receptor−/− mice was generated on 129/SvEv mice with targeted disruption of the CCK1 receptor gene as previously described (19). CCK1 receptor−/− male mice and their wild type male littermates were individually housed and maintained on a 12:12-h light-dark cycle (lights on at 6:00 AM). All procedures were approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University or the Tufts-New England Medical Center Animal Research Committee.
In the first experiment, at 7 weeks of age 20 OLETF and 14 age-matched LETO rats were divided into 6 groups. One group of 6 OLETF rats and one group of 4 LETO rats continued to have ad lib access to a standard chow diet (low-fat/low-caloric diet: 15.8% fat, 65.6% carbohydrate, and 18.6% protein in kcal%, 3.37 kcal/g, Purina Rodent Chow #5P07), and served as regular chow diet controls (named as OLETF Chow and LETO Chow). The second groups of 7 OLETF rats and 5 LETO rats were switched to ad lib access to a medium-fat/medium-caloric diet (45% fat, 35% carbohydrate, and 20% protein in kcal%, 4.7 kcal/g, Research Diets, Inc, New Brunswick, NJ, #D12451) and served as medium-fat diet groups (named as OLETF MF and LETO MF). The last groups of 7 OLETF rats and 5 LETO rats were switched to ad lib access to a high-fat/high-caloric diet (60% fat, 20% carbohydrate, and 20% protein in kcal%, 5.2 kcal/g, Research Diets, Inc #D12492) and served as high-fat diet groups (named as OLETF HF and LETO HF). Dietary regimen was maintained for a 10-week period. Body weights were measured daily and food intake was recorded weekly. All animals had free access to tap water. At the end of 10 weeks, rats were decapitated under ether inhalation anesthesia between 9AM and 11AM. The left side of epidydimal and inguinal subcutaneous white adipose tissue as well as interscapular brown adipose tissue were harvested and weighed. Trunk blood was taken for evaluation of plasma levels of glucose and leptin. Plasma glucose levels were measured by using a blood glucose meter (Glucometer Elite, Bayer) and plasma leptin concentration was determined by rat leptin radioimmunoassay kit (Linco Research, Inc., St. Charles, MO). Brains were removed and rapidly frozen with icy iso-pentane for subsequent analyses of hypothalamic gene expression.
In the second experiment, 9 week old CCK1 receptor−/− mice (n=15) and wild type litter mates (n=13) were assigned to 4 groups: 7 wild type and 7 CCK1 receptor−/− mice with ad lib access to a standard chow diet served as regular diet controls (named as WT Chow and KO Chow) The other 6 wild type and 8 CCK1 receptor−/− mice had ad lib access to a high fat/high caloric diet and served as high-fat diet groups (named as WT HF and KO HF). Both regular chow and high fat diets were the same as those used in rats. Dietary regimens were maintained for a 10-week period. Body weights were measured daily and food intake was recorded weekly. All animals had free access to tap water. At the end of 10 weeks, all mice were killed between 9AM and 11AM. Similar to rats, the left side of epidydimal and inguinal subcutaneous white adipose tissue as well as interscapular brown adipose tissue were harvested and weighed. Trunk blood was taken for evaluation of plasma levels of glucose and leptin. Brains were removed and saved for subsequent analyses of hypothalamic gene expression.
Cryosections and Riboprobes
Fourteen km coronal sections were cut via a cryostat, mounted on superfrost/plus slides (Fisher Scientific), fixed with 4% paraformaldehyde and stored at −70°C for later in situ hybridization determination. In rats, sections for NPY and POMC mRNA determinations were taken at the level of the Arc, 3.1–3.5 mm caudal to bregma and for CRF at the level of the PVN, 1.7–2.0 mm caudal to bregma (25). In mice, sections for NPY and POMC mRNA determinations were taken at the level of the Arc, 1.7–2.0 mm caudal to bregma and for CRF at the level of the PVN, 0.7–1.0 mm caudal to bregma (24).
As previously described (7), 35S-labeled antisense riboprobes of NPY, POMC, or CRF were transcribed from rat NPY precursor cDNA, mouse full length POMC cDNA, or rat CRF precursor cDNA respectively by using in vitro transcription systems (Promega, Madison, WI), and purified by Quick Spin RNA Columns (Roche, Indianapolis, IN).
In Situ Hybridization
Four-six sections (1 in 6 sections for rats or 1 in 5 sections for mice) throughout the brain regions of interest were selected, anatomically matched among animals and used for each in situ hybridization determination. Briefly, frozen tissue sections were allowed to warm to room temperature, treated with acetic anhydride and incubated in hybridization buffer containing 50% formamide, 0.3 M NaCl, 10 mM Tris·Cl, pH 8.0, 1 mM EDTA, pH 8.0, 1X Denhardt’s solution (Eppendorf), 10% dextran sulfate, 10 mM DTT, 500 μg/ml yeast tRNA and 108 cpm/ml of 35S-UTP at 55°C overnight. After hybridization, sections were washed three times with 2 × SSC, treated with 20 kg/ml RNase A (SIGMA) at 37 °C for 30 min, and then rinsed in 2 × SSC twice at 55 °C and washed twice in 0.1 × SSC at 55 °C for 15 min. Slides were dehydrated in an ascending series of ethanol, air-dried and exposed with BMR-2 film (Kodak) for 1–3 days (7).
Quantitation of in situ hybridization
Quantitative analysis of in situ hybridization signals was done with NIH Scion image software (National Institutes of Health). Autoradiographic images were first scanned by EPSON professional scanner (EPSON) and saved in a computer for subsequent analyses with Scion image program using autoradiographic 14C micro scales (Amersham) as a standard. Data for each animal was a mean of the product of hybridization area × density (background density was subtracted) obtained from 4–6 sections. Data from each group were normalized to LETO Chow rats or WT Chow mice as 100 %. All data are presented as mean ± SEM.
For statistical analysis, data were analyzed using two-way ANOVA for changes in body weight, food intake, fat pad weight, plasma glucose and leptin concentration, and levels of hypothalamic gene expression across the six rat or four mouse experimental groups. ANOVA’s were followed by pairwise multiple Fisher least significant difference (LSD) comparisons. P < 0.05 was interpreted as a significant difference.
RESULTS
Effects of high fat diet on body weight and food intake
As shown in Figure 1A, high fat diets increased body weight gain in both OLETF and LETO rats. Overall, ANOVA demonstrated significant effects of strain [_F_(1,28)=160.285, P<0.001] and high fat diet [_F_(2,28)=18.817, _P_<0.001], as well as a significant interaction between strain and diet across the 10-week period of access to high fat diets [_F_(20,380)=1.749, _P_=0.0245], indicating that high fat diets resulted in a greater increase in body weight in OLETF rats. Fisher LSD comparisons revealed that within the LETO rats, the two groups of LETO MF and HF rats gained more body weight than LETO Chow animals, with 13.3% (_P_ =0.013) and 16.2% increases in body weight (_P_ =0.001) relative to LETO Chow controls respectively. The changes in body weight between the LETO MF and HF groups did not differ (_P_>0.05). As compared with LETO Chow animals, OLETF Chow rats became heavier at 6 weeks of age and gained 41% more body weight at 17 weeks of age (Figure 1A). OLETF rats on MF and HF diets had an even greater increase in body weight, gaining 16.7% more on the MF diet and 21.2% more on the HF diet relative to OLETF Chow animals (P<0.001 in both comparisons). Again, the magnitude of weight gain on the MF and HF diets did not differ (_P_>0.05, Figure 1A).
Figure 1.
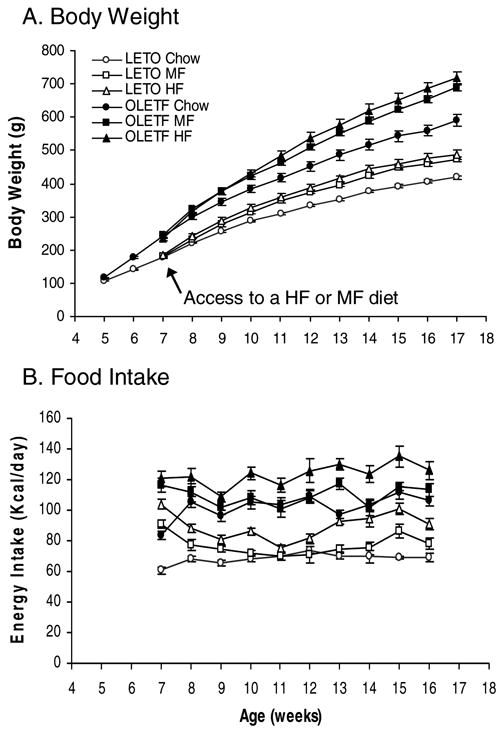
Effects of high fat diet on body weight and food intake in OLETF rats. A. Body Weight: High fat feeding resulted in greater increases in body weight in obese OLETF rats than in lean LETO control rats. B. Caloric Intake: Access to a high fat diet leads to greater overconsumption in OLETF rats than in LETO rats. LETO Chow: Long-Evans Tokushima Otsuka control rats with access to a regular chow diet; LETO MF: LETO rats with access to a medium fat diet; LETO HF: LETO rats with access to a high fat diet; OLETF Chow: Otsuka Long-Evans Tokushima Fatty rats with access to a regular chow diet; OLETF MF: OLETF rats with access to a medium fat diet; and OLETF HF: OLETF rats with access to a high fat diet. Values are means ± SEM. n = 4–7/group.
OLETF rats had altered food intake in response to high fat diet. ANOVA demonstrated significant effects of strain [_F_(1,28)=258.666, P<0.001] and diet [_F_(2,28)=38.034, _P_<0.001] as well as a significant interaction between strain and diet [_F_(2,28)=6.265, _P_<0.01] over the 10 week period (Table 1). The finding of a strain and diet interaction indicated that high fat diet had a greater effect on food intake in OLETF rats. Fisher LSD comparisons revealed that LETO rats initially increased their caloric intake in response to high fat diet (_P_<0.05), but the intake was normalized by the second week on the MF diet (_P_>0.05) and by the fifth week on the HF diet (_P_>0.05). This normalization was then followed by a second period of overconsumption at week 13 on the HF diet (P<0.05) and at week 15 on the MF diet (_P_<0.05) (Figure 1B). Overall, LETO HF animals ate the greatest amount of food and LETO MF rats had more food intake than LETO Chow rats (Table 1). In contrast to LETO rats, OLETF rats with access to MF and HF diets had sustained overconsumption (Figure 1B). Over the 10 week period, OLETF HF rats consumed more calories than OLETF MF and Chow animals (Table 1, _P_<0.05, respectively), but intake did not differ between OLETF MF and Chow rats (Table 1, _P_>0.05).
Table 1.
Effects of high and medium-fat diets on energy intake, fat mass, plasma glucose and leptin levels in rats.
| Cumulative Intake, Kcal | Epididymal Fat, g | Subcutaneous Fat, g | Interscapular Brown Fat, g | Glucose mg/dl | Plasma Leptin ng/ml | |
|---|---|---|---|---|---|---|
| LETO Chow | 4790 ± 77 | 3.76 ± 0.22 | 3.45 ± 0.38 | 0.75 ± 0.04 | 187 ± 5.9 | 10.1 ± 1.43 |
| LETO MF | 5196 ± 85a | 7.81 ± 0.54a | 6.77 ± 0.76 | 0.96 ± 0.06 | 188 ± 12.9 | 18.0 ± 1.75a |
| LETO HF | 6259 ± 70ab | 8.21 ± 0.33a | 9.58 ± 0.83a | 0.94 ± 0.09 | 224 ± 15.4 | 25.0 ± 3.9a |
| OLETF Chow | 7150 ± 214a | 8.67 ± 0.97a | 11.95 ± 2.88a | 1.62 ± 0.13a | 285 ± 30.2a | 29.1 ± 4.19a |
| OLETF MF | 7545 ± 139b | 12.66 ± 0.78bd | 19.97 ± 1.75bd | 2.01 ± 0.19bd | 300 ± 19.5b | 36.4 ± 2.59b |
| OLETF HF | 8490 ± 309cde | 12.60 ± 0.57cd | 22.63 ± 1.56cd | 2.08 ± 0.05cd | 319 ± 20.2c | 49.1 ± 5.88cd |
In contrast to the results in OLETF rats, CCK1 receptor−/− mice had the same body weight as wild-type littermates (Figure 2A). High fat feeding resulted in increases in body weight in both CCK1 receptor−/− and wild type animals. By the end of 10 weeks of access to a high fat diet, ANOVA demonstrated a significant effect of diet [_F_(1,24)=19.294, P<0.001], but no significant effect of stain [_F_(1,24)=0.00387, _P_=0.951] and no interaction between strain and diet [_F_(1,24)=0.00363, _P_=0.952]. Overall, KO and WT mice gained the same amount of body weight on the HF diet (Figure 2A).
Figure 2.
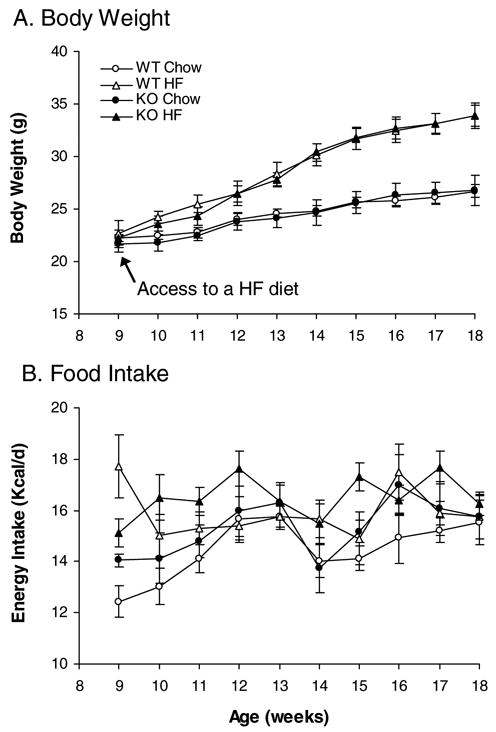
Effects of high fat diet on body weight and food intake in CCK1 receptor−/− mice. High fat feeding induced obesity (A) and increased food intake (B) at an equal degree in wild type and CCK1 receptor−/− mice. WT Chow: wild type mice with access to a regular chow diet; WT HF: WT mice with access to a high fat diet; KO Chow: CCK1 receptor knockout mice with access to a regular chow diet; KO HF: CCK1 receptor knockout mice with access to a high fat diet. Values are means ± SEM. n = 6–8/group.
Feeding response to high fat diet in rats and mice also differed. CCK1 receptor disruption did not affect daily food intake in mice [_F_(1,24)=1.531, _P_=0.228](Figure 2B and Table 2). KO Chow mice consumed the same cumulative amount of food as WT Chow mice over the 10-week period (Table 2). High fat feeding increased food intake in both CCK1 receptor−/− and wild type mice [_F_(1,24)=7.805, P<0.05], but food intake did not differ between KO HF and WT HF mice (_P_>0.05). ANOVA did not reveal any interaction between strain and diet [_F_(1,24)=0.0678, _P_=0.797] over the 10-week observation period.
Table 2.
Effects of high fat diet on energy intake, fat mass, plasma glucose and leptin levels in mice.
| Cumulative Intake, Kcal | Epididymal Fat, g | Subcutaneous Fat, g | Interscapular Brown Fat, g | Plasma Glucose mg/dl | Plasma Leptin ng/ml | |
|---|---|---|---|---|---|---|
| WT Chow | 1014 ± 46 | 0.49 ± 0.045 | 0.23 ± 0.036 | 0.24 ± 0.023 | 170 ± 8.6 | 4.3 ± 0.8 |
| WT HF | 1112 ± 56a | 0.93 ± 0.042a | 0.47 ± 0.072a | 0.21 ± 0.02 | 198 ± 13.8 | 15.7 ± 2.8a |
| KO Chow | 1071 ± 56 | 0.41 ± 0.053 | 0.17 ± 0.042 | 0.21 ± 0.022 | 185 ± 6.8 | 2.9 ± 0.5 |
| KO HF | 1155 ± 45b | 0.92 ± 0.088b | 0.37 ± 0.063b | 0.19 ± 0.013 | 172 ±10.2 | 10.02 ± 0.2b |
Effects of high fat diet on body fat mass, plasma glucose and leptin levels
In the rats, there were significant effects of strain on the amount of epididymal fat [_F_(1,28)=67.147, P<0.001], subcutaneous fat [_F_(1,28)=72.349, _P_<0.001] and brown adipose tissue [_F_(1,28)=97.189, _P_<0.001], as well as significant diet effects on epididymal fat mass [_F_(2,28)=21.199, _P_<0.001], subcutaneous fat mass [_F_(2,28)=12.406, _P_<0.001] and brown fat mass [_F_(2,28)=3.843, _P_=0.034]. Fisher LSD comparisons demonstrated that both LETO MF and HF rats had significantly increased epididymal fat mass relative to LETO Chow rats, but only LETO HF rats had increased subcutaneous fat mass and there were no significant differences in brown fat mass among the three LETO groups (Table 1). As compared with LETO Chow rats, OLETF Chow rats had increased fat mass in all three adipose tissues (Table 1). High fat diets resulted in increases in epididymal, subcutaneous and brown fat mass in both OLETF MF and HF rats relative to OLETF Chow animals (Table 1). The effects of the MF and HF fat diets did not differ in OLETF rats. Although OLETF rats gained more fat mass in response to high fat diet, ANOVA did not reveal a significant interaction between strain and diet in epididymal [_F_(2,28)=0.0851, _P_>0.05], subcutaneous [_F_(2,28)=1.252, _P_>0.05], or brown adipose tissue [_F_(2,28)=0.567, _P_>0.05].
As presented in Table 1, plasma glucose levels were significantly increased in the OLETF rat as compared with the LETO animal [_F_(1,28)=33.816, P<0.001]. Although there was a tendency for high fat diets to increase plasma glucose levels in both strains, the effects were not significant [_F_(2,28)=1.559, _P_=0.230] and there was no significant interaction between strain and diet [_F_(2,28)=0.0956, _P_=0.909].
Consistent with the alterations in fat mass, plasma leptin levels were significantly increased in OLETF rats as compared to LETO rats [_F_(1,28)=36.340, P<0.001]. High fat diets increased plasma leptin levels in both strains [_F_(2,28)=8.657, _P_=0.001], but there was no significant interaction between strain and diet [_F_(2,28)=0.301, _P_=0.742]. OLETF Chow rats had a 2.9-fold increase in plasma leptin levels relative to LETO Chow rats (Table 1). High fat diets resulted in 1.8-fold and 2.5-fold increases in plasma leptin levels of LETO MF and HF rats as compared to LETO Chow animals, and 1.3 and 1.7 folds in OLETF rats in response to MF and HF diets respectively (Table 1).
In contrast to the findings in rats, CCK1 receptor deficiency did not result in alterations in epididymal ([_F_(1,24)=0.372, _P_=0.548], subcutaneous [_F_(1,24)=2.566, _P_=0.122], or brown adipose tissue [_F_(1,24)=1.658, _P_=0.211] in mice (Table 2). In response to high fat diet, both CCK1 receptor−/− and wild type mice had increased epididymal [_F_(1,24)=64.840, P<0.001] and subcutaneous adipose tissues [_F_(1,24)=19.752, P<0.001], but had no alterations in brown adipose tissue [_F_(1,24)=1.315, _P_=0.263] (Table 2). The increases in epididymal and subcutaneous fat mass did not differ between KO HF and WT HF mice as evidenced by the lack of a significant interaction between strain and diet for epididymal ([_F_(1,24)=0.490, _P_=0.491], subcutaneous [_F_(1,24)=0.00367, _P_=0.952] and brown adipose tissue [_F_(1,24)=0.0103, _P_=0.920] (Table 2).
Analysis of plasma glucose levels did not reveal any significant effects of strain [_F_(1,24)=0.428, _P_>0.05] and diet [_F_(1,24)=1.030, _P_>0.05]. At sacrifice, KO Chow mice had normal plasma glucose levels (Table 2, _P_>0.05). Although body weight and fat mass were significantly increased in response to high fat diets, plasma glucose levels were not altered in both KO HF and WT HF mice (Table 2, _P_>0.05).
Consistent with alterations in body weight and fat mass, plasma leptin levels were not affected by the absence of CCK1 receptors [_F_(1,24)=0.957, _P_>0.05], but were significantly increased by high fat diet [_F_(1,24)=33.691, P<0.001]. As presented in Table 2, both KO HF and WT HF mice had significantly increased plasma leptin levels, but the levels did not differ between KO HF and WT HF mice. There was no significant interaction between strain and diet [_F_(1,24)=0.00425, _P_>0.05].
Effects of high fat diet on hypothalamic NPY, POMC and CRF gene expression
In situ hybridization determination revealed that Arc NPY gene expression was significantly affected by strain [_F_(1,28)=4.284, P<0.05], and high fat diet access [_F_(1,28)=3.611, _P_<0.05], but there was no significant interaction between strain and diet [_F_(1,28)=1.524, _P_>0.05]. As compared toLETO Chow rats, Arc NPY gene expression was decreased 31% and 32% in LETO MF and HF rats respectively, and the changes in Arc NPY gene expression did not differ between LETO MF and HF rats (Figure 3A). Relative to LETO Chow controls, OLETF rats on Chow, MF and HF diets had a 32%, 36% and 50% reduction in Arc NPY gene expression respectively (Figure 3A, P<0.05). OLETF HF rats had further reduced Arc NPY gene expression by 27 % as compared to OLETF Chow rats (Figure 3A, P<0.05).
Figure 3.
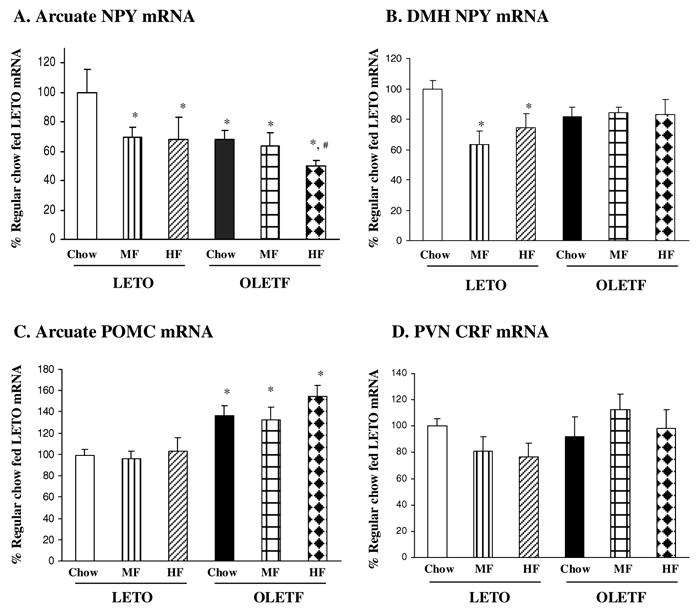
Effects of high fat diet on hypothalamic NPY, POMC and CRF gene expression in OLETF rats. (A) In response to high fat diet, arcuate (Arc) NPY gene expression was decreased in both LETO and OLETF rats. (B) Dorsomedial hypothalamic (DMH) NPY gene expression was decreased in LETO rats but not altered in OLETF rats. (C) OLETF rats had increased Arc POMC gene expression, and high fat feeding did not affect Arc POMC. (D) Paraventricular (PVN) CRF gene expression was not affected by high fat feeding in either LETO or OLETF rats. Values are means ± SEM. n = 4–7/group. *P < 0.05 as compared to LETO Chow values, and #P < 0.05 as compared to OLETF Chow values.
While NPY gene expression was significantly decreased in the DMH of LETO rats in response to high fat diet, it was not affected by high fat diet in OLETF rats (Figure 3B). There were no main effects of strain [_F_(1,26)=0.377, _P_>0.05] and diet [_F_(2,26)=2.786, _P_>0.05], but there was a significant strain and diet interaction [_F_(2,26)=3.562, P<0.05], indicating differential regulation of DMH NPY gene expression by the high fat diets. Fisher LSD comparisons revealed that high fat diet access resulted in a significant reduction in DMH NPY gene expression in LETO MF and HF rats with a 37% and 26% decrease respectively relative to LETO Chow rats (_P_<0.05, Figure 3B). DMH NPY gene expression did not differ between LETO MF and HF rats (_P_>0.05). In contrast, there were no differences in DMH NPY gene expression among three groups of OLETF rats (_P_>0.05, Figure 3B).
Figure 3C shows Arc POMC gene expression in LETO and OLETF rats. ANOVA revealed a significant effect of strain [_F_(1,28)=22.234, P<0.001], but no significant effects of diet [_F_(2,28)=1.006, _P_>0.05] and no strain by diet interaction [_F_(2,28)=0.341, _P_>0.05]. Relative to LETO Chow rats, OLETF Chow, MF and HF rats had a 36%, 32% and 54% increase in Arc POMC gene expression respectively (P<0.05), but the degree of elevation did not differ among the three OLETF groups (_P_>0.05, Figure 3C).
As presented in Figure 3D, in situ hybridization determination did not detect any significant effects of strain [_F_(1,27)=3.991, _P_=0.056] or diet [_F_(2,27)=0.277, _P_=0.761] on PVN CRF gene expression. As well, there was not a significant strain by diet interaction in CRF gene expression in the PVN [_F_(2,27)=2.381, _P_=0.112].
Figure 4 shows the results of Arc NPY, Arc POMC and PVN CRF gene expression in the KO and WT mice. Consistent with their normal body weight, KO Chow mice had normal NPY gene expression in the Arc [_F_(1,18)=2.227, _P_=0.153](Figure 4A). In response to high fat diet, NPY gene expression was significantly decreased in the Arc by 37% and 31% in wild type and CCK1 receptor−/− mice respectively [_F_(1,18)=7.802, _P_=0.012]. There was no significant interaction between strain and diet [_F_(1,18)=0.0000383, _P_>0.05]. In contrast to rats, NPY gene expression was not detected in the DMH of wild type and CCK1 receptor−/− mice with/without access to a high fat diet.
Figure 4.
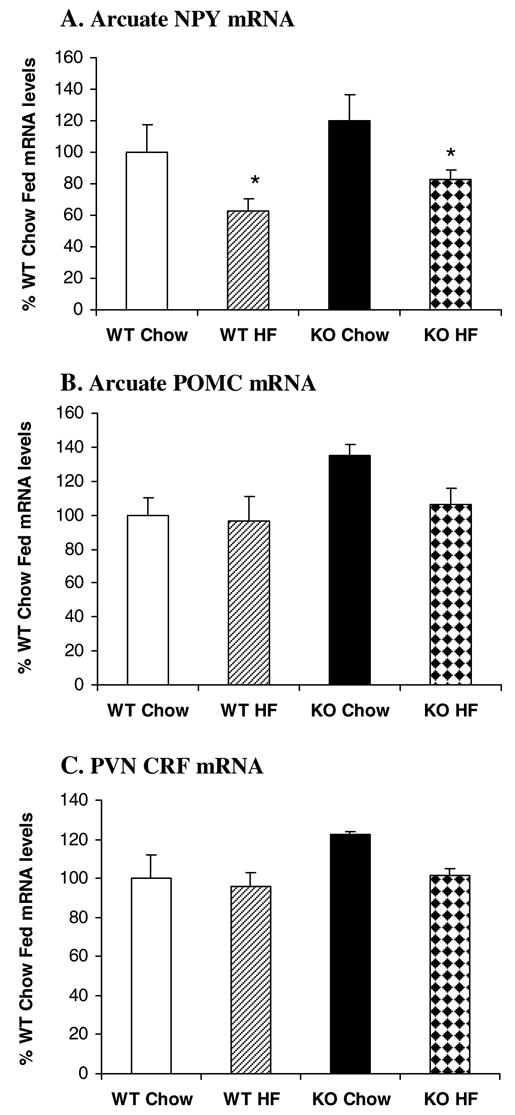
Effects of high fat diet on hypothalamic NPY, POMC and CRF gene expression in CCK1 receptor−/− mice. In response to high fat diet, Arc NPY gene expression was decreased in both WT and KO mice (A). High fat feeding did not affect Arc POMC (B) and PVN CRF gene expression (C) in either WT or KO mice. Values are means ± SEM. n = 6–8/group. *P < 0.05 as compared to Chow controls.
Although there was a trend for increased POMC gene expression (35%) in KO Chow mice relative to WT Chow mice, this increase did not reach statistical significance [_F_(1,21)=4.188, _P_>0.05](Figure 4B). High fat diet access also did not affect Arc POMC gene expression [_F_(1,21)=2.166, _P_>0.05]. Overall, there was no significant interaction between strain and diet [_F_(1,21)=1.293, _P_>0.05].
Similar to rats, PVN CRF gene expression was not affected by strain [_F_(1,19)=2.488, _P_>0.05] or diet [_F_(1,19)=2.039, _P_>0.05] in mice (Figure 4C) and there was no significant strain by diet interaction [_F_(1,19)=0.946, _P_>0.05].
DISCUSSION
The current results demonstrate high fat diet induced increases in daily food intake and obesity in OLETF rats lacking CCK1 receptors. As compared with LETO controls, high fat diets resulted in greater increases in caloric consumption and body weight in OLETF rats. OLETF rats gained more body fat mass and had higher plasma levels of glucose and leptin. Analysis of hypothalamic gene expression revealed that OLETF rats had alterations in the regulation of DMH NPY gene expression. In response to high fat diet, Arc NPY gene expression was decreased in both LETO and OLETF rats, whereas DMH NPY gene expression was significantly decreased in LETO rats but not in OLETF rats. In contrast to the results in OLETF rats, CCK1 receptor disruption did not alter body weight and feeding responses to high fat diet in mice. High fat diet induced obesity and increased daily food intake to an equal degree in both wild type and CCK1 receptor−/− mice. As well, high fat feeding increased body fat mass and plasma leptin levels to the same degree in both mouse strains. In response to high fat diet, NPY gene expression was decreased in the Arc but was not detectable in the DMH in both wild type and CCK1 receptor−/− mice. Together, these data demonstrate differential body weight and feeding responses to high fat diet in rats and mice lacking CCK1 receptors. Dysregulation of DMH NPY gene expression in OLETF rats but not in CCK1 receptor KO mice may account for their differential responses to high fat diets.
The role of CCK1 receptors in mediating the feeding inhibitory actions of peripheral CCK is well documented (5). Exogenous peripheral CCK administration produces dose related suppressions of food intake and results in the earlier appearance of a behavioral satiety sequence (30). The feeding inhibitory effect of CCK is blocked by CCK1 receptor antagonists, but unaffected by CCK2 receptor antagonists (22). Endogenous peripheral CCK is released from the duodenum and jejunum in response to intestinal nutrients. CCK1 receptor antagonists attenuate or block suppression of food intake produced by intra-intestinal nutrient infusions (33). Consistent with such a role of CCK1 receptors, CCK1 receptor deficiency prevents a feeding inhibitory response to peripheral CCK administration and increases meal size in both rats and mice (7, 19, 23). OLETF rats also have a deficit in feeding response to dietary fat and intestinal nutrient preloads. Relative to LETO controls, OLETF rats did not compensate for the increased calories when corn oil was added to their chow diet, and they did not appropriately respond to gastric and duodenal fat preloads (10, 28). To extend these findings, the current data demonstrate a greater impact of high fat diet on food intake and body weight in OLETF rats. High fat feeding for 10 weeks resulted in greater increases in body weight and food intake in OLETF rats. While LETO rats exhibited what appeared to be a transitory compensatory response to high fat diet during which they temporally normalized their daily food intake, OLETF rats had sustained overconsumption over the 10-week period. Although CCK1 receptor−/− mice exhibit a similar peripheral CCK satiety deficit to that of OLETF rats (7, 19), they maintain normal body weight and daily food intake and do not have altered body weight and feeding responses to high fat diet. CCK1 receptor−/− and wild type mice with access to a high fat diet had equal increases in food intake and body weight. Thus, the findings of difference in responses to high fat diet in rats and mice lacking CCK1 receptors implies that a deficit in the satiety actions of peripheral CCK is not sufficient to explain the lack of compensatory responses to high fat diet in the OLETF rat.
Differential roles for CCK1 receptors in maintaining energy balance in rats and mice have been suggested (7). We have previously demonstrated that while pair feeding normalizes the obesity of OLETF rats, pair feeding results in a large increase in DMH NPY gene expression, similar to levels found in young preobese OLETF rats (4). We have suggested that elevated DMH NPY gene expression may be a direct result of a CCK signaling deficit and serve as a major contributing factor to the hyperphagia and obesity of OLETF rats (5). Consistent with this view, we have demonstrated that DMH NPY neurons contain CCK1 receptors and that local CCK administration lowers DMH NPY mRNA levels and inhibits food intake in intact rats (7). In the current study, we demonstrate that in response to high fat diet LETO rats had decreased NPY gene expression in the DMH, reflecting a compensatory change for increased energy intake, whereas OLETF rats did not have the same pattern of reduction in NPY gene expression in the DMH as LETO rats. These data suggest that in the absence of DMH CCK signaling, the regulation of DMH NPY gene expression goes awry in OLETF rats. Such a deficit in the regulation of NPY gene expression in the DMH of OLETF rats may cause their greater increases in food intake and body weight in response to high fat diet. In contrast, CCK1 receptors do not appear to play a role in the control of DMH NPY gene expression in mice. CCK1 receptor−/− mice do not have elevated DMH NPY gene expression, and there is no evidence that the DMH contains CCK1 receptors in normal mice (7). We have suggested that the absence of hyperphagia and obesity in mice lacking CCK1 receptors is due to their ability to compensate for the absence of peripheral CCK satiety signaling (7). The current data further suggest that the regulation of DMH NPY gene expression differs in rats and mice. Although high fat feeding down regulates DMH NPY gene expression in LETO rats, NPY mRNA is not detectable in the DMH in either CCK1 receptor−/− or wild type mice regardless of diets.
The current findings of no induction of DMH NPY gene expression in diet-induced obese mice are different from the previous results reported by Guan and colleagues. They found that NPY gene expression was elevated in the DMH of diet-induced obese mice and the elevation gradually decreased to lean control levels when switched to a regular diet (15). We do not know the reasons for this difference. It could be the result of different diets used in the two experiments. Although both studies used a high fat diet with 60% of the calories from fat, the fat source differed. The dietary fat used in our current experiments was derived from lard (Research Diets, Inc., #D12492), whereas the diet in Guan et al.’s study used hydrogenated vegetable shortening and coconut oil (Harlan Teklad, TD97019) (15). This diet supplies trans fatty acids (from the shortening) and also shorter chain saturated fatty acids (from the coconut oil). Neither trans fatty acids nor these shorter chain saturated fatty acids are found in lard. Secondarily, it could simply be due to a strain difference. In Guan and colleagues’ experiments, C57BL/6J mice were used in the model of diet-induced obesity (15), but the CCK1 receptor−/− mice were generated on 129/SvEv mice (19).
Leptin, a hormone produced in the adipose tissue, is an adiposity signal and plays an important role in maintaining energy homeostasis through regulating hypothalamic peptide signaling pathways (1, 12, 29). Although the DMH contains the functional long-form of leptin receptors (14) and intravenous administration of leptin activates neurons in the DMH (13), the regulation of DMH NPY signaling is likely leptin independent. We have previously demonstrated that leptin receptors and NPY are not co-localized in the DMH neurons and that NPY gene expression is differentially regulated in the Arc and the DMH in response to acute food deprivation and chronic food restriction (6). We found that circulating leptin levels were equally reduced by both food deprivation and restriction, but only chronic restriction affected DMH NPY gene expression (6). Williams et al. have reported that while chronic food restriction resulted in increases in both Arc and DMH NPY levels in lean rats, leptin receptor deficient (cp/cp) rats showed no change in Arc NPY levels, but did have increased DMH NPY levels in response to chronic food restriction (32). Our current findings further support the view that leptin does not appear to regulate NPY gene expression in the DMH. We found that while high fat diet increased fat mass and leptin levels in both LETO and OLETF rats, high fat diet access resulted in a decrease in NPY gene expression in the DMH in LETO rats, but not in OLETF rats. Together, these data suggest that OLETF rats have a deficit in the regulation of DMH NPY gene expression in response to high fat diet, but this deficit is independent of leptin or adiposity signaling.
Within the hypothalamus, Arc NPY and POMC signaling play important roles in the control of food intake and body weight, and their mRNA expression is regulated by leptin. Leptin downregulates Arc NPY and up-regulates Arc POMC gene expression to control energy balance (1, 12, 29). Prior findings from OLETF rats have suggested that their CCK1 receptor deficiency does not result in alterations in the regulation of Arc NPY and POMC gene expression (4). We have demonstrated that hyperphagic and obese OLETF rats have decreased Arc NPY and increased Arc POMC gene expression and pair feeding normalizes their elevated body weight and altered Arc NPY and POMC gene expression (4). We have suggested that alterations in Arc NPY and POMC gene expression in OLETF rats are an appropriate response to their hyperphagia and increased body weight. In the current study, we replicate these previous results demonstrating that OLETF rats have decreased Arc NPY and increased Arc POMC gene expression. Moreover, we find that the patterns of Arc NPY and POMC gene expression in rats and mice lacking CCK1 receptors in response to high fat diet are similar to those of wild type controls. High fat feeding resulted in decreased Arc NPY gene expression and did not affect Arc POMC gene expression in both genotypes of rats and mice. Together, these data suggest that altered Arc NPY and POMC signaling in rats and mice lacking CCK1 receptors do not appear to account for their differential responses to high fat diet.
Alterations in Arc NPY expression or content in animals with high fat diet-induced obesity have been documented. In response to high fat diet, Arc NPY expression is initially decreased at day 2 (34), unchanged at weeks 1, 2 and 7 (17, 18, 20, 34), significantly reduced again at 8 weeks (20), and after 9 weeks both NPY expression and content levels are consistently decreased (3, 15, 16, 20). The current data demonstrating that a 10 week exposure to a high fat diet decreases Arc NPY gene expression are consistent with these previous results. Thus, it is likely that while high fat feeding increases caloric intake, the reduction of Arc NPY gene expression appears to be a compensatory response aimed at restoring energy balance. The similar effects of high fat diet in wild type and CCK1 receptor deficient animals suggest that Arc NPY gene expression is appropriately regulated in both rats and mice lacking CCK1 receptors.
High fat diet induced alterations in Arc POMC gene expression have not been consistently found. Ziotopoulou and colleagues reported that Arc POMC gene expression was unchanged at day 1, 2 and week 1 but significantly elevated at week 2 in response to high fat diet (34). Kinzig et al. found that Arc POMC gene expression was increased in Sprague Dawley rats with access to a high fat diet for 7 weeks (18). In contrast, Lin et al. found that high fat feeding did not affect Arc POMC gene expression at the first and eighth week time points but decreased its expression levels at 19 weeks (20). It has also been reported that Arc POMC gene expression was not significantly altered in animals with access to a high fat diet for 12 weeks or 6 months (9, 15). The reasons for such variance are not clear. Nevertheless, diminished Arc POMC signaling is likely linked to high fat diet-induced obesity. POMC deficient mice are hypersensitive to a high fat diet exhibiting greater increases in food intake and body weight (8). High fat feeding attenuates the anorexic effects of intracerebroventricular injections of a-MSH agonist MTII in rats (9). Arc POMC gene expression would be expected to increase as a compensatory response to positive energy balance and increased plasma leptin levels. Thus, the current findings are consistent with the view that a high fat diet may impair Arc POMC signaling resulting in diet induced obesity.
In summary, the present results demonstrate differential feeding and body weight responses to high fat diet in rats and mice lacking CCK1 receptors. OLETF rat have deficits in their long-term response to high fat diet resulting in an exacerbation of their hyperphagia and obesity. A deficit in DMH CCK actions in the control of DMH NPY gene expression may contribute to this phenotype. In contrast, CCK1 receptor−/− mice, that do not have a central deficit in the control of DMH NPY gene expression, have normal long-term response to high fat diet and regulation of NPY gene expression that resembles wild type animals.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK057609 (to T.H.M.), DK074269 (to S.B.) and DK046767 (to A.S.K.). The OLETF and LETO rats were a generous gift of Otsuka Pharmaceutical.
References
- 1.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 2.Antin J, Gibbs J, Holt J, Young RC, Smith GP. Cholecystokinin elicits the complete behavioral sequence of satiety in rats. J Comp Physiol Psychol. 1975;89:784–790. doi: 10.1037/h0077040. [DOI] [PubMed] [Google Scholar]
- 3.Beck B, Stricker-Krongrad A, Burlet A, Max JP, Musse N, Nicolas JP, Burlet C. Macronutrient type independently of energy intake modulates hypothalamic neuropeptide Y in Long-Evans rats. Brain Res Bull. 1994;34:85–91. doi: 10.1016/0361-9230(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 4.Bi S, Ladenheim EE, Schwartz GJ, Moran TH. A role for NPY overexpression in the dorsomedial hypothalamus in hyperphagia and obesity of OLETF rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R254–260. doi: 10.1152/ajpregu.2001.281.1.R254. [DOI] [PubMed] [Google Scholar]
- 5.Bi S, Moran TH. Actions of CCK in the controls of food intake and body weight: lessons from the CCK-A receptor deficient OLETF rat. Neuropeptides. 2002;36:171–181. doi: 10.1054/npep.2002.0895. [DOI] [PubMed] [Google Scholar]
- 6.Bi S, Robinson BM, Moran TH. Acute food deprivation and chronic food restriction differentially affect hypothalamic NPY mRNA expression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1030–1036. doi: 10.1152/ajpregu.00734.2002. [DOI] [PubMed] [Google Scholar]
- 7.Bi S, Scott KA, Kopin AS, Moran TH. Differential roles for cholecystokinin a receptors in energy balance in rats and mice. Endocrinology. 2004;145:3873–3880. doi: 10.1210/en.2004-0284. [DOI] [PubMed] [Google Scholar]
- 8.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O’Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36) Proc Natl Acad Sci U S A. 2004;101:4695–4700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clegg DJ, Benoit SC, Air EL, Jackman A, Tso P, D’Alessio D, Woods SC, Seeley RJ. Increased dietary fat attenuates the anorexic effects of intracerebroventricular injections of MTII. Endocrinology. 2003;144:2941–2946. doi: 10.1210/en.2002-0218. [DOI] [PubMed] [Google Scholar]
- 10.Covasa M, Ritter RC. Attenuated satiation response to intestinal nutrients in rats that do not express CCK-A receptors. Peptides. 2001;22:1339–1348. doi: 10.1016/s0196-9781(01)00461-2. [DOI] [PubMed] [Google Scholar]
- 11.Dourish CT, Ruckert AC, Tattersall FD, Iversen SD. Evidence that decreased feeding induced by systemic injection of cholecystokinin is mediated by CCK-A receptors. Eur J Pharmacol. 1989;173:233–234. doi: 10.1016/0014-2999(89)90528-1. [DOI] [PubMed] [Google Scholar]
- 12.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 13.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- 15.Guan XM, Yu H, Trumbauer M, Frazier E, Van der Ploeg LH, Chen H. Induction of neuropeptide Y expression in dorsomedial hypothalamus of diet-induced obese mice. Neuroreport. 1998;9:3415–3419. doi: 10.1097/00001756-199810260-00015. [DOI] [PubMed] [Google Scholar]
- 16.Hansen MJ, Jovanovska V, Morris MJ. Adaptive responses in hypothalamic neuropeptide Y in the face of prolonged high-fat feeding in the rat. J Neurochem. 2004;88:909–916. doi: 10.1046/j.1471-4159.2003.02217.x. [DOI] [PubMed] [Google Scholar]
- 17.Kim EM, Welch CC, Grace MK, Billington CJ, Levine AS. Effects of palatability-induced hyperphagia and food restriction on mRNA levels of neuropeptide-Y in the arcuate nucleus. Brain Res. 1998;806:117–121. doi: 10.1016/s0006-8993(98)00755-0. [DOI] [PubMed] [Google Scholar]
- 18.Kinzig KP, Scott KA, Hyun J, Bi S, Moran TH. Altered hypothalamic signaling and responses to food deprivation in rats fed a low-carbohydrate diet. Obes Res. 2005;13:1672–1682. doi: 10.1038/oby.2005.205. [DOI] [PubMed] [Google Scholar]
- 19.Kopin AS, Mathes WF, McBride EW, Nguyen M, Al-Haider W, Schmitz F, Bonner-Weir S, Kanarek R, Beinborn M. The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight. J Clin Invest. 1999;103:383–391. doi: 10.1172/JCI4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin S, Storlien LH, Huang XF. Leptin receptor, NPY, POMC mRNA expression in the diet-induced obese mouse brain. Brain Res. 2000;875:89–95. doi: 10.1016/s0006-8993(00)02580-4. [DOI] [PubMed] [Google Scholar]
- 21.Melville LD, Smith GP, Gibbs J. Devazepide antagonizes the inhibitory effect of cholecystokinin on intake in sham-feeding rats. Pharmacol Biochem Behav. 1992;43:975–977. doi: 10.1016/0091-3057(92)90435-i. [DOI] [PubMed] [Google Scholar]
- 22.Moran TH, Ameglio PJ, Schwartz GJ, McHugh PR. Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK. Am J Physiol. 1992;262:R46–50. doi: 10.1152/ajpregu.1992.262.1.R46. [DOI] [PubMed] [Google Scholar]
- 23.Moran TH, Katz LF, Plata-Salaman CR, Schwartz GJ. Disordered food intake and obesity in rats lacking cholecystokinin A receptors. Am J Physiol. 1998;274:R618–625. doi: 10.1152/ajpregu.1998.274.3.R618. [DOI] [PubMed] [Google Scholar]
- 24.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2. Elsevier Academic Press; San Diego, California: 2001. [Google Scholar]
- 25.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. Elsevier Academic Press; San Diego, California: 2005. [Google Scholar]
- 26.Reidelberger RD, O’Rourke MF. Potent cholecystokinin antagonist L 364718 stimulates food intake in rats. Am J Physiol. 1989;257:R1512–1518. doi: 10.1152/ajpregu.1989.257.6.R1512. [DOI] [PubMed] [Google Scholar]
- 27.Reidelberger RD, Varga G, Solomon TE. Effects of selective cholecystokinin antagonists L364,718 and L365,260 on food intake in rats. Peptides. 1991;12:1215–1221. doi: 10.1016/0196-9781(91)90197-w. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz GJ, Whitney A, Skoglund C, Castonguay TW, Moran TH. Decreased responsiveness to dietary fat in Otsuka Long-Evans Tokushima fatty rats lacking CCK-A receptors. Am J Physiol. 1999;277:R1144–1151. doi: 10.1152/ajpregu.1999.277.4.R1144. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 30.Smith GP, Gibbs J. Satiating effect of cholecystokinin. Ann N Y Acad Sci. 1994;713:236–241. doi: 10.1111/j.1749-6632.1994.tb44071.x. [DOI] [PubMed] [Google Scholar]
- 31.Takiguchi S, Takata Y, Funakoshi A, Miyasaka K, Kataoka K, Fujimura Y, Goto T, Kono A. Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats. Gene. 1997;197:169–175. doi: 10.1016/s0378-1119(97)00259-x. [DOI] [PubMed] [Google Scholar]
- 32.Williams G, Shellard L, Lewis DE, McKibbin PE, McCarthy HD, Koeslag DG, Russell JC. Hypothalamic neuropeptide Y disturbances in the obese (cp/cp) JCR:LA corpulent rat. Peptides. 1992;13:537–540. doi: 10.1016/0196-9781(92)90085-h. [DOI] [PubMed] [Google Scholar]
- 33.Yox DP, Brenner L, Ritter RC. CCK-receptor antagonists attenuate suppression of sham feeding by intestinal nutrients. Am J Physiol. 1992;262:R554–561. doi: 10.1152/ajpregu.1992.262.4.R554. [DOI] [PubMed] [Google Scholar]
- 34.Ziotopoulou M, Mantzoros CS, Hileman SM, Flier JS. Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2000;279:E838–845. doi: 10.1152/ajpendo.2000.279.4.E838. [DOI] [PubMed] [Google Scholar]