Cystic fibrosis (original) (raw)
Summary points
- Cystic fibrosis is the commonest inherited disease in white populations, with an incidence of 1 in 2500 newborns; over 7000 people in the United Kingdom currently have the disease
- Until recently, the diagnosis has been largely clinical, although the widespread implementation of a screening programme for newborns is now complete in the UK
- Cystic fibrosis is a multiorgan disease best managed in a multidisciplinary setting in conjunction with a specialist centre for cystic fibrosis, with treatment tailored to the individual
- The cornerstones of management are proactive treatment of airway infection and encouragement of good nutrition and an active lifestyle
- Conventional treatment has improved greatly over the past few decades; however, current treatments at best slow the decline in lung function. Newer approaches such as gene and small molecule based treatments may have more potential to halt disease progression
Over 7000 people have cystic fibrosis in the United Kingdom. It is the commonest genetically inherited disease in white populations (1 in 2500 newborns), although it is increasingly recognised as being important in non-white populations. However, most general practitioners have only one or two patients on their list, and as management generally takes place in specialist centres, many general paediatricians will be involved in the care of only a small number of patients.
Progress in our understanding of the disease and the impact of this on management has been rapid over the past 20 years. Cystic fibrosis used to be a digestive and lung disease of young children but more recently has become a complex, multisystem disease extending into adulthood; there will soon be more adults than children with the condition. The predicted median survival for babies born in the 21st century is now more than 50 years.1 This increased survival—together with changes in standard treatment, the increasing implementation of newborn screening, and the focus on new therapeutic strategies—leads us to consider that an update on this albeit relatively rare disease may be of general interest.
What is the cause of cystic fibrosis?
Cystic fibrosis is an autosomal recessive disease. It is caused by mutations in the CFTR (cystic fibrosis transmembrane conductance regulator) gene.2 The commonest mutation is the deletion of phenylalanine at codon 508 (phe508del, until recently known as ΔF508). This occurs in about 70% of patients with cystic fibrosis (www.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/cftr.shtml). Over 1600 mutations of the CFTR gene have been described. Different mutations in this gene have varying effects on CFTR function and can result in different phenotypes of the disease. Some mutations will result in milder forms of the disease, although there is not enough evidence about these rarer mutations to counsel patients about their prognosis. The CFTR protein is expressed in many cells and has several functions, not all of which have been linked with disease. The primary function of the CFTR protein is as an ion channel that regulates liquid volume on epithelial surfaces through chloride secretion and inhibition of sodium absorption.
The commonly accepted explanation for airway disease in cystic fibrosis is the “low volume” hypothesis. A reduced volume of airway surface liquid causes failure of mucociliary clearance, the lungs’ innate defence mechanism.3 The mucociliary dysfunction means that a patient with cystic fibrosis cannot effectively clear inhaled bacteria. In addition, there is an excessive inflammatory response to pathogens. For a given bacterial load, a person with cystic fibrosis will have up to 10 times more inflammation than a person with a lower respiratory tract infection but without the disease. This may also be the case for other insults such as viruses or even for airborne particulate matter and pollutants. The reasons for the excessive inflammatory response to pathogens are not fully understood. The abnormal composition and secretion of mucus may also be important. At birth, the airway is uninfected and probably uninflamed, although some controversy exists in this area,4 but the end result of the abnormalities described above is irreversible airway damage with bronchiectasis and respiratory failure in most patients (fig 1). Ion and water abnormalities may also cause disease in other epithelia-lined organs (see tables 1, 3, 4).”

Fig 1 Severe bronchiectasis in end stage cystic fibrosis shown in chest radiograph (top) and computed tomogram (bottom). For reasons that are not fully understood, the upper lobes are often most severely affected, although the patient has severe bronchiectasis throughout the whole of the right lung. Note presence of indwelling intravenous catheter (a “port-a-cath”) on the right lateral chest wall
Table 1.
Age related presentations of cystic fibrosis
| Age group | Common presentations | Less common presentations |
|---|---|---|
| Antenatal | Chorionic villous sampling or amniocentesis in high risk family; echogenic bowel on ultrasound | Perforated meconium ileus |
| Neonatal | Diagnosis made on newborn screening; meconium ileus (10% of patients with cystic fibrosis) causing bowel obstruction with or without perforation and peritonitis | Gut atresias; obstructive jaundice; fat soluble vitamin deficiencies (bleeding disorder, vitamin K; haemolytic anaemia, vitamin E; raised intracranial pressure, vitamin A) |
| Infants and young children | Recurrent respiratory symptoms (cough, wheeze, pneumonias); failure to thrive (exocrine pancreatic insufficiency present in 85-90% of cases leads to steatorrhoea, diarrhoea, and abdominal distension) | Rectal prolapse; dehydration and electrolyte disturbance (pseudo-Bartter’s syndrome); anaemia, oedema, and hypoprotenaemia |
| Older children and adults | Recurrent respiratory symptoms as above (may be labelled asthmatic) with or without finger clubbing (fig 2); nasal polyps or sinusitis; male infertility (congenital bilateral absence of the vas deferens) | Acute pancreatitis; liver disease; malabsorption; dehydration and electrolyte disturbance (pseudo-Bartter’s syndrome); pulmonary infection with atypical mycobacteria |
Table 3.
Gastrointestinal problems and their management
| Organ | Manifestation | Management | Comments |
|---|---|---|---|
| Pancreas | Exocrine insufficiency (85-90% of newborns): malabsorption, steatorrhoea, poor growth | Supplementation with pancreatic enzymes† and fat soluble vitamins | May be aided by alkaline environment (H2 blockers or proton pump inhibitors)* |
| Pancreatitis | As for other causes; Pancrex powder | Uncommon; occurs only in patients with pancreatic excocrine sufficiency | |
| Oesophagus | Gastro-oesophageal reflux | Prokinetic plus antacid; surgery if recalcitrant | Probably common; reported incidence variable |
| Small bowel | Meconium ileus | Gastrograffin enemas; surgery (with or without resection) | About 10% of newborns with cystic fibrosis |
| Distal intestinal obstruction syndrome | Bowel cleaning agents such as Gastrograffin or kleanprep | Review dose of and adherence to enzymes | |
| Coeliac disease; malabsorption despite adequate enzymes | Gluten-free diet | Incidence seems to be increased in cystic fibrosis | |
| Crohn’s disease | Usual treatment | Incidence seems to be increased in cystic fibrosis | |
| Colon | Constipation | Dietary advice, laxatives | Check no malabsorption, if present, check use of pancreatic enzyme replacement therapy carefully |
| Rectum | Rectal prolapse | Usually resolves with pancreatic enzymes; rarely surgery required | |
| Liver17 | Fatty liver (usually not symptomatic); cirrhosis (variceal bleeding, hypersplenism) | Ursodeoxycholic acid, taurine*; severe cases may need transplantation | Liver disease in up to 30% of patients by adulthood; liver cell failure late, with ominous prognosis |
Table 4.
Management of other common complications of cystic fibrosis
| Organ | Manifestation of cystic fibrosis | Treatment | Comments |
|---|---|---|---|
| Upper airway | Polyps18 | Topical steroids; antibiotics; surgery if medical management fails | Surgery may have medium term benefit, but recurrence common |
| Sinusitis | Topical steroids; antibiotics; surgery if medical management fails | Most cases are asymptomatic (changes seen on x ray films or computed tomograms almost universal): no treatment required | |
| Endocrine pancreas19 | Insulin deficiency; frank diabetes | Insulin; continue high fat diet; oral hypoglycaemic agents rarely useful | Deleterious impact on respiratory health and nutrition even before diabetes diagnosed |
| Bones | Osteopenia; pathological fracture | Prevention: weight bearing exercise, high dairy intake, vitamin D and K therapy* (bisphosphonates if severe*) | |
| Cystic fibrosis arthropathy | Anti-inflammatory agents | ||
| Sweat gland | Electrolyte depletion leading to failure to thrive, acute collapse | Sodium and potassium chloride supplementation | |
| Male reproductive tract | Bilateral absence of vas deferens | Sperm aspiration, and assisted fertilisation techniques | |
| Female reproductive tract | Vaginal candidiasis; stress incontinence | Topical antifungal agents | Seek gynaecological advice |
What are the clinical features and when should the diagnosis be considered?
Disease manifests in many organs, but most notably the upper and lower airways, pancreas, bowel, and reproductive tracts (table 1).5 For most patients, lung disease is the most important problem in terms of symptoms and the treatment required and the fact that it is the most likely cause of death. Table 1 outlines the clinical presentation, which varies according to age.
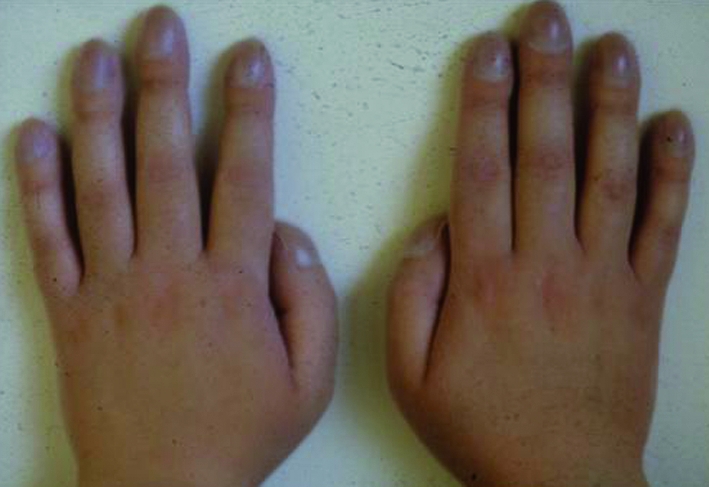
Fig 2 Finger clubbing indicates advance suppurative lung disease. It is not characteristic of asthma, with which older patients have sometimes been misdiagnosed
How is cystic fibrosis diagnosed?
The optimal diagnostic test for cystic fibrosis is the measurement of sweat electrolyte levels.6 Patients with the disease have raised concentrations of sodium and chloride (>60 mmol/l, diagnostic; 40-60 mmol/l, intermediate (but more likely to be diagnostic in infants); <40 mmol/l, normal). However, undoubted cases of cystic fibrosis with normal sweat electrolytes have been described. Newer techniques have reduced the amount of sweat needed (fig 3). The test needs to be done by someone trained and experienced. For this reason the diagnosis will usually be made in secondary and tertiary centres, although primary care professionals play a vital role in identifying the patients who need investigation. In the rare cases where the diagnosis remains in doubt, other diagnostic tests are available—for example, measurement of the nasal potential difference to assess altered salt transport is available in a few specialist centres in the UK.7

Fig 3 Child’s arm during sweat test with the macroduct system. After pilocarpine iontophoresis to stimulate sweating, the closed capillary collecting system is applied to the skin of the forearm. Sweat can be seen entering the tubing (blue); electrolyte analysis can be reliably performed on as little as 50 μl of sweat
The UK now has a programme for screening all newborns for cystic fibrosis using the Guthrie blood spot test.8 The initial screen is for raised concentrations of immunoreactive trypsinogen. Positive samples will be tested for common CFTR gene mutations followed by a second screen for immunoreactive trypsinogen if required. Screen positive infants will be referred for sweat testing.
Screening programmes have been in place in some parts of the world for many years, but they may be inappropriate in countries with a low prevalence of CFTR gene mutations. The advantages of early diagnosis include nutritional benefits; early access to specialised care; a reduction in the time of diagnostic uncertainty; and the ability to counsel parents for prenatal testing.
Screening programmes have some negative aspects, however. Programmes will identify some healthy heterozygote carriers as potential patients. This may have psychological implications and stress for the family until the diagnosis is excluded. Moreover, some patients, even those with classic cystic fibrosis, will be missed.
Once a diagnosis has been confirmed, other family members may be offered screening. All siblings need to be screened for the disease, which may be presymptomatic or unrecognised. Asymptomatic adult relatives, may wish to be screened for carrier status to enable them to make informed choices about prenatal screening. In our experience, screening and counselling of other family membersis most readily facilitated through primary care but requires coordination between genetic laboratories to ensure rapid and cost efficient testing.
Management
Most patients in the UK and Europe receive care coordinated by a tertiary cystic fibrosis centre, which improves outcomes. However, patients benefit greatly from links with and access to local care, in many cases having formalised “shared care” with local clinics. Primary care teams can provide valuable help with surveillance and early treatment of infection; dietary and nutritional support; and social and psychological support for patients and families. Primary care also provides continuity during the difficult transition from paediatric to adult care; an informative patient’s perspective of the issues encountered during this period has recently been published.9
Much of the current clinical practice has evolved over decades without being subjected to high quality randomised controlled trials. Tables 2-4 outline the various treatments and indicate those that are based on randomised controlled trials, meta-analyses, or systematic review and those that are treatments for which we consider consensus is lacking.
Table 2.
Management of cystic fibrosis lung disease
| Disease stage | Pulmonary status | Aim | Management | Comments |
|---|---|---|---|---|
| Early | Preinfection | Mucus clearance; prevent infection; maintain good lung function | Segregation and cohorting to prevent cross infection*; airway clearance techniques (physiotherapy and adjuncts, mucolytics such as rhDNase,†10 hypertonic saline†11); prophylactic antibiotics (usually against _Sa_†12; most commonly flucloxacillin or co-amoxiclav in UK); influenza vaccination usually recommended | Segregation of patients with organisms such as Bcc or epidemic strains of Pa is common (practice more variable with regard to other strains of Pa, Sm, or Hi); for both rhDNase and hypertonic saline evidence favours short to medium term benefit (no long term or survival data); prophylactic antibiotics decrease incidence of infection with Sa (long term benefits not well defined); increase in infection with Pa seems limited to trials including broad spectrum cephalosporins |
| Intermittent isolation of organisms | Eradication of infection | Pa eradication protocols† include both topical (nebulised) and systemic (usually oral ciprofloxacin) | Eradication achieved in 80-90%,13 but uncertain long term benefit | |
| Intermediate | Chronic infection with usual organisms (Pa, Sa, Hi) | Suppression of bacterial load and thus limitation of inflammatory response | Depends on organism (Pa: nebulised tobramycin or colomycin) | Pa: medium term benefit,† uncertain effects on survival; new, faster nebuliser devices (such as e-flow and iNeb) available |
| Treat infective exacerbations | Oral or intravenous antibiotics appropriate for culture | Elective v symptomatic use* | ||
| Reduce inflammation | Ibuprofen*; macrolide antibiotics (azithromycin)†14 | Ibuprofen: limited use in much of Europe15 (used more often in US); azithromycin: good evidence for short/medium term benefit, but mechanism of action uncertain (anti-inflammatory properties thought likely); no evidence supporting a role for corticosteroids except in treating allergic bronchopulmonary aspergillosis | ||
| Infection with less common organisms (Bcc, Sm, Ax) | Eradication if early; suppression of bacterial load most commonly | Treat on an individual basis; seek specialist microbiological advice | Confirm diagnosis in a reference laboratory | |
| Allergic bronchopulmonary aspergillosis | Reduce allergic response; prevent bronchiectasis | Oral corticosteroids; consider addition of an antifungal agent* | Long course often required | |
| Non-tuberculous mycobacterial infection | Eradication | Usually prolonged combination treatment: ethambutol, rifampicin, azithromycin, amikacin | Can be difficult to determine whether isolates are contributing to disease manifestations; most would treat if recurrent positive cultures | |
| End stage with complications | Severe haemoptysis | Prevent bleeding, which may be fatal | Bronchial artery embolisation (rarely lobectomy) | |
| Pneumothorax | Drainage. Pleurodesis if persistent/recurrent | May affect suitability for transplantation in future | ||
| Respiratory failure | Lung or heart and lung transplantation16 |
Lung disease
The aims of treating the lungs at different stages of disease vary; table 2 outlines the conventional management at each of these stages. Many of the treatment options have been discussed in systematic reviews. Respiratory treatments represent the greatest challenge to patients and families: doing physiotherapy and taking inhaled drugs such as antibiotics often takes up a lot of time—more than an hour a day during periods of good health and much longer during a respiratory exacerbation.
Extrapulmonary disease
Patients with cystic fibrosis often have gastrointestinal problems; table 3 outlines the nature and management of these (management should be in close collaboration with a specialist dietician). Table 4 lists other complications of the disease, plus their management strategies.
Psychological issues
Cystic fibrosis clearly poses a huge burden to patients and families in terms of the life shortening nature of the disease, the time consuming treatments prescribed, and the ongoing morbidity. Times of particular stress include diagnosis, adolescence (when adherence to treatment can often be poor), and end of life. Support and coping strategies from clinical psychologists with experience of the disease are often invaluable.20
Sources and selection criteria
We searched PubMed for the terms “cystic fibrosis”, “therapy”, “treatment”, “management”, “complications”, and “diagnosis” in various combinations. We also searched all entries under “cystic fibrosis” in the Cochrane Library. From this search, we selected randomised controlled trials, high quality journal reviews and meta-analyses. We also drew from our own personal archives of references from known leaders in this field.
Ongoing research
- Research is being conducted into gene therapy that aims at introducing a normal copy of CFTR into lung epithelial cells.21 Achieving expression after repeat administration of viral vectors has been a major problem owing to immune recognition. Because of this, the UK Cystic Fibrosis Gene Therapy Consortium, which has been formed in recent years to develop cystic fibrosis gene therapy for clinical benefit is focusing current efforts on a non-viral approach
- New drugs to improve ion transport22 and osmotic agents23 to increase airway surface liquid are currently in phase II clinical trials, as are anti-inflammatory agents, mucolytics, and pseudomonas vaccines
- New methods for administering current agents, such as the development of dry powder formulations (ease of administration) and of liposomal preparations (enhanced activity) of antibiotics are being developed
Additional educational resources
- Kerem E, Conway S, Elborn S, Heijerman H; Consensus Committee. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 2005;4:7-26.
- Bush A, Alton E, Davies JC, Griesenbach U, Jaffe A. Cystic fibrosis in the 21st century (progress in respiratory research). Basel: Karger, 2005.
- Cystic Fibrosis Trust, 11 London Road, Bromley BR1 1BY (www.cftrust.org.uk/)—Charity whose work includes research into the disease and support to patients and their families
- Cystic Fibrosis Foundation (www.cff.org/)—US non-profit organisation whose work includes research into the disease and support to patients and their families
- Genetics Home Reference (http://ghr.nlm.nih.gov/condition=cysticfibrosis)—US government website supplying general scientific information on the disease
- Association of Clinical Biochemistry. Guidelines for the performance of the sweat test for the investigation of cystic fibrosis in the UK. 2003. http://acb.org.uk/docs/sweat.pdf
- Cystic Fibrosis Mutation Database (www.genet.sickkids.on.ca/cftr/)—Aims to provide researchers and other professionals with up to date information about individual mutations in the CFTR gene and phenotypic data
- Information on the UK newborn screening programme (www.ich.ucl.ac.uk/newborn/cf/index.htm)
- Breathing Room (www.thebreathingroom.org/cg)— Illustrations of many issues affecting patients and their carers
- UK Cystic Fibrosis Gene Therapy Consortium (www.cfgenetherapy.org.uk)—The research programme of three leading gene therapy groups in the UK
Contributors: JCD wrote the original draft of this paper and all authors contributed to subsequent drafts. Jackie Francis provided certain photographic images. JCD acts as guarantor for the article.
Competing interests: JCD and EWFWA are members of the UK Cystic Fibrosis Gene Therapy Consortium.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1.Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J 2007;29:522-6. [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245:1066-73. [DOI] [PubMed] [Google Scholar]
- 3.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998;95:1005-15. [DOI] [PubMed] [Google Scholar]
- 4.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918-51. [DOI] [PubMed] [Google Scholar]
- 5.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med 2006;173:475-82. [DOI] [PubMed] [Google Scholar]
- 6.Rosenstein BJ, Cutting GR, for the Cystic Fibrosis Foundation Consensus Panel. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998;132:589-95. [DOI] [PubMed] [Google Scholar]
- 7.Middleton PG, Geddes DM, Alton EWFW. Protocols for in vivo measurement of the ion transport defects in cystic fibrosis nasal epithelium. Eur Respir J 1994;7:2050-6. [PubMed] [Google Scholar]
- 8.Southern KW, Munck A, Pollitt R, Travert G, Zanolla L, Dankert-Roelse J, et al. A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros 2007;6:57-65. [DOI] [PubMed] [Google Scholar]
- 9.Wicks E. Cystic fibrosis. BMJ 2007;334:1270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones AP, Wallis CE. Recombinant human deoxyribonuclease for cystic fibrosis. Cochrane Database Syst Rev 2003;(3):CD001127. [DOI] [PubMed] [Google Scholar]
- 11.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354:229-40. [DOI] [PubMed] [Google Scholar]
- 12.Smyth A, Walters S. Prophylactic anti-staphylococcal antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2003;(3):CD001912. [DOI] [PubMed] [Google Scholar]
- 13.Wood DM, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev 2006;(1):CD004197. [DOI] [PubMed] [Google Scholar]
- 14.Southern KW, Barker PM, Solis A. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2004;(2):CD002203. [DOI] [PubMed] [Google Scholar]
- 15.Lands LC, Dezateux C, Crighton A. Oral non-steroidal anti-inflammatory drug therapy for cystic fibrosis. Cochrane Database Syst Rev 1999;(2):CD001505. [DOI] [PubMed] [Google Scholar]
- 16.Orens JB, Estenne M, Arcasoy S, Conte JV, Corris P, Egan JJ, et al. International guidelines for the selection of lung transplant candidates: 2006 update—a consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2006;25:745-55. [DOI] [PubMed] [Google Scholar]
- 17.Colombo C, Russo MC, Zazzeron L, Romano G. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr 2006;43(suppl 1):S49-55. [DOI] [PubMed] [Google Scholar]
- 18.Yung MW, Gould J, Upton GJ. Nasal polyposis in children with cystic fibrosis: a long-term follow-up study. Ann Otol Rhinol Laryngol 2002;111:1081-6. [DOI] [PubMed] [Google Scholar]
- 19.Onady GM, Stolfi A. Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst Rev 2005;(3):CD004730. [DOI] [PubMed] [Google Scholar]
- 20.Glasscoe CA, Quittner AL. Psychological interventions for cystic fibrosis. Cochrane Database Syst Rev 2003;(3):CD003148. [DOI] [PubMed] [Google Scholar]
- 21.Davies JC, Alton EW. Airway gene therapy. Adv Genet 2005;54:291-314. [DOI] [PubMed] [Google Scholar]
- 22.Deterding R, Retsch-Bogart G, Milgram L, Gibson R, Daines C, Zeitlin PL, et al. Safety and tolerability of denufosol tetrasodium inhalation solution, a novel P2Y2 receptor agonist: results of a phase 1/phase 2 multicenter study in mild to moderate cystic fibrosis. Pediatr Pulmonol 2005;39:339-48. [DOI] [PubMed] [Google Scholar]
- 23.Daviskas E, Anderson SD. Hyperosmolar agents and clearance of mucus in the diseased airway. J Aerosol Med 2006;19:100-9. [DOI] [PubMed] [Google Scholar]