Molecular Requirements for Bi-directional Movement of Phagosomes Along Microtubules (original) (raw)
Abstract
Microtubules facilitate the maturation of phagosomes by favoring their interactions with endocytic compartments. Here, we show that phagosomes move within cells along tracks of several microns centrifugally and centripetally in a pH- and microtubuledependent manner. Phagosome movement was reconstituted in vitro and required energy, cytosol and membrane proteins of this organelle. The activity or presence of these phagosome proteins was regulated as the organelle matured, with “late” phagosomes moving threefold more frequently than “early” ones. The majority of moving phagosomes were minus-end directed; the remainder moved towards microtubule plus-ends and a small subset moved bi-directionally. Minus-end movement showed pharmacological characteristics expected for dyneins, was inhibited by immunodepletion of cytoplasmic dynein and could be restored by addition of cytoplasmic dynein. Plus-end movement displayed pharmacological properties of kinesin, was inhibited partially by immunodepletion of kinesin and fully by addition of an anti-kinesin IgG. Immunodepletion of dynactin, a dynein-activating complex, inhibited only minus-end directed motility. Evidence is provided for a dynactin-associated kinase required for dyneinmediated vesicle transport. Movement in both directions was inhibited by peptide fragments from kinectin (a putative kinesin membrane receptor), derived from the region to which a motility-blocking antibody binds. Polypeptide subunits from these microtubule-based motility factors were detected on phagosomes by immunoblotting or immunoelectron microscopy. This is the first study using a single in vitro system that describes the roles played by kinesin, kinectin, cytoplasmic dynein, and dynactin in the microtubule-mediated movement of a purified membrane organelle.
In vertebrates, phagocytosis serves to clear invading parasites recognized by the immune response and cells killed by infection or having died by apoptosis. During phagocytosis a cell, such as a macrophage, takes up particles that are too large to be internalized by endocytosis, thereby forming a new membrane compartment, the phagosome (Silverstein et al., 1989). Once formed, the phagosome is the site of a series of biochemical reactions leading to the killing of any live organism it contains. It eventually matures, by sequential fusions with endocytic compartments, into a phagolysosome (Muller et al., 1980; Pitt et al., 1992_a_ ; Jahraus et al., 1994; Desjardins et al., 1994_a_ ). This process allows the phagosome to acidify and acquire the hydrolases, contained within endocytic organelles, required for degradation of the internalized particle.
Early evidence suggested that microtubules facilitate fusions between phagosomes and endocytic organelles (Goldstein et al., 1973; Pesanti and Axiline, 1975; D'Arcy Hart et al., 1983). Recent video analysis has shown that in macrophages, phagosomes and late endosomes encounter one another repeatedly (Desjardins et al., 1994_a_ ). We have also shown that the acquisition by phagosomes of membrane protein- and fluid phase-markers of late endocytic organelles is inhibited by microtubule depolymerization (Desjardins et al., 1994_a_ ; Blocker et al., 1996). Phagosomes and a variety of endocytic compartments interact directly with microtubules (Gruenberg et al., 1989; Hopkins et al., 1990; Scheel and Kreis, 1991; Pierre et al., 1992; Blocker et al., 1996). Toyohara and Inaba (1989) have shown that microtubules are required for transport of phagosomes to the cell center, but the molecular details of this movement remain obscure.
Microtubule-based motors such as cytoplasmic dynein and kinesin are widely thought to drive directed migration of membrane organelles across long cytoplasmic distances (Vale, 1987; Schroer, 1994). These motors were discovered as ATPases generating polarized microtubule gliding along a coverglass and movement of latex beads along microtubules (Lasek and Brady, 1985; Brady, 1985; Vale et al., 1985_b_ ; Lye et al., 1987; Paschal and Vallee, 1987). Since then, much has been learned about how these mechano-enzymes function (Bloom and Endow, 1994; Holzbaur and Vallee, 1994; Gilbert et al., 1995). In vitro, they have been shown to be required for motility of crude membrane vesicle preparations along microtubules (Vale et al., 1985_a_ ; Schroer et al., 1988, 1989; Schnapp and Reese, 1989). Recently, a putative membrane receptor for kinesin, kinectin, has been described (Toyoshima et al., 1992; Yu et al., 1995). The way in which cytoplasmic dynein binds organelles is unclear, but it is unable to move membrane vesicles in vitro unless accompanied by dynactin and another unidentified factor (Schroer and Sheetz, 1991; Gill et al., 1991). Much less is known of the specific in vivo cargoes of these microtubule motors. Their immunolocalization by light microscopy resulted in detergent-extractable, punctate patterns within cytoplasm which do not fully colocalize with markers of known organelles (Hollenbeck, 1989; Hirokawa et al., 1991; Lin and Collins, 1992; Gill et al., 1991; Paschal et al., 1993).
Of all the motility factors, the function of kinesin is by far the best understood. Recent anti-sense mRNA experiments and function-blocking antibodies have demonstrated a role for kinesin in microtubule plus end–directed movements of the endoplasmic reticulum, the _trans_-Golgi network, endosomes, and some cell-type specific organelles (Hollenbeck and Swanson, 1990; Feiguin et al., 1994; Allan, 1996; Haimo and Thaler, 1994; Burkhardt et al., 1993).
Kinectin was first identified as a membrane protein that bound to a kinesin affinity column (Toyoshima et al., 1992). On the basis of its sequence, kinectin is predicted to be a 160-kD coiled-coil forming dimer with a large cytoplasmic portion, a single transmembrane region and a very short lumenal domain (Yu et al., 1995). An antibody that binds within the predicted coiled-coil domain blocks motor binding to microsomes and both kinesin- and dynein-mediated vesicle motility (Kumar et al., 1995). The latter result has led to the idea that kinectin may be a “dual motor receptor” (Sheetz and Yu, 1996). By immunofluorescence microscopy, the protein was localized to the endoplasmic reticulum (Toyoshima et al., 1992; Fütterer et al., 1995). Its possible involvement in other kinesin-driven organelle movements is an open question.
The minus-end directed motor cytoplasmic dynein has been attributed several functions in mitosis (Vaisberg et al., 1993; Pfarr et al., 1990; Steuer et al., 1990) and roles in maintenance of Golgi structure (Allan, 1996), transport of internalized material to late endosomes (Bomsel et al., 1990; Aniento et al., 1993), and aggregation of pigment granules (Haimo and Thaler, 1994). However, a complete list of its cytoplasmic cargoes has not yet been established.
The role(s) played by motor-associated factors such as the dynein-activator, dynactin, in these processes is even less clear. Dynactin, which increases the frequency of cytoplasmic dynein-mediated vesicle movements in vitro (Schroer and Sheetz, 1991), is a large multi-protein complex (Gill et al., 1991; Paschal et al., 1993) with an intriguing shape (Schafer et al., 1994). Evidence for genetic interactions of dynactin with dynein is available from yeast, filamentous fungi and Drosophila (for review see Schroer, 1996). Perturbation of dynactin function in vivo (Echeverri et al., 1996; Burkhardt, J.K., C.J. Echeverri, and R.B. Vallee. 1996. Mol. Biol Cell. 6:266a.) or in vitro (Waterman-Storer, C.M., S.A. Kuznetsov, G.M. Langtord, D.G. Weiss, and E.L.F. Holzbaur. 1994. Mol. Biol. Cell. 5:H68) disrupts a number of processes thought to involve dynein. In vitro data support a direct interaction between dynein and dynactin (Vaughan and Vallee 1995; Karki and Holzbaur 1995). However, essentially nothing is known about how all these soluble motility factors interact with each other and their receptor(s) on target organelles.
To approach these problems, we have chosen to focus on the motile interaction of phagosomes with microtubules. We routinely use 1-μm latex beads as a marker for phagosomes (Wetzel and Korn, 1969; Stossel et al., 1971; Muller et al., 1980). In living cells, latex-bead containing phagosomes move along linear tracks, predominantly towards the cell center. We show here that this movement is microtubule-dependent and sensitive to cytosolic pH. The low buoyant density of latex allows the purification of intact phagosomes, by flotation on a sucrose step gradient, from J774 mouse macrophages that have internalized beads (Desjardins et al., 1994_a_ , b). We use a novel in vitro assay for movement of purified phagosomes along polarity-marked microtubules to show directly that phagosomes move bidirectionally, but predominantly toward microtubule minus-ends, similar to their behavior in vivo. Movement requires regulatable membrane proteins, one of which is likely to be kinectin, as well as kinesin, cytoplasmic dynein, dynactin, and at least one other component that could be a dynactin-associated kinase modulating dynein-mediated organelle motility.
Materials and Methods
Cell Culture
J774 mouse macrophages and normal rat kidney (NRK)1 cells were maintained as described in Blocker et al. (1996) and Jahraus et al. (1994), respectively. Phagosomes were prepared from J774 cells as previously described in Blocker et al. (1996). The chicken macrophage cell line HD-11 was obtained from Dr. T. Graf (EMBL, Heidelberg, Germany). It was generated and maintained as previously described (Beug et al., 1979).
In Vivo Movement Analysis
NRK cells were fed a 0.1% (wt/vol) solution of 1 μm latex beads for 30 min at 37°C in internalization medium (IM, MEM, 10 mM Hepes, 5 mM d-glucose, pH 7.4, 5% FCS from Sigma Chem. Co., St. Louis, MO) and chased, before the experiment, for 3 h in culture medium. Where indicated the cells were exposed to nocodazole (Sigma) at 10 μM in IM or in culture medium with a 20-min pre-incubation in IM on ice to depolymerize microtubules. The cells were acidified using acetate Ringers as described in Heuser (1989) for 20 min at 37°C. Cells were allowed to return to physiological pH in Ringers (Heuser, 1989) for 15 min at 37°C. Cells were then fixed in methanol at −20°C for 3 min. Microtubules were labeled using a mouse monoclonal anti-α tubulin (Amersham Corp., Arlington Heights, IL) at 1:1,000 and a rhodamine conjugated goat anti–mouse (Dianova, Germany) at 1:300. Cells were photographed on a Axiophot microscope (Zeiss) using 63× Plan-APOCHROMAT lens, a 2.5× magnifier and a Rhodamine filter set (Zeiss) on 400 ASA Kodak black and white film.
Motility Assay
Preparation.
Glass coverslips (22 × 22 mm; No. 1 Gold Seal, Clay Adams) were coated with poly-l-ornithine (PLO) as described in Blocker et al. (1996) at least one night before the experiment to insure uniform attachment and reduce gliding of microtubules along the coverglass. Rhodaminelabeled polarity marked microtubules were polymerized immediately before the experiment from stable GMP-CPP seeds as described in Howard and Hyman (1993) but in the presence of a 1:1 molar ratio of N-ethylmaleimide (Sigma) modified tubulin to unmodified tubulin (Hyman et al., 1991_b_ ). This results in a population of microtubules more homogenous in length and without a minus-end extension. To ensure unambiguous scoring of polarity, microtubules were kept dilute (about five times the concentration needed to obtain a suitable lawn) so as to reduce any end-toend annealing and subsequent breakage that might occur during pipetting.
5-μl perfusion chambers were made by sealing onto a glass slide (KTH 360, Propper Ltd., UK) a PLO-coated glass coverslip using two lines of Apezion M grease (Roth, Karlsruhe, Germany), containing 22-mm glass beads (Duke Scientific) and extruded through a syringe fitted with a 20-g blunted needle. Chambers were first wetted by perfusion of 1 vol of BRB80 (80 mM K-Pipes, 1 mM MgCl2, 1 mM EGTA, pH 6.8) for 2–5 min. This step is essential to eliminate bundling of microtubules during attachment to the coverslip, especially when PLO coverslips are less than a few days old. Two volumes of microtubules, diluted in BRB80, 10 μM taxol (Paclitaxel, Sigma), were then perfused into the chamber and allowed to bind for 5 min at room temperature. Excess microtubules were washed out by perfusion of 1 vol of 10 mg/ml aspartic acid pH 7, 10 μM taxol, to neutralize the free PLO for 3 min at room temperature. Aspartic acid was washed out by perfusion of 1 vol of BRB80, 10 μM taxol. The assay reaction was perfused next and contained (1) 2–4 μl of phagosomes at 2.5 × 109 beads/ml containing 1 μm carboxylated blue latex beads (Seradyn), which fluoresce dimly in the Texas red range, coupled to fish skin gelatin (FSG, Sigma), purified, and stripped (except where otherwise stated) with 2 M NaCl as described in Blocker et al. (1996); (2) 4 μl of 30 mg/ml macrophage cytosol prepared as in Blocker et al. (1996); (3) 1 μl of ATP regenerating system made as in Blocker et al. (1996) but from a stock of 100 mM ATP containing 100 mM MgS04, at 2 mM final concentration in the assay; (4) 1 μl of 8× antifade (Hyman and Mitchison, 1991_a_ ; Blocker et al., 1996) in PMEE (35 mM K-Pipes, 5 mM MgSO4, 1 mM EGTA, 0.5 mM EDTA, pH 7.4) with 10 mM DTT and 100 μM taxol; (5) where required the assay volume was topped to 10 μl with PMEE. The assay was always performed from frozen stocks of all reagents, including phagosomes and cytosol which were flash frozen and stored in liquid nitrogen (stocks were aliquoted and only thawed once). Reactions were incubated for 5 min at 32.5°C to allow time for the organelles to bind to microtubules and then observed for a maximum of 30 min at 32.5°C.
Under these conditions a few microtubules in each field displayed slow but constant gliding along the plane of the coverslip, the level of which provided an internal control for microtubule motor activity. Of the gliding observed, 80% corresponded to movement towards microtubule plusends. Despite our efforts to block the coverglass surface, small but significant numbers of phagosomes bound nonspecifically to the glass. This binding could not be inhibited by pretreating phagosomes with proteases and did not appear to be affected by other biological parameters. Although movement of phagosomes along microtubules was strongly dependent on the protein concentration of the cytosol in the assay (being undetectable below 5 mg/ml cytosol and increasing linearly till 20 mg/ml cytosol) some motility also occured when 1 mg/ml casein was used in place of cytosol, suggesting that purified phagosomes retained some motor activity on their surfaces. Purified phagosomes pretreated with 2 M NaCl displayed no motility in 1 mg/ml casein but moved normally in cytosol and were thus used throughout this work, except where otherwise stated. When the motility of different preparations of phagosomes needed to be compared, the volume of a given phagosome preparation added to each assay was adjusted so the final bead content of the reactions remained the same: the OD600 of each phagosome preparation, diluted several hundredfold in PBS containing 0.1% TX-100, was measured and compared to an established linear standard curve of uninternalized beads; the bead number for each point on the latter curve having been determined by FACS analysis (Jahraus, A., T.E. Tjelle, O. Ullrich, A. Habermann, and G. Griffiths, manuscript submitted for publication).
Data Acquisition.
Chambers were observed at 32.5°C (using a stageheating device) with 63× Plan-APOCHROMAT lens, a Rhodamine/Texas red filter set (BP546, FT580, LP 590; Zeiss) and a Zeiss Axiovert 10 using a 50 W mercury lamp, appropriate heat absorption and reflection filters, connected to a Zeiss shutter and a CCD-IRIS camera (Sony, Japan). The video signal was fed through a Hamamatsu camera control, into an image processor (Argus 10, Hamamatsu) and saved using a Sony OMDR and large optical disks. The shutter, the image processor, and the OMDR were controlled by a computer program (PC) available from Dr. E. Stelzer (EMBL, Heidelberg, Germany). At least 10 fields (each 160 μm2) were observed per slide, and for each field 60 frames were captured at 1-s intervals, except where otherwise stated. The field size was calibrated by measuring the number of pixels in a 40-μm pitch on a 1-mm graticule.
Image Analysis.
Images were analyzed using NIH image 1.55 on a MacIntosh Quadra connected to a video disk player or, more efficiently, using a dedicated program developed to detect, count, and track phagosomes. It runs on a Series 151 digital image processor (Imaging Technology, Bedford, MA) hosted by a SPARC station 20 (SUN, Mountain View, CA). After thresholding of the gray level image, phagosomes are recognized according to geometrical criteria such as intensity, surface, and eccentricity. Phagosome coordinates are calculated by a central moment algorithm which is also able to discriminate between a single phagosome and an aggregate. After all phagosomes are characterized and their coordinates are stored, a tracking algorithm is used to establish valid trajectories. The algorithm uses a first-order Kalman filtering approach whereby at each frame, and on the basis of the previous ones, predictions of the phagosome positions are established and compared with the computed ones. The best matches are selected as trajectory points. Tracks are analyzed according to several criteria that allow for the supervised selection of true movements along microtubules (linearity along a minimum length, speed over this length). The program yields data tables for each sequence which were used for generating the values in the figures (further details will be published elsewhere and are available from Dr. J.-C. Olivo, EMBL, Heidelberg, Germany). Movement speeds reported in this study are the averages of the maximal speed of many phagosomes; errors are standard deviations of these group estimates. The ability of the organelles to move under different conditions is expressed as movements/min/field. The values reported in this study are the mean values obtained from at least two separate but identical reactions and from at least two separate preparations of phagosomes and cytosol. The error is expressed as the population standard deviation. Where statistical analysis was performed, it was done using paired Student's t tests for testing the significance of the difference in two averages or frequencies.
Antibodies, IgG Purification, and Immunodepletions
Antibodies.
SUK4, a monoclonal antibody against sea urchin kinesin (Ingold et al., 1988) was obtained from Developmental Studies Hybridoma Bank (Rockville, MD). 70.1 and 440.4 monoclonals against chicken cytoplasmic dynein intermediate and heavy chains, respectively (Steuer et al., 1990), were a gift of Dr. M. Sheetz or purchased from Sigma Chem. Co. Monoclonal antibody 150.1 against p150_Glued_ of dynactin is described in Steuer et al. (1990). 45A and 62B monoclonal antibodies against the dynactin complex are described in Schafer et al. (1994). The 150B antibody recognizes p150_Glued_ and will be described elsewhere (Gill, S., and T. Schroer, unpublished observations). The rabbit affinity-purified polyclonal against dynein heavy chain was a generous gift from Dr. E.A. Vaisberg (Vaisberg et al., 1993). The anti-kinesin heavy chain antibody H1 (Hirokawa et al., 1989) was a kind gift from Dr. G.S. Bloom. Dr. M.P. Sheetz generously donated the mouse anti–chicken kinectin monoclonal antibody 160.9.1 (Toyoshima et al., 1992).
Immunodepletions.
Cytosol was depleted of kinesin with ascites of the SUK4 monoclonal IgG bound to protein A–Sepharose through the IgG fraction of rabbit anti–mouse IgG serum (Sigma). Another monoclonal IgG (P5D4), raised against a viral antigen (Kreis, 1986), was used as a control antibody. Dynactin complex was depleted using ascites of the 45A monoclonal IgG (Schafer et al., 1994). The same procedure (Bomsel et al., 1990; Aniento et al., 1993) was used to deplete cytoplasmic dynein with the 70.1 monoclonal IgM (Sigma) coupled to protein A–Sepharose beads via the IgG fraction of a rabbit anti–mouse IgM serum (Sigma). A monoclonal IgM, raised against chondroitin sulphate (Sigma), served as a matched control. For 60 μl of dry bead gel 10–50 μl of antibody was used, stored in 50% gylcerol at −20°C. Beads were washed in PBS, 5 mg/ml bovine serum albumin (Biomol, Hamburg, Germany), and finally into PMEE, 1 mM DTT, and protease inhibitors (Blocker et al., 1996). Immunodepletions were always performed on the day of the experiment using a freshly thawed aliquot of cytosol or ATP release, because it was determined that mock immunodepleted cytosol could not be refrozen without significant loss of motile activity. For 50 μl of cytosol or ATP release, two rounds of immunodepletion were performed for 30 min on ice using 30 μl dry bead gel per round. Before each round and between each round, the bead slurry was carefully aspirated dry using centrifugation and a fine gel loading tip to minimize sample dilution.
Staurosporine (Sigma) was dissolved at 200 μM in DMSO and stored in aliquots at −20°C.
Elution of Immunodepletion Beads.
After immunodepletion, the supernatant was collected as above and the beads from each round were eluted in a total of 50 μl PMEE, 1 M NaCl, 1 mM DTT, and protease inhibitors (Blocker et al., 1996) for 30 min on ice. The eluate was collected, desalted, and concentrated 10-fold in Nanosep centrifugal concentrators with a 3-kD cut-off point (Pall Filtron) according the manufacturer's instructions.
IgG Purification.
P5D4 and SUK4 IgG were purified using caprylic acid clearing and ammonium sulphate precipitation, much as in Ingold et al. (1988) because this method was found to be more suitable for small scale purification and recovery of concentrated IgG fractions than protein A affinity chromatography. Briefly, 200 μl of ascites fluid was diluted in 4 vol of 60 mM sodium acetate, pH 4, and brought to pH 4.5 with 0.1 M NaOH. 25 μl of caprylic acid was added dropwise. After 30 min of stirring at room temperature, the solution was clarified by centrifugation at 10,000 g for 30 min. The supernatant was further cleared by filtering through a nylon mesh, slowly brought to 45% ammonium sulphate, and left to stir for 30 min at 4°C. IgG was pelleted in the cold at 5,000 g for 15 min, resuspended in 50 μl of PBS, and dialyzed in micro-collodion bags (Sartorius) against three changes of PBS. This yielded 10–20 mg/ml essentially pure IgG, as monitored by SDS-PAGE and Coomassie blue staining, which was stored in aliquots at −20°C.
Purified Dynein, Dynactin, and Recombinant Kinectin Fragments
Kinectin Fragments.
The generation and purification of the chicken kinectin protein fragments will be described in detail elsewhere (Yu, H., J. Kumar, V.S. De Serrano, W. Chan, J. Heringa, A. Shendarov, W.J. Strittmatter, and M.P. Sheetz, manuscript submitted for publication). Overlapping DNA fragments corresponding to amino acids 295-612, 568-943, and 9241321 of the published chicken kinectin sequence (Yu et al., 1995) were cloned and expressed in E. coli. The kinectin fragments were purified from bacterial cytosol by ammonium sulphate precipitation, followed by gel filtration, ion-exchange chromatography, and final dialysis into PMEE. They were stored at −80°C.
Dynein and Dynactin.
Details of the procedure for purification of cytoplasmic dynein and dynactin from bovine brain will be published elsewhere (Bingham, J., and T. Schroer, manuscript in preparation). Briefly, both proteins in a high speed supernatant were concentrated and enriched by two successive rounds of ion exchange chromatography on fast-flow S-Sepharose (Pharmacia LKB Biotechnology, Piscataway, NJ) followed by velocity sedimentation into a 5–20% sucrose gradient. Dynein and dynactin copurify through these steps of the purification. The final sucrose gradient fractions enriched for dynein and dynactin were then separated on a Mono Q ion exchange column (Pharmacia). Both proteins had the expected polypeptide compositions and were >98% pure as judged by Coomassie blue staining of overloaded gels (dynactin was very slightly contaminated with dynein). Both proteins were routinely obtained at 5 mg/ml. We established that such preparations, collected straight off the Mono Q column, could be stored at −20°C cryoprotected in 50% glycerol in PMEE, 1 mM DTT, and 0.5 mM Mg2+-ATP or by flash freezing in 1.25 M sucrose also in PMEE, 1 mM DTT, and 0.5 mM Mg2+-ATP in aliquots in liquid nitrogen, followed by storage at −80°C. After such long-term storage, freshly thawed dynein preserved microtubule gliding activity down to 20–50 μg/ml (10–25 nM) in assays performed as described in Middleton and Carbon (1994).
Electrophoresis and Immunoblotting
SDS-PAGE was performed as in Laemmli (1970) using a minigel apparatus according to the manufacturer's instructions (Bio-Rad Labs, Hercules, CA). Blots were performed using a Bio-Rad wet minigel chamber at 70 mA overnight as this led to more complete transfer of large polypeptides such as cytoplasmic dynein heavy chain. Blots were revealed using the ECL detection system (Amersham). Quantitative immunoblotting of dynein and dynactin in J774 macrophage cytosol was performed by calibrating the polyclonal dynein heavy chain antibody and the p150_Glued_ antibody 150B against purified bovine dynein and dynactin of known concentrations. Autoradiograms were optically scanned and the band intensities were measured using NIH Image for the MacIntosh. Densitometry of Coomassie-stained gels was performed similarly.
Electron Microscopy
HD-11 or NRK cells were prepared for cryosectioning and immunogold labeling as previously described (Griffiths, 1993). Briefly, the cells were removed from the dishes by digestion with 50 μg/ml proteinase K (Merck, Darmstadt, Germany) for 1–2 min at 4°C. They were fixed in 8% paraformaldehyde (Merck) in 250 mM Hepes, pH 7.4, infiltrated with 2.1 M sucrose as a cryoprotectant, and frozen in liquid nitrogen. Thawed ultra thin cryosections were labeled with the primary antibody, which was detected with protein–A colloidal gold (Department of Cell Biology, University of Utrecht, The Netherlands). Finally, the grids were embedded in a 9:1 mixture of 2% methylcellulose (Sigma) and 3% uranyl acetate (Serva) to protect from drying and to add contrast.
Results
The Movement of Phagosomes Inside Cells Is Microtubule-dependent and Sensitive to Cytosolic pH
Previous studies have shown that phagosomes move within cells and suggested that this movement is microtubule-mediated. To show more directly that phagosome motility was indeed occurring along microtubules, we carried out a series of in vivo studies. First, the behavior of phagosomes was observed in intact cells by bright-field video microscopy. Latex beads were briefly pulsed and chased into J774 macrophages. The phagosomes thus formed were seen to move along linear tracks over distances of several microns at speeds that approached 1 μm/s. Phagosomes moved mainly towards the cell center, but some also moved back towards the periphery of the cell. After longer chase periods, most phagosomes accumulated in the perinuclear area. Once these organelles had clustered in the center of the cell, they tended to move less frequently (data not shown). These data argue that phagosomes are able to move directionally within J774 cells.
We sought to establish more definitively whether phagosome motility was predominantly microtubule-mediated. Initial attempts to use the microtubule-depolymerizing drug nocodazole were unsuccessful, because even brief treatment with this drug caused J774 cells to round up, making spatial analysis impossible (see also for example Cassimeris et al., 1986). We therefore used normal rat kidney (NRK) cells, a cell type known to undergo phagocytosis (Jahraus et al., 1994; Desjardins et al., 1994_b_ ) whose morphology is relatively insensitive to microtubule depolymerization. After pulse and chase with latex beads, NRK cells also accumulated latex-containing phagosomes in their perinuclear region (Fig. 1 A). In about half the cells, the phagosomes appeared clustered near the microtubule organizing center (MTOC, compare Fig. 1, A and B). Fig. 1, C and D show that if the cells were treated with nocodazole during the chase period, microtubules were entirely depolymerized and phagosomes no longer accumulated in the perinuclear region, but were instead distributed either peripherally or at random. Thus, phagosomes require microtubules to display directed movement within these cells.
Figure 1.
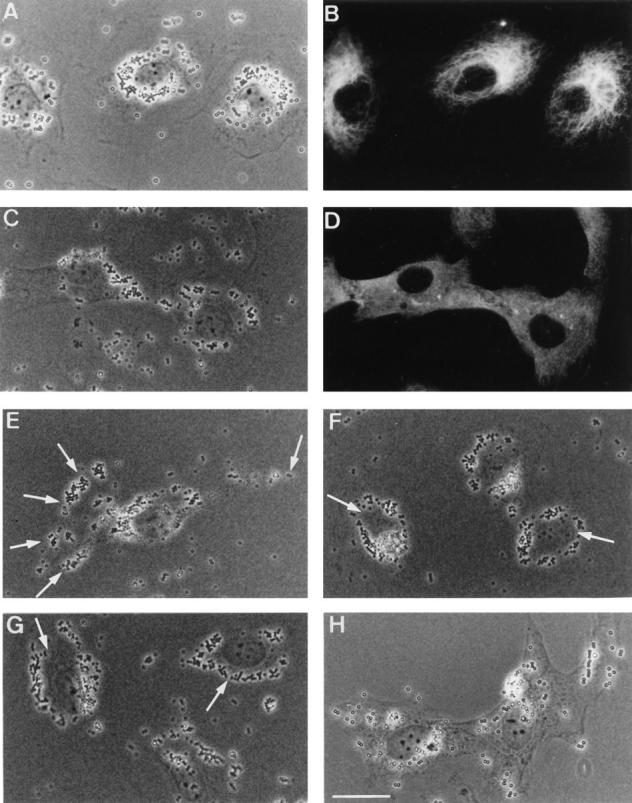
Phagosome movement is microtubule-dependent and sensitive to cytosolic pH. (A) NRK cells were pulsed with latex beads for 30 min and chased for 3 h. At this time all the beads are observed to be in the perinuclear region of the cells. (B) Indirect immunofluorescence labeling of microtubules in the same cells. (C) NRK cells were pulsed with beads as above but subsequently treated and chased in the presence of nocodazole. The phagosome distribution in these cells appears random. (D) The same cells as in D stained using an anti α-tubulin antibody. (E) NRK cells pulsed and chased as above but then treated with acetate Ringers for 20 min. About 30– 50% of the phagosomes were seen to move out to the periphery of the cell (arrows indicate cell boundary). This is observed in ∼70% of the cells. (F) NRK cells loaded with beads, incubated in acetate Ringers for 20 min, and then physiological Ringers for 15 min. Rebound movement of phagosomes to restore tight perinuclear localization of these organelles is observed. Only ∼50% of the cells survive this treatment. (G) NRK cells loaded with beads as above but treated with nocodazole before treatment with acetate Ringers in the presence of nocodazole. Phagosomes do not move to the periphery of the cells but instead appear to be “released” from the perinuclear region (arrows point to the increased space between phagosomes and the nuclear membrane). When cells are treated with nocodazole after the phagosomes have reached the perinuclear region rather than during the chase period, their distribution is very similar (not shown). (H) NRK cells treated with acetate Ringers and then nocodazole before treatment with physiological Ringers containing nocodazole. Phagosomes do not return to a tight perinuclear localization. Bar, 10 μm.
Finally, we wanted to know if phagosome movement along microtubules displayed characteristics similar to those reported for endocytic organelles to which they are functionally related. The direction of movement of endosomes and lysosomes, but not other organelles, is sensitive to acidification of the cytosol in several cell types (Heuser, 1989; Parton et al., 1991; Lin and Collins, 1992). In these experiments, exposure of cells to mildly acidic physiological medium caused microtubule-dependent endocytic organelle dispersal, a phenomenon that could be reversed by returning the cells to a medium at physiological pH. Acidification of NRK cells with acetate Ringers for 20 min caused 30–50% of phagosomes to move to the periphery of the cells (Fig. 1 E). This result was seen in ∼70% of the cells. Fig. 1 F shows that upon return to physiological Ringers for 15 min, the phagosomes returned to a tight cluster at the cell center in the 50% of the cells that survived this treatment. When acidification was performed after treatment of the cells with nocodazole, phagosomes did not move to the periphery of the cells but instead appeared to be “released” from the tight cluster within the perinuclear area seen in the absence of nocodazole (Fig. 1 G). To test if return of phagosomes to the cell center after dispersal was also microtubule-mediated, cells were first acidified to disperse phagosomes, and then treated with nocodazole followed by physiological Ringers. Under these conditions, the reclustering of phagosomes was blocked and they remained peripheral (Fig. 1 H). These results show that the position and movement of phagosomes within cells are dependent on an intact microtubule network. As microtubules mostly emanate from the MTOC with their plus-ends towards the cell periphery in these cells, the ability of phagosomes to display microtubuledependent centripetal and centrifugal movement suggests that these organelles carry both a plus-end and a minusend directed microtubule motor on their surface.
Phagosomes Move Bidirectionally Along Microtubules but Mainly to the Minus End
To dissect the mechanism of phagosome transport in molecular detail, we have reconstituted the movement of purified phagosomes along polarity-marked microtubules in vitro using a fluorescence video microscopy assay. A uniform lawn of dimly fluorescent microtubules marked at their minus ends by brightly labeled “seeds” (Howard and Hyman, 1993) was laid down on the coverglass of a perfusion chamber. A mixture of purified, salt-stripped phagosomes (containing weakly fluorescent latex beads coupled to fish skin gelatin, FSG; see Materials and Methods), J774 macrophage cytosol, and an ATP regenerating system were then added. In the presence of both ATP and cytosol, phagosomes displayed movements, sometimes many microns in length, along microtubules (Figs. 2 A and 3 A). A small number of microtubules in each field displayed gliding which was essentially plus-end directed (not shown).
Figure 2.
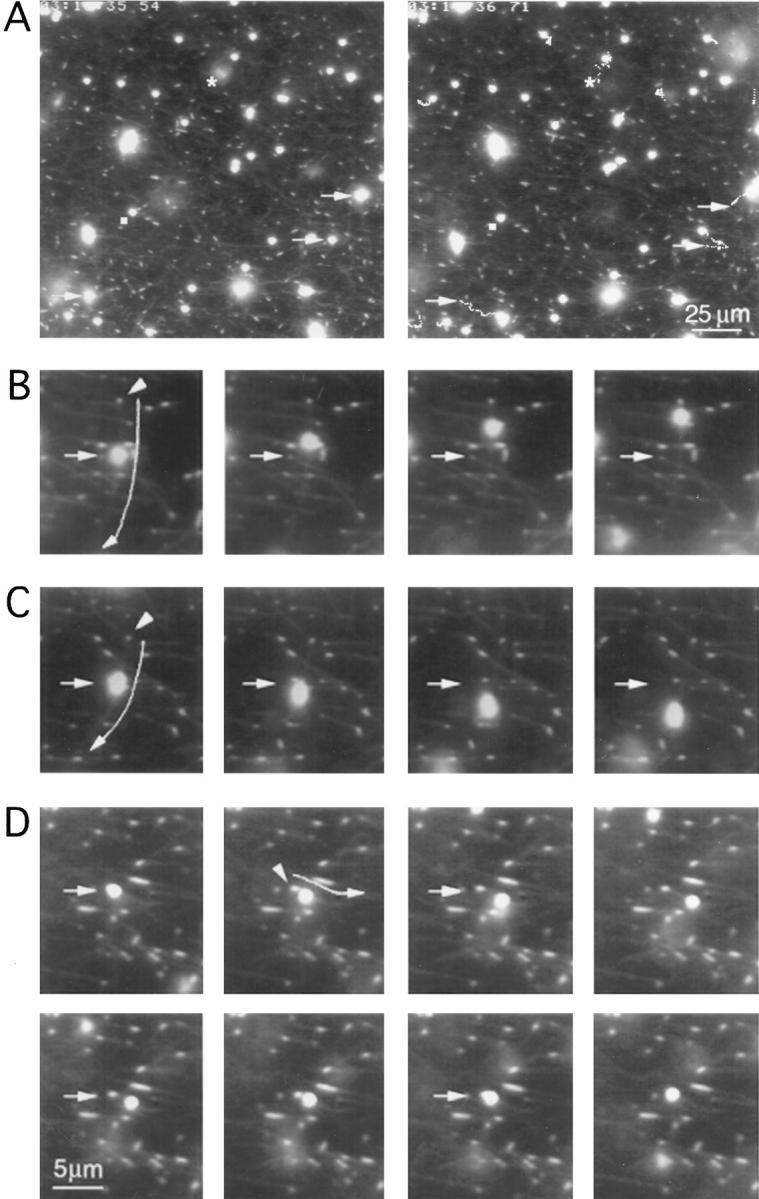
In vitro motility of phagosomes along polarity-marked microtubules. (A) The first (left) and last (right) recorded frames of a typical motility assay field, as analyzed by the tracking program (numbers given by the program to each phagosome have been omitted to allow better visualization of the tracks). Arrows indicate the starting position of three phagosomes that have moved significant distances. By following the tracks starting at these arrows in the left panel, one sees that phagosomes can move relatively long distances and often switch microtubules in the process. The star and square, respectively, indicate phagosomes demonstrating Brownian movement and remaining stationary throughout, for comparison. (B) A close-up of a minus-end directed movement. The arrowhead indicates the brightly labeled rhodamine tubulin “seed” marking the minus end of the microtubule along which the phagosome is moving. The short arrows indicate the original starting point of each phagosome. Long arrows are drawn parallel to the microtubule along which the phagosome is moving to allow better visualization; the arrow points towards the plus end of the microtubule. Frames are shown at 3-s intervals. (C) A plus-end directed movement. (D) A bidirectional movement.
Figure 3.
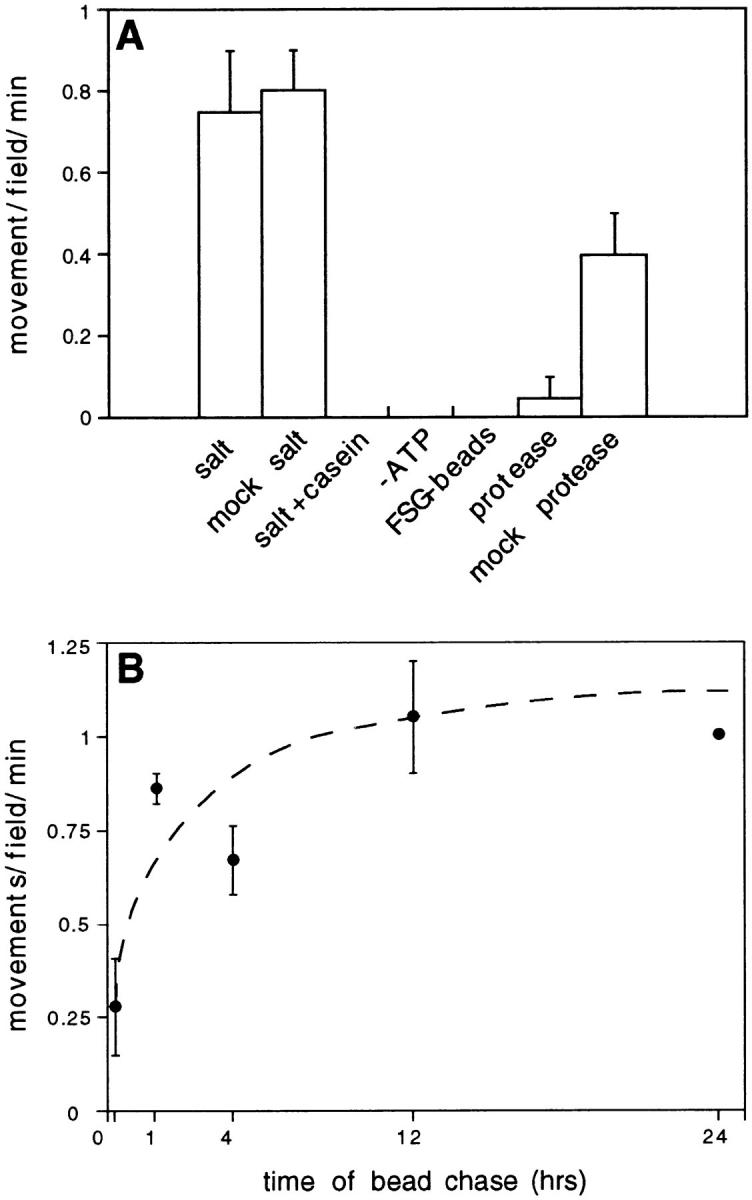
Movement requires energy, cytosol, and proteins of the phagosome membrane, and is regulated during phagosome maturation. (A) The motility assay was performed with 2 M NaCl- or mock-stripped phagosomes in the presence of 15 mg/ml cytosol and an ATP regenerating system (salt and mock salt). Saltstripped phagosomes did not move in the presence of 1 mg/ml casein (salt + casein), nor in the presence of cytosol but in the absence of the ATP regenerating system (−ATP). Uninternalized fish skin gelatin-coated beads (FSG-beads) did not move in the presence of cytosol and ATP. Treatment of phagosomes with 30 μg/ml chymotrypsin for 30 min at 4°C (protease; the protease was then inhibited by treatment with 3,4-dichloroiscoumarin and the phagosomes were separated from residual active protease by flotation into a small sucrose gradient as described in Blocker et al., 1996) reduced their ability to move by eightfold over control (mock protease). (B) Latex bead containing phagosomes were purified from J774 macrophages after various times of internalization: a 20-min pulse, a 1-h pulse followed by a 1-, 4-, 12-, or 24-h chase. The different phagosome preparations were adjusted for bead content in the assay by optical density measurement (see Materials and Methods). This figure was generated using non–saltstripped phagosomes. The dotted line represents an optimized curve fit generated by the computer program KaleidoGraph. Each value represents the mean of the average movements/field/ min of at least two, but often many more, identical motility chambers; errors are population standard deviations. Each experiment was independently repeated at least twice, but often many more times. For each point at least two different preparations of cytosol and phagosomes was tested.
In this system, 10–30% of the phagosomes that became bound to microtubules moved along them at any time. Of the moving phagosomes, 70.8% displayed minus-end directed movement, at the average maximal velocity of 1.21 ± 0.39 μm/s (n = 34, Fig. 2 B). The remaining 29.8% displayed plus-end directed movement, with a maximal velocity of 0.93 ± 0.26 μm/s (n = 14, Fig. 2 C). 8.9% of all moving phagosomes displayed bidirectional movement either along the same or two different microtubules (Fig. 2 D). These results directly demonstrate that phagosomes are able to move in both directions along microtubules but move primarily to the minus-end.
Movement Requires Membrane Protein(s) of the Phagosome
Next, we sought to determine whether the directional movement of phagosomes was dependent upon proteins within their enclosing membrane. Bidirectional phagosome movement did not appear to be a simple reflection of the abundance of different microtubule motor proteins in cytosol, since in the same assay samples microtubules displayed mostly plus-end directed gliding (a phenomenon which is assumed to reflect the activity of motors bound to the coverglass according to their cytosolic abundance). Moreover, when we assayed the cytosol-dependent movement of uninternalized carboxylated latex beads whose naked surface was not blocked with FSG, we observed excellent motility that was again predominantly plus-end directed (3.9 movements/field/min, 70% plus-end directed, n = 19). We next compared the motility of uninternalized FSGcoupled latex beads with the behavior of salt-stripped and protease-treated phagosomes. In the presence of cytosol latex beads covalently coupled to FSG did not move at all along microtubules, in contrast to their predominantly minus-end directed movement after internalization into phagosomes. Protease treatment of phagosomes reduced their ability to move along microtubules by nearly eightfold over control (Fig. 3 A), but had no effect on microtubule gliding in the assay (not shown). As stripping of the phagosomes with 2 M NaCl did not affect their motility in the presence of cytosol (Fig. 3 A), the proteins required to recruit the cytosolic motility factors to the phagosome membrane are likely to be either tightly associated peripheral- or integral-membrane proteins of this organelle.
Phagosomes are de novo assembled, maturing organelles. Although latex is nonbiodegradable, latex bead–containing phagosomes display some degree of maturation (Desjardins et al., 1994_a_ ; Blocker et al., 1996; Jahraus, A., T.E. Tjelle, O. Ullrich, A. Habermann, and G. Griffiths, manuscript submitted for publication) and we wanted to know whether their ability to move along microtubules changed as a function of time. As shown in Fig. 3 B, “early” phagosomes (isolated after 20 min of bead internalization) displayed only a third of the motility of “later” phagosomes (isolated after 1 h pulse/1 h chase or longer). The directional preference of early and late phagosomes appeared identical (data not shown). Since both cytosol and microtubules were identical in these assays, it follows that the increase in motility observed with phagosome maturation reflects a biological difference in proteins exposed on the cytoplasmic surface of these organelles.
Pharmacological Characterization of Phagosome Motility
To better define the microtubule motors involved in phagosome movement, we performed a pharmacological characterization of motility using reagents that allow some separation of the activity of cytoplasmic dynein and kinesin-like motors (McIntosh and Porter, 1989; Shimizu et al., 1991). To test the effect of some nucleotides and nucleotide analogues in a controlled manner (Table I), we first desalted macrophage cytosol (as described in Blocker et al., 1996) and measured motility in the presence of 10 mM Mg2+ATP. Under these conditions, phagosomes displayed a normal level of motility which was 81.8% minus-end directed (Table I). The spectrum of response of the plus-end directed motor was identical to that of conventional, but not of kinetochore-kinesins (Hyman and Mitchison, 1991_a_ ), because AMP-PNP reduced plus end–directed movements and GTP increased them while decreasing their speed. The minus-end directed motor activity was essentially unaffected by AMP-PNP but inhibited by GTP and vanadate above 1 μM, as expected for dyneins (Gibbons et al., 1978; Lye et al., 1987). Taken together these results are consistent with the notion that bidirectional phagosome movement along microtubules is mediated by motors with properties of cytoplasmic dynein and conventional kinesin.
Table I.
Pharmacological Characterization of Phagosome Motility
| Condition | Number of movements/min/field | Percent plus end–directed movements | n | Velocity (μm/second) |
|---|---|---|---|---|
| no ATP | 0 ± 0 | NA | NA | NA |
| 10 mM MgATP | 1.17 ± 0.51 | 18.2 | 33 | 1.03 ± 0.31 |
| 10 mM MgGTP | 0.13 ± 0.10 | 100 | 10 | 0.38 ± 0.06 |
| 10 mM MgATP | 0.85 ± 0.29 | 6.5 | 31 | ND |
| + 1 mM MgAMP | ||||
| PNP | ||||
| 1 μM VO4+ | 0.79 ± 0.20 | 30.8 | 26 | ND |
| 10 μM VO4+ | 0.27 ± 0.11 | 57.1 | 21 | ND |
| 100 μM VO4+ | 0.17 ± 0.04 | ND | NA | ND |
Cytoplasmic Dynein, Dynactin, and Kinesin Localize to Phagosomes In Vivo
At least three non-isogenic forms of cytoplasmic dynein have been recently identified in various systems (Tanaka et al., 1995; Vaisberg et al., 1996) and several nonkinetochore kinesin-related proteins are known to co-exist within the same cell type (reviewed in Hirokawa, 1996). We therefore wished to determine which member(s) of the kinesin and dynein families were in fact present on this organelle in vivo. Immunoblot analysis on purified phagosomes indicated that some, but not all, subunits of conventional dynein, dynactin, and kinesin could be detected (Blocker, 1995). Since all three proteins are peripherally associated with membranes, we reasoned that they might be partially lost from phagosomes during purification. We therefore decided to examine the association of these proteins with phagosomes in a more in vivo context, by performing immunoelectron microscopy on cells that had internalized latex beads. The antibody 440.4 raised against conventional chicken dynein heavy chain (CD1; however, it is not yet clear whether this antibody also recognizes the other cytoplasmic dynein heavy chains, Vaisberg, E., personal communication) labeled the cytoplasm as well as membrane organelles, in particular phagosomes, in HD11 chicken macrophages (Fig. 4 A). A subunit of the dynactin complex, p150_Glued_, showed a very similar distribution (Fig. 4 B and Burkhardt, J.K., A. Habermann, G. Griffiths, and T.A. Schroer, manuscript in preparation). The monoclonal antibody H1, against bovine kinesin heavy chain, was tested on NRK cells. Kinesin was found on all endocytic organelles including phagosomes (Fig. 4 C), although it was more abundant on other organelles (Blocker, A., A. Habermann, and G. Griffiths, in preparation). Although it was not possible to estimate from this data the approximate number of such molecules present per phagosome (antibody labeling efficiencies using this method are very difficult to estimate), the relative quantitative distribution of these factors on phagosomes and other organelles will be published elsewhere. The key point for the present study is that cytoplasmic dynein (mostly likely CD1), dynactin, and conventional kinesin localize to phagosomes in vivo.
Figure 4.
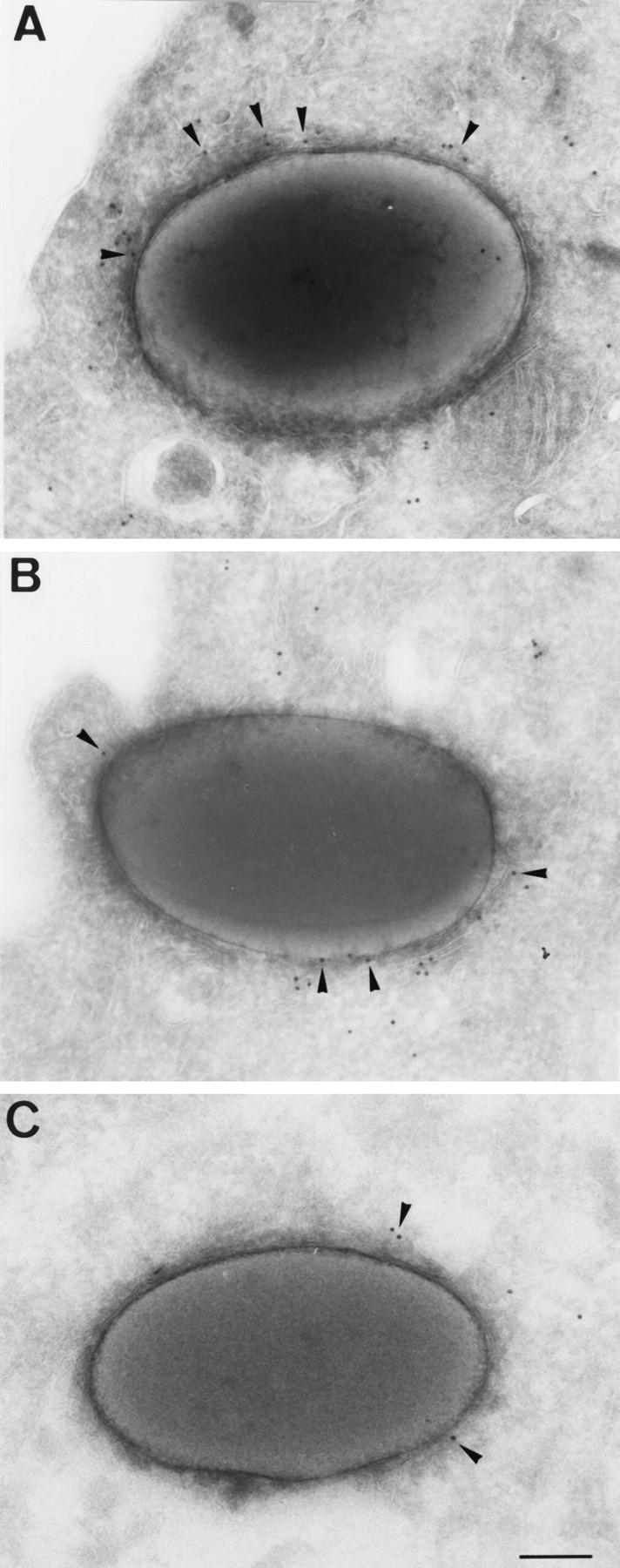
Subunits of cytoplasmic dynein, the dynactin complex, and kinesin are found on phagosomes. HD-11 chicken macrophages (A and B) or NRK cells (C) were pulsed and chased with 1 μm diameter latex beads for 1 h and then processed for cryosectioning and immunoelectron microscopy. Immunolocalization on phagosomes of (A) cytoplasmic dynein heavy chain, using the 440.4 monoclonal antibody; (B) the dynactin subunit p150_Glued_, using the 150.1 monoclonal antibody; and (C) kinesin heavy chain using the H1 monoclonal antibody. Note the labeling (arrowheads) on the limiting membrane of latex-bead containing phagosomes. Bar, 0.25 μm.
Minus End–directed Phagosome Movement Is Mediated by Cytoplasmic Dynein
The previous results suggested that the motility of phagosomes is driven by cytoplasmic dynein (CD1; see Vaisberg et al., 1996), dynactin, and conventional kinesin. To verify this hypothesis, we used immunological reagents specific for these different proteins to determine whether each was functionally involved in mediating phagosome motility in our in vitro assay. Immunodepletion of cytoplasmic dynein was performed using the monoclonal antibody 70.1 against the conventional cytoplasmic dynein 74-kD intermediate chain (however, it is still unclear whether cytoplasmic dynein heavy chains share associated subunits, Vaisberg, E., personal communication). Performing two rounds of immunodepletion removed ∼80% of the original dynein heavy chain (Fig. 5 A). Removal of dynein reduced phagosome motility significantly as compared to a mock-depleted control. Minus end–directed motility was reduced from 70– 25% while plus-end movement was not affected (Fig. 6 A). We estimated the amount of dynein present in cytosol to be 2 nM by quantitative immunoblotting using purified bovine dynein as a standard. Motility of dynein depletedcytosol could be fully restored by addition of 25 nM purified bovine brain dynein (Figs. 5 B and 6 A; this was the lowest concentration that supported microtubule gliding, see Materials and Methods). Thus, cytoplasmic dynein (most likely CD1) is the motor for minus end–directed movement of phagosomes along microtubules.
Figure 5.

(A) (Left) J774 cytosol was immunodepleted of cytoplasmic dynein with the dynein intermediate chain monoclonal antibody 70.1 (70.1), and mock depleted with an anti-chondritin sulphate antibody (control); the blot was probed with a polyclonal anti-cytoplasmic dynein heavy chain (DHC). The blot was also probed with 150B, an antibody to p150_Glued_ of dynactin (below). (Right) Macrophage cytosol was depleted of dynactin complex with the Arp1 monoclonal antibody 45A (45A), and mock depleted with P5D4 (control); the blot was probed with 150B. The blot was reprobed with anti-DHC (below). (B) Pure bovine brain cytoplasmic dynein (dynein), dynactin (dynactin), and J774 macrophage ATP release (ATP release) were run on a 6–15% gradient gel and stained with Coomassie blue. (C) Phagosomes were purified from HD11 chicken monocyte after different chase periods within cells: 20 min pulse, no chase and 1 h pulse followed by a 4- or 12-h chase. The fractions were normalized for bead content, blotted along side cytosol (Cyt) and a crude membrane fraction (Memb) from HD11 monocytes, at the same protein concentration, and probed with 160.9.1, a monoclonal antibody against chicken kinectin.
Figure 6.
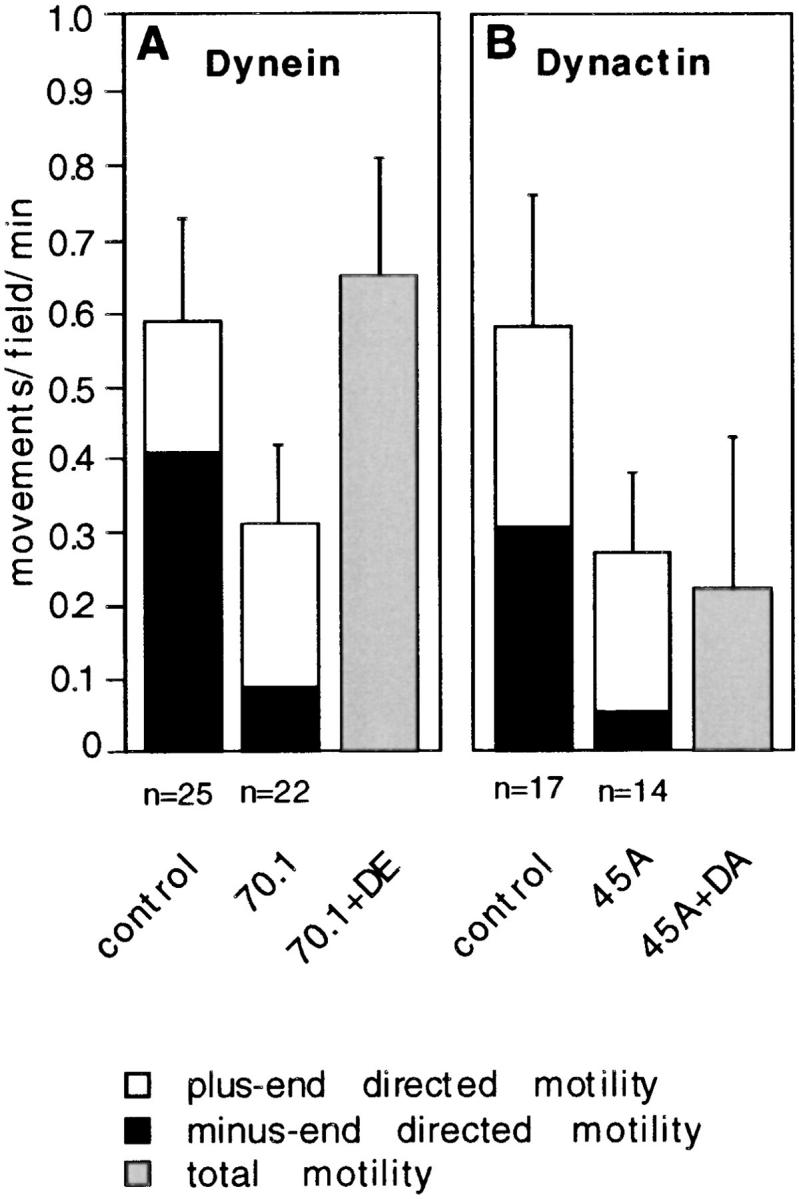
Phagosome motility requires cytoplasmic dynein and dynactin complex. (A) Phagosome motility driven by cytosol subjected to two rounds of immunodepletion with the anti-cytoplasmic dynein intermediate chain monoclonal antibody 70.1 (70.1) or mock adsorbed with an isotype-matched monoclonal antibody (anti-chondroitin sulfate; control). Bar 70.1+DE shows the activity of dynein-depleted cytosol to which 25 nM bovine brain dynein was added. (B) Cytosol was immunodepleted of dynactin by two rounds of adsorption with the anti-Arp1 of dynactin monoclonal antibody 45A (45A) or mock depleted with an isotypematched control monoclonal antibody (P5D4; control) and then assayed for motility. The bar labeled 45A+DA shows the activity of dynactin-depleted cytosol to which 25 nM dynactin was added. n is the total number of movements of clear polarity scored for each condition. The percents of plus end–directed movements observed in dynein and dynactin depleted cytosols are significantly different from those seen in the control depleted cytosols at P = 0.01 and P = 0.10, respectively. Black bars correspond to the percent of minus end–directed movements; white bars correspond to the percent of plus end–directed movements; and gray bars represent total motility. Each value represents the mean of the average movements/field/min of at least two, but often many more identical motility chambers; errors are population standard deviations. Each experiment was independently repeated at least twice, but often many more times. For each point at least two different preparations of cytosol, phagosomes, and purified protein or ATP release were tested.
Dynein-mediated Movement Requires Dynactin and at Least One Other Dynactin-regulating Component (Activator X)
To investigate cytoplasmic dynein function in more detail, we immunodepleted dynactin complex from cytosol using 45A, a monoclonal antibody against its most abundant subunit, the actin-related protein Arp1. Blotting with 150B, a monoclonal antibody against the dynactin subunit p150_Glued_, verified that this procedure removed 95% of the complex (Fig. 5 A). It should be noted that nearly quantitative removal of dynactin from cytosol did not detectably reduce the amount of dynein heavy chain (Fig. 5 A). Immunodepletion of dynactin complex inhibited minus-end directed motility almost completely while leaving plus end– directed motility unaffected (Fig. 6 B). Using quantitative immunoblotting, we estimated the concentration of dynactin in 30 mg/ml cytosol to be ∼2 nM, similar to that of dynein. Thus, in the most dilute cytosol that supports normal levels of phagosome movement (15 mg/ml), endogenous dynactin is present at a concentration of ∼1 nM. Surprisingly, motility could not be restored by addition of chick brain dynactin at 5 nM (the highest concentration available; not shown) or by bovine brain dynactin at concentrations between 2.5 and 25 nM (Fig. 6 B), or even as high as 250 nM. This suggested that an additional component of cytosol, that was immunodepleted in conjunction with dynactin, was necessary for motility.
To test this hypothesis, we determined whether dynactin-enriched fractions taken from different steps of the purification could restore phagosome motility to dynactindepleted cytosol. A partially purified mixture of dynactin and cytoplasmic dynein did not restore motility (the 20S sucrose gradient pool, see Schroer and Sheetz, 1991; data not shown). However, phagosome motility could be fully restored by macrophage microtubule “ATP release” (prepared as in Blocker et al., 1996; Fig. 7 A). This fraction is enriched in microtubule-binding proteins that dissociate from microtubules in the presence of ATP and contains a number of abundant, defined polypeptides (including dynein and dynactin), as well as many other minor, unidentified proteins (its composition is shown in Fig. 5 B). Dynactin represents ∼5% of the total protein in this fraction, as estimated by quantitative immunoblotting. That a crude, ATP release fraction, but not partially or highly purified dynactin could restore activity to dynactin-depleted cytosol suggested that ATP release contained an additional “dynactin-activating” component necessary for dyneinmediated vesicle motility.
Figure 7.

Dynactin activity requires at least Activator X, which may be a dynactinassociated kinase. All assay samples, with the exception of bar 17 (mock-depleted cytosol; control), contained dynactin-depleted cytosol prepared as in Fig. 6. (A) Effect of different concentrations of ATP release on dynactindependent motility. Dynactin-depleted cytosol was reconstituted with different concentrations of macrophage ATP release (0.4 mg, 4 μg, or 0.8 μg/ml) and assayed for motility. The sample containing 0.8 μg/ml ATP release also contained 25 nM bovine brain dynactin. (B) Removal of the “dynactin-activating” activity from ATP release by immunoadsorption of dynactin. ATP release samples were immunoadsorbed with monoclonal antibody 45A (dynactin depleted ATP release) or mock-adsorbed with a control monoclonal antibody (control depleted ATP release), then tested for ability to restore activity to dynactin-depleted cytosol in combination with exogenous bovine brain dynactin (± dynactin). A high salt eluate of the 45A immunoadsorbent (ATP release dynactin eluate; 1.5 μg/ml), control immunoadsorbent (ATP release control eluate; 1.5 μg/ml), or the eluate of 45A immunoadsorbent from the original depletion of cytosol (cytosol dynactin eluate; 20 μg/ml) were also tested for activity. Bar 5 represents the same data as in bar 1 for comparison. Values for bars 9 and 10 are significantly different at P = 0.05. (C) Effect of the general protein kinase inhibitor, staurosporine, on the dynactin-activating activity of the ATP release. 50 nM staurosporine was added to dynactin-depleted cytosol immediately before addition of bovine dynactin and ATP release or to mock-depleted cytosol (control; motility is similar to control cytosol alone, compare with Fig. 6 B; and was also unaffected by addition of DMSO alone, not shown). Bar 13 illustrates the same data as in bars 1 and 5; bar 14 illustrates the same data as bar 3; and bar 15 illustrates the same data as bar 6; these three values are included for comparison. Each value in this figure represents the mean of the average movements/field/min of at least two, but often many more, identical motility chambers; errors are population standard deviations. Each experiment was independently repeated at least twice, but often many more times. For each point at least two different preparations of cytosol, phagosomes, and purified protein or ATP release were tested.
To verify this, and exclude that the hypothetical dynactin-activating component of macrophage ATP release was simply “activated” dynactin, we diluted ATP release to a concentration at which endogenous dynactin was present in amounts below the 1 nM normally required for dyneinbased motility in our assay (4 μg/ml ATP release provides 0.2 nM dynactin). As expected, at this concentration the ATP release did not restore activity to dynactin-depleted cytosol. However, further addition of 25 nM bovine brain dynactin fully restored activity to the sample (Fig. 7 A). Moreover, we found that ATP release could be diluted to concentrations as low as 0.8 μg/ml and still restore activity to dynactin-depleted cytosol in the presence of exogenously added dynactin (Fig. 7 A). These data strongly support the notion that the ATP release contains at least one other factor, which we term Activator X, that is required for minus-end directed phagosome motility driven by dynein and dynactin.
Activator X May be a Dynactin-associated Kinase
That exogenously added bovine dynactin by itself could not restore activity to dynactin-depleted cytosol suggested that Activator X might be immunoprecipitated along with dynactin by the 45A monoclonal antibody. To explore this possibility further, we took advantage of the fact that ATP release contains both dynactin and the putative Activator X. ATP release was immunodepleted of dynactin using the 45A monoclonal antibody or mock-depleted and tested for its ability to restore motility to dynactin-depleted cytosol in the presence of bovine dynactin (Fig. 7 B). Dynactin-depleted ATP release was unable to restore motility to dynactin-depleted cytosol containing bovine dynactin, whereas control-depleted ATP release continued to function normally (Fig. 7 B, compare bars 7 and 8). This was similar to our initial findings with cytosol and supported the hypothesis that Activator X interacted with dynactin such that it could be co-precipitated with dynactin by the 45A antibody.
As a further test of our hypothesis we attempted to elute Activator X from the immunoadsorbent. Control and 45A antibody beads were washed with 1 M NaCl and the eluates desalted and concentrated. The salt eluates were then tested for the ability to restore motility to dynactin- depleted cytosol containing exogenously added bovine dynactin. Fig. 7 B shows that the eluate of the 45A antibody beads (bar 9) fully restored motility to dynactin-depleted cytosol in the presence of bovine dynactin (but not in its absence, bar 10), whereas the eluate from the control immunoadsorbent did not (bar 11). However, we were unable to restore motility to dynactin-depleted cytosol with a similar eluate from 45A beads that had been adsorbed with cytosol (Fig. 7 B, bar 12). We found that the recovery of X activity from the 45A immunoadsorbent incubated with ATP release was very low, at best 5% (in terms of the volume of ATP release and concentrated eluate that had to be added to obtain half-maximal motility) of the initial activity in the ATP release. The poor recovery was most likely due to incomplete elution of X activity from the beads or inactivation of X by high salt and the desalting/ concentration process. As ATP release is 100-fold enriched for dynactin (and presumably also Activator X) relative to cytosol (based on quantitative immunoblotting; not shown), it is likely that we were unable to recover enough cytosolic X to activate dynactin. We were also unable to identify any specifically enriched polypeptides on silver-stained gels of eluates of 45A immunoadsorbent exposed to ATP release (not shown). Nevertheless, these data strongly suggest that Activator X is associated with dynactin in both cytosol and ATP release.
Our initial observation that Activator X activity could be detected in ATP release at very low concentrations (Fig. 7 A) suggested that X was acting on dynactin catalytically, in contrast to dynactin which is believed to act stochiometrically with dynein. If Activator X is an enzyme, what might it be? Data are available suggesting that dynactin subunits and in particular p150_Glued_ are phosphorylated (Schafer et al., 1994; Farshori, P., and E.L.F. Holzbaur. 1994. Mol. Biol. Cell. 5:287a). For this reason we sought to determine whether Activator X was a protein kinase using the general protein kinase inhibitor staurosporine. This particular inhibitor was chosen because, unlike most other kinase inhibitors, it is not an ATP analogue that must be used at mM concentrations and therefore might inhibit the activity of microtubule motor proteins. Rather, staurosporine effectively inhibits protein kinase activity when used in the nM range. When added directly to the assay, 50 nM staurosporine completely blocked the ability of ATP release to restore motility to dynactin-depleted cytosol in the presence of bovine dynactin, whereas it did not effect the motility of control-depleted cytosol (Fig. 7 C). However, in preliminary experiments, we were unable to demonstrate any specific, direct phosphorylation of bovine dynactin by ATP release or eluates of 45A immunoadsorbent exposed to ATP release nor any motility of purified phagosomes in 1 mg/ml casein in the presence of only bovine dynein, dynactin, and macrophage ATP release (not shown). These negative results could indicate that Activator X is acting indirectly via yet a further cytosolic component(s). Nevertheless, the simplest interpretation of these data is that ATP release contains a kinase or a kinase-activating factor essential for vesicle motility mediated by dynein and dynactin and that its substrate(s) is already phosphorylated in normal cytosol.
Plus-end Directed Movement Requires Kinesin
Although phagosomes moved predominantly in the minus-end direction, we observed a significant number of movements (∼30%) toward the plus-end of microtubules. Our immunolocalization data (Fig. 4 C) indicated that conventional kinesin was present on the phagosome surface, suggesting that it was serving as the plus-end motor. To explore this question further, we used the mouse monoclonal antibody against sea urchin kinesin heavy chain, SUK4, which is known to inhibit the kinesin ATPase (Ingold et al., 1988). When SUK4 IgG was added at 100 μg/ml to the assay, bidirectional motility was nearly completely inhibited whereas addition of control IgG had no effect; 10 μg/ml SUK4 had little effect (Fig. 8 A). However, when we depleted 95% of kinesin heavy chain using the SUK4 antibody, we saw no significant decrease in minus-end directed motility but a 51% decrease in plus-end directed motility (n = 21, statistically significant at P = 0.01). These results strongly suggest that plus end–directed phagosome movement is driven by kinesin.
Figure 8.
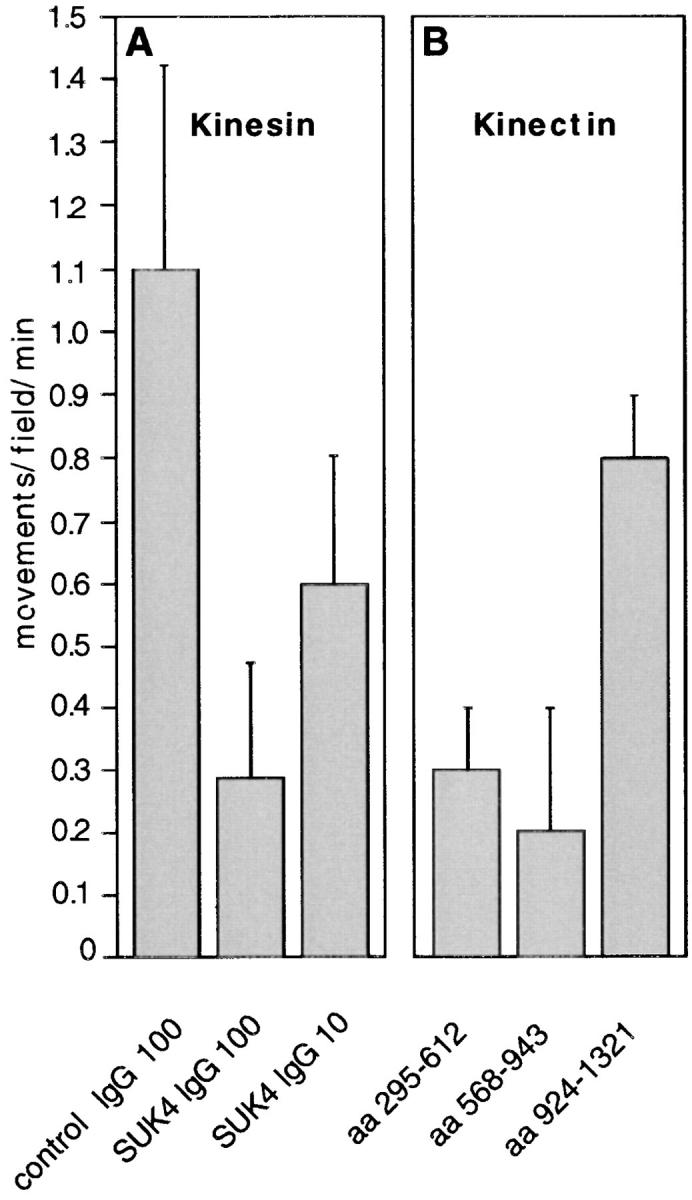
Phagosome motility requires kinesin and kinectin. (A) Effects of kinesin antibodies on phagosome motility. The function-blocking kinesin antibody, SUK4, was tested for its effects on phagosome motility when added directly to the assay at two different concentrations (100 μg/ml, SUK4 IgG 100 and 10 μg/ml, SUK4 IgG 10). Bar control IgG 100 shows the effect on motility of 100 μg/ml control isotype-matched IgG (P5D4). (B) Effects of kinectin peptides on phagosomes motility. 10 μM chicken kinectin fragments corresponding to amino acids 295-612 (aa 295-612), 568-943 (aa 568-943), and 924-1321 (aa 924-1321) were added directly to the phagosome motility assay. For both A and B, each value represents the mean of the average movements/field/min of at least two, but often many more, identical motility chambers; errors are population standard deviations. Each experiment was independently repeated at least twice, but often many more times. For each experiment at least two different preparations of cytosol and phagosomes were tested. Only one preparation of purified IgGs or protein fragments were tested.
Kinectin Function Is Required for Phagosome Motility
As kinesin appeared to be the motor involved in plus end–directed phagosome motility, we wanted to know whether kinectin was (part of) the phagosome membrane receptor for this motor. Kinectin, as a doublet of 120–160-kD bands, was found on phagosomes (purified from the HD11 chicken monocyte cell line) by immunoblot using the mouse monoclonal anti-chicken kinectin antibody 160.9.1 (Fig. 5 C). The smaller isoform is believed to lack the NH2-terminal lumenal and transmembrane domains, as a result of either alternative splicing or proteolysis (Yu, H., and M. Sheetz, personal communication). As phagosome motility increases with maturation time (Fig. 3), we have determined the abundance of kinectin on phagosome membranes as a function of time (Fig. 5 C). Although kinectin was not enriched on phagosomes relative to total cellular membranes, at the oldest phagosome time point it showed a 7.5 enrichment over cytosol suggesting that it is an endogenous, if poorly abundant, protein of the phagosome membrane (Fig. 5 C). Moreover, the protein showed a 10fold increase on phagosomes with time (from a 20-min pulse, no chase to a 1-h pulse followed by 12-h chase).
We next sought to determine whether kinectin was participating functionally in phagosome motility. A kinectin function-blocking antibody was available which inhibits motility of a microsome preparation along microtubules in vitro (Kumar et al., 1995) and adversely affects Golgi function and transport through the secretory pathway in cells (Yu, H., J. Kumar, V.S. De Serrano, W. Chan, J. Heringa, A. Shendarov, W.J. Strittmatter, and M.P. Sheetz, manuscript submitted for publication). However, this antibody is specific for chicken kinectin and could not be used in our assay derived from mouse cells. Instead, we determined whether protein fragments derived from a region within chicken kinectin's cytoplasmic coiled-coil domain would interfere with movement. Two of these peptides (amino acids 295-612 and 568-943 of chicken kinectin) prevent dynein and kinesin binding to membranes and bidirectional motility of crude membrane preparations along microtubules in vitro, whereas a third peptide has little effect in that assay (Yu, H., J. Kumar, V.S. De Serrano, W. Chan, J. Heringa, A. Shendarov, W.J. Strittmatter, and M.P. Sheetz, manuscript submitted for publication). The function-blocking antibody has been shown to bind to the region covered by peptide 568-943, which has therefore been postulated to be the motor-binding domain (Yu, H., J. Kumar, V.S. De Serrano, W. Chan, J. Heringa, A. Shendarov, W.J. Strittmatter, and M.P. Sheetz, manuscript submitted for publication).
The two bacterially produced and purified protein fragments of 30 kD which inhibited the assay of Yu and coworkers were added to our motility assay. Both peptides strongly inhibited phagosome motility in both plus and minus end directions (Fig. 8 B). Another kinectin-derived peptide of similar size, but from a different part of the coiled-coil region (amino acids 924-1321) had no effect. Peptide 568-943 appeared to be a more potent inhibitor: it was active at 5 and 10 μM but not at 2 μM, while peptide 295-612 was only active at 10 μM (data not shown). In summary, our data argue that kinectin plays an important role in the interaction of phagosomes with the microtubule motor machinery.
Discussion
We have reconstituted the motility of phagosomes, a specialized type of endocytic organelle, along microtubules in vitro. The bidirectional movement of phagosomes along microtubules that we observed in vitro displays similar characteristics to the microtubule-dependent phagosome motility we saw in intact cells, in terms of speed and relative directionalities. Using the in vitro assay we showed that phagosome movement along microtubules minimally requires the presence of cytoplasmic dynein (probably CD1), dynactin complex, a dynactin-associated activity which may be a protein kinase, kinesin, and kinectin.
Phagosomes move bidirectionally but predominantly towards the minus-end of microtubules. Our pharmacological characterization and immunodepletion/re-addition experiments indicate that the major phagosome microtubule motor is most likely to be conventional cytoplasmic dynein. However, at this point, we cannot exclude that the monoclonal antibodies against CD1 that we used in this work also recognize the other cytoplasmic dyneins expressed at lower levels in mammalian cells. The pharmacological data further argue that conventional kinesin is the motor for plus-end directed phagosome motility along microtubules. Moreover, immunodepletion of 95% of kinesin with the SUK4 monoclonal antibody reduced plus-end directed motility by 50% while leaving minus-end motility unaffected. We believe that immunodepletion of the great majority of kinesin and cytoplasmic dynein did not totally abolish minus or plus-end directed motility simply because these motors are in vast excess in cytosol. However, we cannot exclude that they were partially replaced functionally by other cytoplasmic dyneins and kinesin-related proteins whose activities may be somewhat redundant in vivo. In particular, should the reagents we used against CD1 turn out to recognize the minor dynein forms as well, it would remain to be investigated whether CD2 and CD3 perhaps share with CD1 this particular localization/function or are uniquely responsible for it.
In contrast to our immunodepletion results, addition of purified SUK4 IgG to the assay inhibited bidirectional motility. However, this large molecule binds the motor domain of kinesin and could prevent dynein function by steric hindrance provided dynein and kinesin bind closely adjacent sites on the phagosome membrane. Inhibition of bidirectional vesicle motility along microtubules was also observed upon addition of a dynactin or kinesin antibody to extruded squid axoplasm (Waterman-Storer et al., 1994; Brady et al., 1990), whereas selective immunodepletion of either of these proteins results in inhibition of only dynein or kinesin-mediated motility.
That cytoplasmic dynein and kinesin act as motors for phagosome motility along microtubules in vitro agrees with our immunolocalization results showing these molecules on phagosomes in vivo. These data are also consistent with the work of Hollenbeck and Swanson (1990) who found that kinesin drives radial extension of tubular lysosomes in macrophages and with the work of Bomsel et al. (1990) and Aniento et al. (1993), showing that dynein and kinesin are required for microtubule-stimulated fusion of endocytic carrier vesicles with late endosomes in vitro. Our results directly show that phagosomes display bidirectional motility along microtubules. Since phagosomes display in vivo-motility properties similar to those reported for endosomes and lysosomes (Heuser, 1989; Parton et al., 1991; Lin and Collins, 1992), it appears that most endocytic organelles are capable of regulated bidirectional movement along microtubules. A physiological role for plus-end directed movement has recently been demonstrated for lysosomes involved in Trypanosoma cruzi entry into cells (Rodriguez et al., 1996).
The data presented here, and previously (Schroer et al., 1985; Vale et al., 1985_a_ ; Burkhardt et al., 1993), argue that motors require proteinaceous receptor(s) within the organelle membrane to move vesicles along microtubules. One of the required membrane proteins is kinectin, as bidirectional phagosome movement was nearly completely inhibited by kinectin peptides that have been shown to inhibit kinesin/dynein-dependent microtubule motility of chick brain microsomes (Yu, H., J. Kumar, V.S. De Serrano, W. Chan, J. Heringa, A. Shendarov, W.J. Strittmatter, and M.P. Sheetz, manuscript submitted for publication). It is not clear why both of these peptides, which share little overlap, have inhibitory effect in the same concentration range in both our assay and that of Yu and coworkers. The latter have speculated that motors bind to kinectin via relatively long range but weak interactions rather than by high affinity interaction with a small number of amino acids. Our data also show that kinectin is on phagosomes in vivo, providing the first evidence that this protein participates in the motility of an organelle other than the endoplasmic reticulum.
The activity or presence of these motor receptor(s) is regulated on the phagosome surface, as “early” phagosomes are three times less likely to move along microtubules than “late” phagosomes. We also found that phagosomes retain the capacity for movement in vivo, even after many hours in the cell (Fig. 1) and similar findings have been reported for late endosomes (Swanson et al., 1987; Heuser, 1989; Hollenbeck and Swanson, 1990; Knapp and Swanson, 1990). The time-dependent increase in phagosome motility may be explained, at least in part, by the progressive accumulation of kinectin on maturing phagosomes, perhaps leading to increased recruitment of soluble motility factors.
Our data further support the notion that kinectin is a membrane receptor for kinesin. However, they provide no further evidence either for or against the dual motor receptor hypothesis (Sheetz and Yu, 1996). Indeed, the use of antibodies against kinectin, as well as kinectin peptides, both of which inhibit motility bidirectionally, suffers from the likelihood of steric hindrance of dynein activity (which we observed using the anti-kinesin antibody SUK4). Either a single, dual motor receptor or two, tightly linked motor receptors would most simply explain both the coordinate regulation of plus and minus end–directed motility observed by Hamm-Alvarez et al. (1993), and the parallel increase in frequency of minus and plus end–directed movement that we measured during phagosome maturation (not shown). Nevertheless, the mode of membrane association of cytoplasmic dynein remains to be established. It has been proposed that dynactin acts as a “receptor” for cytoplasmic dynein (Vaughan and Vallee, 1995). How this might occur is not clear as these complexes are largely soluble. In addition, although the interaction of dynein and dynactin has been demonstrated through in vitro (Vaughan and Vallee, 1995; Karki and Holzbaur, 1995) and genetic studies (reviewed in Schroer, 1996), we and others did not find dynein and dynactin stochiometrically associated in cytosol (Steuer, E., and M. Sheetz, unpublished results; Gill et al., 1991; Waterman-Storer et al., 1996). The possibility that dynein and dynactin form a complex at the surface of organelles has not been investigated. The question remains: how are dynein and dynactin recruited to the membrane?
Once on the organelle, how dynactin activates cytoplasmic dynein for vesicle motility is also not understood. In investigating this further, we found that immunodepletion of dynactin inhibits only minus-end directed motility as predicted from its discovery as an activator of dyneinmediated vesicle motility in vitro (Schroer and Sheetz, 1991). We found however that to activate dynein, dynactin required a putative protein kinase (Activator X), since purified dynactin could not restore motility to dynactindepleted cytosol unless we also added a highly diluted microtubule “ATP-release” fraction whose effect was inhibited by the general kinase inhibitor staurosporine. We also showed that Activator X was coprecipitated with dynactin from ATP release, suggesting why dynactin-depleted cytosol is unable to activate purified dynactin (if one assumes that X is lost or inactived during the purification of bovine dynactin). It is possible that Activator X is the previously characterized Activator 2 (Schroer and Sheetz, 1991), although the latter was originally reported to stimulate both kinesin- and dynein-driven motility while Activator X seems to be required for minus end–directed motility only.
Although the precise identity as well as the direct substrate(s) of Activator X remain to be established, the notion of dynactin regulation by phosphorylation fits into a general pattern of regulation of motors by the same posttranslational modification. Indeed, phosphorylation has been shown to control motor-organelle binding (Lin et al., 1994; Niclas et al., 1996; Sato-Yoshitake et al., 1992; Leopold et al., 1992; Hollenbeck, 1993). It also regulates motor activity directly by modification of motor subunits (Dillman and Pfister, 1994; Allan, 1995; Hirokawa et al., 1991) or indirectly via modification of motor-associated polypeptides (McIlvain et al., 1994; Schroer et al., 1988; Haimo and Thaler, 1994).
More generally, phosphorylation appears to be a mechanism for physiological control of the direction (as in melanophores, see Rozdzial and Haimo, 1986) and frequency (Hamm-Alvarez et al., 1993) of organelle movement by external signals or by the cell cycle machinery (Allan and Vale, 1991). The experiments presented here and those of Burkhardt et al. (1993) show that regulation of movement frequency and polarity can also be autonomous to the membrane organelle. The use of purified organelles in these systems now allows investigation of polarity and frequency regulation at the level of the organelle membrane.
In theory, regulation of the motile activity of each organelle is all that is required for their steady-state localization within cells relative to the microtubule network. Yet, we and others (Rickard and Kreis, 1996) have identified nonmotor microtubule-associated proteins (MAPs) which are involved in creating a link between the organelle and the microtubule. It appears that such factors are not required for organelle motility along microtubules as the removal of the phagosome-microtubule linker from cytosol, along with other MAPs, actually stimulates motility threefold (Blocker, 1995). In fact, these MAP-like linkers may actually be inhibitors of organelle motility. In support of this hypothesis, “young” phagosomes bind well to microtubules but move relatively poorly, while “older” phagosomes show opposite behaviors (Blocker et al., 1996; Fig. 3 B).
Detailed biochemical studies of how motility factors interact with/on the phagosome membrane along with biophysical studies including force measurements of phagosome-microtubule interactions (Askin et al., 1990; reviewed in Vale, 1994) and high precision tracking (Gelles et al., 1988; Wang et al., 1995) of phagosome movements should yield more direct information as to how frequency and polarity regulation (Vale et al., 1989, 1992) is achieved. The challenge for the future is to determine how the MAP-linker proteins coordinate with opposing motor proteins, their regulators, and microtubule dynamics (Rodionov et al., 1994; Desai and Mitchison, 1995) to determine organelle positioning and trafficking within cells.
Note added in proof:
Since this paper was submitted, two articles (Holleran, E.A., M.K. Tokito, S. Karki, E.L.F. Holzbaur. 1996. J. Cell Biol. 135: 1815–1829 and Muresan, V., C.P. Godek, T.S. Reese, and B.J. Schnapp. 1996. J. Cell Biol. 135:383–397) have been published that provide evidence suggesting that spectrin is a link between the Arp1 filament of dynactin and the organelle membrane.
Acknowledgments
This work was supported by a grant from the Human Frontiers Science Programme to J.K. Burkhardt, G. Griffiths, and T.A. Schroer. This article is dedicated to Zacharie Cazes, born November 12th 1996, who was such encouraging company for his mother during her spring and summer “late nights on the scope.”
Abbreviations used in this paper
AMP-PNP
5′-adenylylimidodiphosphate
DIC
3,4-dichlorisocoumarin
CD
cytoplasmic dynein
FSG
fish skin gelatin
GTP
guanosine-5′-triphosphate
IM
internalization medium
MTOC
microtubule organizing center
NRK
normal rat kidney cells
PLO
poly-l-ornithine
Footnotes
We are particularly indebted to Mike Sheetz for inspiring discussions and unpublished reagents against kinectin protein and to Anja Habermann for outstanding technical assistance throughout this work. We thank Steve Gill for kindly providing the unpublished antibody 150B. We thank Heinz Horstmann for initial electron microscopy, Frédérique Briquet-Laugier for help in developing the movement analysis programme, Alan Sawyer for advice on antibodies, Angel Nebreda for a guided tour of “kinase country,” and Jacomine Krinjse-Locker for minimizing the radiation exposure of A. Blocker's unborn child. We are grateful to George Bloom and Eugeni Vaisberg for generous gifts of antibodies and discussions of work in progress. Finally, we thank George Bloom, Mike Sheetz, Kai Simons, Gert Vriend, and two anonymous reviewers for considerably improving the manuscript.
Please address all correspondence to A. Blocker, Unité de Pathogénie Microbienne Moléculaire, INSERM U389, Institut Pasteur, 28 rue du Dr. Roux, 75724 Paris cedex France. Tel.: 33 1 40 61 32 47. Fax: 33 1 45 68 89 53. E-Mail: ablocker@pasteur.fr
References
- Allan VJ. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J Cell Biol. 1995;128:879–891. doi: 10.1083/jcb.128.5.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan VJ. Role of motor proteins in organizing the endoplasmic reticulum and Golgi apparatus. Semin Cell Biol. 1996;7:335–342. [Google Scholar]
- Allan VJ, Vale RD. Cell cycle control of microtuble-based membrane transport and tubule formation in vitro. J Cell Biol. 1991;113:347–359. doi: 10.1083/jcb.113.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkin A, Schütze K, Dziedzic JM, Euteneuer U, Schliwa M. Force generation of organelle transport measured in vivo by an infrared laser trap. Nature (Lond) 1990;348:346–348. doi: 10.1038/348346a0. [DOI] [PubMed] [Google Scholar]
- Beug H, von Kirchbach A, Derlin G, Conscience J-F, Graf T. Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinctive phenotypes of differentiation. Cell. 1979;18:375–390. doi: 10.1016/0092-8674(79)90057-6. [DOI] [PubMed] [Google Scholar]
- Blocker, A. 1995. Analyse des interactions entre phagosomes et microtubules. Ph.D. thesis. University of Paris XI, Orsay, France.
- Blocker A, Severin FF, Habermann A, Hyman AA, Griffiths G, Burkhardt JK. MAP-dependent binding of phagosomes to microtubules. J Biol Chem. 1996;271:3803–3811. doi: 10.1074/jbc.271.7.3803. [DOI] [PubMed] [Google Scholar]
- Bloom, G., and S. Endow. 1994. Motor Proteins 1: kinesins. Vol. 1. P. Sheterline, editor. Academic Press, London.
- Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature (Lond) 1985;317:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, McIlvain JM, Jr, Hamm-Alvarez S, Argon Y, Sheetz MP. Lytic granules from cytotoxic T cells exhibit kinesin-dependent motility on microtubules in vitro. J Cell Sci. 1993;104:151–162. doi: 10.1242/jcs.104.1.151. [DOI] [PubMed] [Google Scholar]
- Cassimeris LU, Wadsworth P, Salmon ED. Dynamics of microtubule depolymerization in monocytes. J Cell Biol. 1986;102:2023–2032. doi: 10.1083/jcb.102.6.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcy Hart, P., M.R. Young, M.M. Jorgan, W.J. Perkins, and M.J. Geisow. Chemical inhibitors of phagosome-lysosome fusion in cultured macrophages also inhibit saltatory lysosomal movements. J Exp Med. 1983;158:477–492. doi: 10.1084/jem.158.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. A new role of motor proteins as couplers to depolymerizing microtubules. J Cell Biol. 1995;128:1–4. doi: 10.1083/jcb.128.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol. 1994a;124:677–688. doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Celis JE, van Meer G, Dieplinger H, Jahraus A, Griffiths G, Huber LA. Molecular characterization of phagosomes. J Biol Chem. 1994b;269:32194–32200. [PubMed] [Google Scholar]
- Dillman III, J.F., and K.K. Pfister. Differential phosphorylation in vivoof cytoplasmic dynein associated with anterogradely moving organelles. J Cell Biol. 1994;127:1671–1681. doi: 10.1083/jcb.127.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri CJ, Paschal BM, Vaughan KT, Vallee R. Molecular characterisation of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J Cell Biol. 1996;132:617–633. doi: 10.1083/jcb.132.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Ferreira A, Kosik KS, Caceres A. Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J Cell Biol. 1994;127:1021–1039. doi: 10.1083/jcb.127.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fütterer A, Kruppa G, Krämer B, Lemke H, Krönke M. Molecular cloning and characterization of human kinectin. Mol Biol Cell. 1995;6:161–170. doi: 10.1091/mbc.6.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelles J, Schnapp BJ, Sheetz MP. Tracking kinesin-driven movements with nanometer-scale precision. Nature (Lond) 1988;331:450–453. doi: 10.1038/331450a0. [DOI] [PubMed] [Google Scholar]
- Gibbons IR, Cosson MP, Evans JA, Gibbons BH, Houck B, Martinson KH, Sale WS, Tang W-JY. Potent inhibition of dynein andenosintriphophatase and of the motility of cilia and sperm flagella by vanadate. Proc Natl Acad Sci USA. 1978;79:2220–2224. doi: 10.1073/pnas.75.5.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SP, Webb MR, Martin B, Johnson KA. Pathway of processive ATP hydrolysis by kinesin. Nature (Lond) 1995;373:671–718. doi: 10.1038/373671a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Schroer TA, Szilak I, Steuer ER, Sheetz MP, Cleveland DW. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J Cell Biol. 1991;115:1636–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein IS, Hoffstein J, Gallin J, Weissmann G. Mechanisms of lysosomal enzyme release from human leukocytes: microtubule assembly and membrane fusion induced by a component of complement. Proc Natl Acad Sci USA. 1973;70:2916–2920. doi: 10.1073/pnas.70.10.2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G. 1993. Fine Structure Immunocytochemistry. Springer Verlag, Heidelberg, Germany.
- Gruenberg J, Griffiths G, Howell KE. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle function in vitro. J Cell Biol. 1989;108:1301–1316. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimo LT, Thaler CD. Regulation of organelle transport: lessons from color change in fish. BioEssays. 1994;16:727–733. [Google Scholar]
- Hamm-Alvarez SF, Kim PY, Sheetz MP. Regulation of vesicle transport in CV-1 cells and extracts. J Cell Sci. 1993;106:955–966. doi: 10.1242/jcs.106.3.955. [DOI] [PubMed] [Google Scholar]
- Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. Organelle transport along microtubules-the role of KIFs. Trends Cell Biol. 1996;6:135–140. doi: 10.1016/0962-8924(96)10003-9. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Pfister KK, Yorifugi H, Wagner MC, Brady ST, Bloom GS. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56:867–878. doi: 10.1016/0092-8674(89)90691-0. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Yoshida Y, Sato-Yoshitake R. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. The distribution, abundance and subcellular localization of kinesin. J Cell Biol. 1989;108:2335–2342. doi: 10.1083/jcb.108.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck P J. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem. 1993;60:2265–2275. doi: 10.1111/j.1471-4159.1993.tb03513.x. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Swanson JA. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature (Lond) 1990;346:864–866. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]
- Holzbaur ELF, Vallee RB. Dyneins: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Holzbaur ELF, Hammarback JA, Paschal BM, Kravit NG, Pfister KK, Vallee RB. Homology of a 150K cytoplasmic dynein-associated polypeptide with the Drosophila gene Glued. . Nature (Lond) 1991;351:579–583. doi: 10.1038/351579a0. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Gibson A, Shipman M, Miller K. Movement of internalized ligand-receptor complexes along a continuous endosomal reticulum. Nature (Lond) 1990;346:335–339. doi: 10.1038/346335a0. [DOI] [PubMed] [Google Scholar]
- Howard, J., and A.A. Hyman. 1993. Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence microscopy. In Motility Assays for Motor Proteins. Vol. 39. J.M. Scholey, editors. Academic Press, Inc., San Diego. 105–113. [DOI] [PubMed]
- Hyman AA, Mitchison T. Two different microtubule-based motor activities with opposite polarities in kinetochores. Nature (Lond) 1991a;351:206–211. doi: 10.1038/351206a0. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison T. Preparation of modified tubulins. Methods Enzymol. 1991b;196:478–485. doi: 10.1016/0076-6879(91)96041-o. [DOI] [PubMed] [Google Scholar]
- Ingold AL, Cohn SA, Scholey JM. Inhibition of kinesin-driven motility by monoclonal antibodies to kinesin heavy chains. J Cell Biol. 1988;107:2657–2667. doi: 10.1083/jcb.107.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahraus A, Storrie B, Griffiths G, Desjardins D. Evidence for retrograde traffic between terminal lysosomes and the prelysosomal/late endosome compartment. J Cell Sci. 1994;107:145–157. doi: 10.1242/jcs.107.1.145. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur ELF. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270:28806–28811. doi: 10.1074/jbc.270.48.28806. [DOI] [PubMed] [Google Scholar]
- Knapp PE, Swanson JA. Plasticity of the tubular lysosomal compartment in macrophages. J Cell Sci. 1990;95:433–439. doi: 10.1242/jcs.95.3.433. [DOI] [PubMed] [Google Scholar]
- Kreis TE. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein, block its transport to the cell surface. EMBO (Eur Mol Biol Organ) J. 1986;5:931–941. doi: 10.1002/j.1460-2075.1986.tb04306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. Kinectin, an essential anchor for kinesin-driven motility. Science (Wash DC) 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Kypta RM, Hemming A, Courtneidge SA. Identification and characterization of p59 fyn(a src-like protein kinase) in normal and polyoma virus transformed cells. EMBO (Eur Mol Biol Organ) J. 1988;7:3837–3844. doi: 10.1002/j.1460-2075.1988.tb03269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the heads of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lasek RJ, Brady ST. Attachment of transported vesicles to microtubules in axoplasm is facilitated by AMP-PNP. Nature (Lond) 1985;316:645–647. doi: 10.1038/316645a0. [DOI] [PubMed] [Google Scholar]
- Leopold PL, McDowall AW, Pfister KK, Bloom GS, Brady ST. Association of kinesin with characterized membrane-bound organelles. Cell Motil Cytoskeleton. 1992;23:19–33. doi: 10.1002/cm.970230104. [DOI] [PubMed] [Google Scholar]
- Lin SXH, Ferro KL, Collins CA. Cytoplasmic dynein undergoes intracellular redistribution concomitant with phosphorylation of the heavy chain in response to serum starvation and okadaic acid. J Cell Biol. 1994;127:1009–1019. doi: 10.1083/jcb.127.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SXH, Collins CA. Immunolocalization of cytoplasmic dynein to lysosomes in cultured cells. J Cell Sci. 1992;101:125–137. doi: 10.1242/jcs.101.1.125. [DOI] [PubMed] [Google Scholar]
- Lye RJ, Porter ME, Scholey JM, McIntosh JR. Identification of a microtubule-based motor in the nematode C. elegans. . Cell. 1987;51:309–318. doi: 10.1016/0092-8674(87)90157-7. [DOI] [PubMed] [Google Scholar]
- McIlvain JM, Jr, Burkhardt JK, Hamm-Alvarez S, Argon Y, Sheetz MP. Regulation of kinesin activity by phosphorylation of kinesinassociated proteins. J Biol Chem. 1994;269:19176–19182. [PubMed] [Google Scholar]
- McIntosh JR, Porter ME. Enzymes for microtubule-dependent motility. J Biol Chem. 1989;264:6001–6004. [PubMed] [Google Scholar]
- Middleton K, Carbon J. KAR3-encoded kinesin is a minus-enddirected motor that functions with centromere binding proteins (CBF3) on an in vitroyeast kinetochore. Proc Natl Acad Sci USA. 1994;91:7212–7216. doi: 10.1073/pnas.91.15.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Steinman RM, Cohn ZA. The membrane proteins of the vacuolar system I. Analysis by a novel method of intralysosomal iodination. J Cell Biol. 1980;86:304–413. doi: 10.1083/jcb.86.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclas J, Allan VJ, Vale RD. Cell cycle regulation of dynein association with membranes modulates microtubule-based organelle transport. J Cell Biol. 1996;133:585–593. doi: 10.1083/jcb.133.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal B, Vallee R. Retrograde transport by the microtubule-associated protein MAP1C. Nature (Lond) 1987;330:181–183. doi: 10.1038/330181a0. [DOI] [PubMed] [Google Scholar]
- Paschal BM, Holzbaur ELF, Pfister KK, Clark S, Meyer DI, Vallee RB. Characterization of a 50-kDa polypeptide in cytoplasmic dynein preparations reveals a complex with p150Gluedand a novel actin. J Biol Chem. 1993;268:15318–15323. [PubMed] [Google Scholar]
- Parton RG, Dotti CG, Bacallao R, Kurtz I, Simons K, Prydz K. pH-induced microtubule-dependent redistribution of late endosomes in neuronal and epithelial cells. J Cell Biol. 1991;113:261–274. doi: 10.1083/jcb.113.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesanti EL, Axiline SG. Phagolysosome formation in normal and colchicine treated macrophages. J Exp Med. 1975;142:903–913. doi: 10.1084/jem.142.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfarr CM, Coue M, Grissom PM, Hays TS, Porter ME, McIntosh JR. Cytoplasmic dynein is localized to kinetochores during mitosis. Nature (Lond) 1990;345:263–265. doi: 10.1038/345263a0. [DOI] [PubMed] [Google Scholar]
- Pierre P, Scheel J, Rickard JE, Kreis TE. CLIP-170 links endocytic vesicles to microtubules. Cell. 1992;70:887–900. doi: 10.1016/0092-8674(92)90240-d. [DOI] [PubMed] [Google Scholar]
- Pitt A, Mayorga LS, Schwartz AL, Stahl PD. Transport of phagosomal components to an endosomal compartment. J Biol Chem. 1992a;267:126–132. [PubMed] [Google Scholar]
- Pitt A, Mayorga LS, Stahl PD, Schwartz AL. Alterations in the protein composition of maturing phagosomes. J Clin Invest. 1992b;90:1978–1983. doi: 10.1172/JCI116077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JE, Kreis TE. CLIPs for organelle-microtubule interactions. Trends Cell Biol. 1996;6:178–183. doi: 10.1016/0962-8924(96)10017-9. [DOI] [PubMed] [Google Scholar]
- Rodionov VI, Lim S-S, Gelfand VI, Borisy GG. Microtubule dynamics in fish melanophores. J Cell Biol. 1994;126:1455–1464. doi: 10.1083/jcb.126.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Samoff E, Rioult MG, Chung A, Andrews N. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesinmediated transport. J Cell Biol. 1996;134:349–362. doi: 10.1083/jcb.134.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozdzial MM, Haimo LT. Bidirectional pigment granule movements of melanophores are regulated by protein phosphorylation and dephosphorylation. Cell. 1986;47:1061–1070. doi: 10.1016/0092-8674(86)90821-4. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifugi H, Inagaki M, Hirokawa N. The phophorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles F-actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel J, Kreis TE. Motor protein-independent binding of endocytic carrier vesicles to microtubules in vitro. J Biol Chem. 1991;266:18141–18148. [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA. 1989;86:1548–1552. doi: 10.1073/pnas.86.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA. Structure, function and regulation of cytoplasmic dynein. Curr Opin Cell Biol. 1994;6:69–73. doi: 10.1016/0955-0674(94)90118-x. [DOI] [PubMed] [Google Scholar]
- Schroer TA. Structure and function of dynactin. Sem Cell Dev Biol. 1996;7:321–328. [Google Scholar]
- Schroer TA, Sheetz MP. Two activators of microtubule-based vesicle transport. J Cell Biol. 1991;115:1309–1318. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, Schnapp BJ, Reese TS, Sheetz MP. The role of kinesin and other soluble factors in organelle movement along microtubules. J Cell Biol. 1988;107:1785–1792. doi: 10.1083/jcb.107.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, Steuer ER, Sheetz MP. Cytoplasmic dynein is a minus-end directed motor for membranous organelles. Cell. 1989;56:937–946. doi: 10.1016/0092-8674(89)90627-2. [DOI] [PubMed] [Google Scholar]
- Severin FF, Shanina NA, Kuznetsov SA, Gelfand VI. MAP2mediated binding of chromaffin granules to microtubules. FEBS Lett. 1991;282:65–68. doi: 10.1016/0014-5793(91)80445-9. [DOI] [PubMed] [Google Scholar]
- Sheetz MP, Yu H. Regulation of kinesin and cytoplasmic dyneindriven organelle motility. Sem Cell Dev Biol. 1996;7:329–334. [Google Scholar]
- Shimizu T, Furusawa K, Ohashi S, Toyoshima YY, Okuno M, Malik F, Vale RD. Nucleotide specificity of the enzymatic and motile activities of dynein, kinesin and heavy meromyosin. J Cell Biol. 1991;112:1189–1197. doi: 10.1083/jcb.112.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein, S.C., S. Greenberg, F. Di Virgilio, and T. Steinberg. 1989. Phagocytosis. In Fundamental Immunology. W.E. Paul, editor. Raven Press Ltd., New York. pp. 703–720.
- Steuer E, Wordeman L, Schroer TA, Sheetz MP. Localization of cytoplasmic dynein to mitotic spindles and the kinetochores. Nature (Lond) 1990;345:266–268. doi: 10.1038/345266a0. [DOI] [PubMed] [Google Scholar]
- Stossel TP, Pollard TD, Mason RJ, Vaughan M. Vesicles and properties of phagocytic vesicles from polymorphonuclear leukocytes. J Clin Invest. 1971;50:1745–1757. doi: 10.1172/JCI106664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, Bushnell A, Silverstein SC. Tubular lysosome morphology and distribution within macrophages depend on the integrity of cytoplasmic microtubules. Proc Natl Acad Sci USA. 1987;84:1921–1925. doi: 10.1073/pnas.84.7.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Zhang Z, Hirokawa N. Identification and molecular evolution of new dynein-like protein sequences in rat brain. J Cell Sci. 1995;108:1883–1893. doi: 10.1242/jcs.108.5.1883. [DOI] [PubMed] [Google Scholar]
- Toyohara A, Inaba K. Transport of phagosomes in mouse peritoneal macrophages. J Cell Sci. 1989;94:143–153. doi: 10.1242/jcs.94.1.143. [DOI] [PubMed] [Google Scholar]
- Toyoshima I, Yu H, Steuer ER, Sheetz MP. Kinectin, a major kinesin-binding protein on the ER. J Cell Biol. 1992;118:1121–1131. doi: 10.1083/jcb.118.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Grissom PM, McIntosh JR. Mammalian cells express three distinct dynein heavy chains that are localized to different cytoplasmic organelles. J Cell Biol. 1996;133:831–842. doi: 10.1083/jcb.133.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Koonce MP, McIntosh JR. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J Cell Biol. 1993;123:849–858. doi: 10.1083/jcb.123.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. Intracellular transport using microtubule-based motors. Annu Rev Cell Biol. 1987;3:347–378. doi: 10.1146/annurev.cb.03.110187.002023. [DOI] [PubMed] [Google Scholar]
- Vale RD. Getting a grip on myosin. Cell. 1994;78:733–737. doi: 10.1016/s0092-8674(94)90402-2. [DOI] [PubMed] [Google Scholar]
- Vale RD, Malik F, Brown D. Directional instability of microtubule transport in the presence of kinesin and dynein, two opposite polarity motor proteins. J Cell Biol. 1992;119:1589–1596. doi: 10.1083/jcb.119.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Organelle, bead and microtubule translocations promoted by soluble factors from the squid giant axon. Cell. 1985a;40:449–454. doi: 10.1016/0092-8674(85)90204-1. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel forcegenerating protein, kinesin, involved in microtubule-based motility. Cell. 1985b;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Soll DR, Gibbons IR. One-dimensional diffusion of microtubules bound to flagellar dynein. Cell. 1989;59:915–925. doi: 10.1016/0092-8674(89)90614-4. [DOI] [PubMed] [Google Scholar]
- Vaughan, K.T., and R.B. Vallee. 1995. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150_Glued. J. Cell Biol._ 131:1507–1516. [DOI] [PMC free article] [PubMed]
- Wang Z, Khan S, Sheetz MP. Single cytoplasmic dynein molecules movements: characterization and comparison with kinesin. Biophys J. 1995;69:2011–2023. doi: 10.1016/S0006-3495(95)80071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Holzbaur ELF. The product of the Drosophila gene, Glued, is the functional homologue of the p150Gluedcomponent of the vertebrate dynactin complex. J Biol Chem. 1996;271:1153–1159. doi: 10.1074/jbc.271.2.1153. [DOI] [PubMed] [Google Scholar]
- Wetzel MG, Korn ED. Phagocytosis of latex beads by Acanthamoeba castellanii(NEFF). III Isolation of the phagocytic vesicles and their membranes. J Cell Biol. 1969;43:90–104. doi: 10.1083/jcb.43.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Nicchitta CV, Kumar J, Becker M, Toyoshima I, Sheetz MP. Characterization of kinectin, a kinesin-binding protein: primary sequence and N-terminal topogenic signal analysis. Mol Cell Biol. 1995;6:171–183. doi: 10.1091/mbc.6.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]