Comparative Genomic Analyses of Seventeen Streptococcus pneumoniae Strains: Insights into the Pneumococcal Supragenome (original) (raw)
Abstract
The distributed-genome hypothesis (DGH) states that pathogenic bacteria possess a supragenome that is much larger than the genome of any single bacterium and that these pathogens utilize genetic recombination and a large, noncore set of genes as a means of diversity generation. We sequenced the genomes of eight nasopharyngeal strains of S_treptococcus pneumoniae_ isolated from pediatric patients with upper respiratory symptoms and performed quantitative genomic analyses among these and nine publicly available pneumococcal strains. Coding sequences from all strains were grouped into 3,170 orthologous gene clusters, of which 1,454 (46%) were conserved among all 17 strains. The majority of the gene clusters, 1,716 (54%), were not found in all strains. Genic differences per strain pair ranged from 35 to 629 orthologous clusters, with each strain's genome containing between 21 and 32% noncore genes. The distribution of the orthologous clusters per genome for the 17 strains was entered into the finite-supragenome model, which predicted that (i) the S. pneumoniae supragenome contains more than 5,000 orthologous clusters and (ii) 99% of the orthologous clusters (∼3,000) that are represented in the S. pneumoniae population at frequencies of ≥0.1 can be identified if 33 representative genomes are sequenced. These extensive genic diversity data support the DGH and provide a basis for understanding the great differences in clinical phenotype associated with various pneumococcal strains. When these findings are taken together with previous studies that demonstrated the presence of a supragenome for Streptococcus agalactiae and Haemophilus influenzae, it appears that the possession of a distributed genome is a common host interaction strategy.
Streptococcus pneumoniae is a gram-positive bacterium commonly found in the nasopharynges of healthy persons, predominantly children. In addition to its commensal form, S. pneumoniae is also a major cause of morbidity and mortality worldwide. S. pneumoniae infection can cause meningitis and bacteremia, as well as many mucosal diseases such as pneumonia, sinusitis, and otitis media (OM). Worldwide, S. pneumoniae infections are estimated to result in 1.1 million deaths a year, predominantly from pneumonia, and even in the United States pneumococcus disease is one of the top 10 causes of death (21). The economic burden associated with S. pneumoniae infections is tremendous, because it is the causative agent for 30 to 50% of OM infections worldwide, and in the United States alone the cost of OM, which is the most prevalent infectious disease among children, is estimated at $5 billion annually (5, 12).
S. pneumoniae played a critical role in the demonstration that DNA is the hereditary genetic material. In 1944 Avery and colleagues showed that DNA is the transforming factor identified by Griffith as being capable of making avirulent S. pneumoniae lethal (1). Since then, S. pneumoniae has served as a model organism for the study of bacterial transformation. It contains an inducible system for the uptake of DNA from the environment that allows for extensive recombination (3, 29, 34). Previous work from our laboratory using genomic libraries from eight clinical strains identified novel genes not present in the TIGR4 reference strain and showed a nonuniform distribution of many S. pneumoniae genes, suggesting a significant degree of genomic plasticity among the isolates (37).
A large amount of intraspecies genic variation (which pertains to the absence or presence of genes and should not be confused with allelic variation) has been observed for several bacteria. Tettelin and colleagues analyzed the genomes of six Streptococcus agalactiae strains of multiple serotypes and showed that ∼20% of the genes are not shared among all strains (43). A similar trend was found for 13 Haemophilus influenzae genomes, where only ∼50% of the genes are conserved among all strains (18). Both of these studies support the distributed-genome hypothesis, which states that for some bacteria, the full complement of genes available to a given species exists in a “supragenome” pool, one that each member of a population of naturally transformable bacterial strains contributes to and draws genes from, resulting in a high degree of genic diversity (7).
In this study we sequenced the genomes of eight clinical S. pneumoniae isolates and combined these data with sequences from nine other publicly available S. pneumoniae strains. We present a global comparative analysis of the genes and genomes, which demonstrates great genic diversity among the strains. In addition, we use a mathematical model to predict the number of genomes that must be sequenced to provide coverage of the S. pneumoniae supragenome (18).
MATERIALS AND METHODS
DNA sequencing.
We obtained eight clinical S. pneumoniae isolates, with different serotypes and multilocus sequence typing (MLST) types, from pediatric patients participating in a Fluzone vaccine trial at the Children's Hospital of Pittsburgh. The genomes of these strains were sequenced at the Center for Genomic Sciences (CGS) using a 454 Life Sciences GS-20 sequencer (26). Strains were sequenced to a depth of 16-fold or greater and were assembled de novo by the 454 Newbler de novo assembler to 281 contigs per genome, on average. Lander-Waterman statistics predict that more than 99.9% of each genome was sequenced. Regions of repetitive sequence caused most of the assembly gaps. Informal comparison between high-quality Sanger reads and 454 data suggest an error rate of less than 1 in 1,000 bases in each assembly. Most base call errors are single-base insertions or deletions in homo-nucleotide repeats, which can result in artifactual frameshifts.
Sequence data for the other nine strains examined came from various sources. Four isolates were sequenced by the S. pneumoniae Sequencing Group at the Sanger Institute: 23F, INV104B, INV200, and OXC141. The sequences of the latter three strains are incomplete and can be obtained from ftp://ftp.sanger.ac.uk/pub/pathogens/spn/. The published genome sequence of strain R6 (GenBank accession number AE007317) was produced by Eli Lilly & Co (19). The Institute for Genomic Research (TIGR) sequenced strains TIGR4 (GenBank accession number AE005672), TIGR670-6B (project information available at http://www.ncbi.nlm.nih.gov/sites/entrez?db=genomeprj&cmd=Retrieve&dopt=Overview) (23, 44), and strain D39 (s_pneumoniae_d39_1; GenBank accession number CP000410) (23). Unfinished sequence data for strain PAT6420135 are available from reference 22, and the strain from which the sequence was obtained is now an ATCC patent deposit bacterial strain (ATCC 55840). The Entrez Nucleotide database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide) query “patent[TITL] AND 6420135[TITL]” retrieves the 391 DNA sequences that make up this genome assembly, which was produced by Human Genome Sciences, Inc.
Genome assembly of CGS S. pneumoniae sequences.
The 454-assembled genomic contigs were ordered and oriented into scaffolds by alignment, using Nucmer software (6), against each of the nine non-CGS S. pneumoniae genomes, which indicated the closest reference strain. Using a maximum-parsimony approach, each genome was reduced to about 60 contigs by a combination of (i) Sanger sequencing of PCR amplicons targeted to fill gaps between neighboring contigs, as inferred from the scaffolding, and (ii) paired-end Sanger sequencing of clones from a library and identification of clones that spanned gaps in the 454 sequence. Gap closure experiments were designed by a custom Perl script, and PCR primers were designed by Primer3 (36). Clones and PCR amplicons were assembled along with 454 contigs by a modified Phred-Phrap-Consed pipeline where 454 contigs were converted to PHD format files and were input into Phrap as long reads (9, 10, 13, 14). Data were manipulated and visualized using CONSED.
CDS prediction.
Prediction of putative coding sequences (CDSs) and gene annotation were done by NCBI using the Microbial Genome Annotation Tools and Genome Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). Briefly, gene predictions were performed using GeneMark, GeneMark.hmm, and Glimmer 2. To detect any genes missed by this method, a six-frame translation of slices of the nucleotide sequence was done. The predictions and the slices were then searched against the NCBI Entrez Protein Cluster database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=proteinclusters) as well as proteins from all complete microbial genomes. Annotation was supplied from both sets and supplemented with information from the Conserved Domain Database (42) and from Clusters of Orthologous Groups (25). rRNAs were predicted using BLAST to search against an RNA sequence database (4), and tRNAs were predicted using tRNAscan-SE (24).
Clustering algorithm.
A complete description of the algorithms used to create the orthologous clusters is given by Hogg et al. (18). Briefly, to allocate the CDSs into core, unique, or distributed clusters, tfasty34 (Fasta package, version 3.4) was used for six-frame translation homology searches of all predicted proteins against all possible translations (33). Proprietary software designed at the CGS was used to parse this output, grouping the genes into clusters. A cluster was defined as a group of genes which share at least 70% identity, over 70% of its length, with one or more of the other genes in the group, and where at least one sequence in the cluster is equal to or longer than 120 residues. These parameters were selected because they minimize the change in the number of clusters per change in parameters and thus may represent a good estimate to distinguish orthologues from paralogues (18). Multiple genes from the same strain can be included in one cluster; therefore, in most strains the number of clusters is lower than the number of CDSs. To account for the cases where CDSs were missed in the gene-calling step, fasta34 (Fasta package, version 3.4) was used to align all predicted genes against the contigs. If an alignment with the required score was detected in the contig, the gene was considered present even if the gene was not identified.
Phylogenetic-tree building.
For the genic-difference-based dendrogram, a gene possession-based phylogenetic tree of the 17 S. pneumoniae strains was constructed by defining the distance between a pair of genomes, i and k, to be 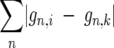 where gn,i is 1 if gene n is present in strain i and zero otherwise. The strains were clustered using tools available at http://www.let.rug.nl/∼kleiweg/clustering/#cluster. “difftbl” was used for conversion of vector data into a difference table using city block as a method, and “cluster” was used for data clustering of the difference table using the group average method. For the relationship among MLST types, we used the batch allelic profile from the pneumococcal MLST database (http://spneumoniae.mlst.net/).
where gn,i is 1 if gene n is present in strain i and zero otherwise. The strains were clustered using tools available at http://www.let.rug.nl/∼kleiweg/clustering/#cluster. “difftbl” was used for conversion of vector data into a difference table using city block as a method, and “cluster” was used for data clustering of the difference table using the group average method. For the relationship among MLST types, we used the batch allelic profile from the pneumococcal MLST database (http://spneumoniae.mlst.net/).
Nucleotide sequence accession numbers.
The GenBank accession numbers for the following eight clinical isolates, sequenced in this study, are given in parentheses after the isolate designations: CGSSp3BS71 (AAZZ00000000), CGSSp6BS73 (ABAA00000000), CGSSp9BS68 (ABAB00000000), CGSSp11BS70 (ABAC00000000), CGSSp14BS69 (ABAD00000000), CGSSp18BS74 (ABAE00000000), CGSSp19BS75 (ABAF00000000), and CGSSp23BS72 (ABAG00000000).
RESULTS
Background on all publicly available S. pneumoniae strains and genome sequences of eight novel clinical isolates.
In this analysis we compared 17 S. pneumoniae strains (Table 1). The genomes for eight clinical strains, each with a unique serotype and MLST type (CGSSp3BS71 [AAZZ00000000], CGSSp6BS73 [ABAA00000000], CGSSp9BS68 [ABAB00000000], CGSSp11BS70 [ABAC00000000], CGSSp14BS69 [ABAD00000000], CGSSp18BS74 [ABAE00000000], CGSSp19BS75 [ABAF00000000], and CGSSp23BS72 [ABAG00000000]), were sequenced at the CGS. The serotype for each of these strains is indicated by the number following the CGSSp prefix with the exception of CGSSp18BS74, which has recently been retyped. Each of these strains is a low-passage-number isolate obtained from the nasopharynx of a child who developed respiratory symptoms at any time over the course of a year and who was a participant in a Fluzone vaccine trial at the Children's Hospital of Pittsburgh (17, 37). Table S1 in the supplemental material shows the extent of genomic coverage and the number of contigs after assembly with Newbler and after gap closure using PCR.
TABLE 1.
Bacterial strains and sources used for the genomic comparison of S. pneumoniae strains
| Strain name | Sequence source | Serotype | Source | MLST type |
|---|---|---|---|---|
| CGSSp11BS70 | CGS | 11 | Pittsburgh, PA | 62 |
| CGSSp14BS69 | CGS | 14 | Pittsburgh, PA | 124 |
| CGSSp18BS74 | CGS | 6 | Pittsburgh, PA | New |
| CGSSp19BS75 | CGS | 19 | Pittsburgh, PA | 485 |
| CGSSp23BS72 | CGS | 23 | Pittsburgh, PA | 37 |
| CGSSp3BS71 | CGS | 3 | Pittsburgh, PA | 180 |
| CGSSp6BS73 | CGS | 6 | Pittsburgh, PA | 460 |
| CGSSp9BS68 | CGS | 9 | Pittsburgh, PA | 1269 |
| D39 | TIGR | 2 | United States | |
| R6 | Eli Lilly and Company | No capsule | Derivative of D39 | |
| 23F | Sanger | 23 | Spanish pandemic | |
| INV104B | Sanger | 1 | Oxford, United Kingdom | |
| INV200 | Sanger | 14 | Oxford, United Kingdom | |
| OXC141 | Sanger | 3 | Oxford, United Kingdom | |
| TIGR4 | TIGR | 4 | Norway | |
| TIGR670-6B | TIGR | 6B | Spain | |
| PAT6420135 (ATCC 55840) | Human Genome Sciences, Inc. | Unknown | Unknown |
The Sanger Institute sequenced four strains of serotypes 1, 14, 3, and 23F. The first three strains are from patients in the United Kingdom, while the last strain is from a multiple-antibiotic-resistant strain from a pandemic in Spain. Strain TIGR670-6B was also isolated in Spain in 1988 and is serotype 6B. TIGR4 was isolated from Norway. The clinical strain D39 was isolated about 90 years ago and gave rise to R36 (an unencapsulated, avirulent mutant that was much easier to transform), which in turn gave rise to R36A, the strain used by Avery and colleagues for their landmark experiments demonstrating that genetic information is contained in DNA (1). Strain R6 is a derivative of R36A isolated approximately 40 years ago; thus, R6 is derived from D39 (19, 23). To our knowledge, there is no publicly available information on the origins of the patented strain PAT6420135.
Identification of CDSs and core, distributed, and unique orthologous clusters.
Prediction of CDSs and annotations were done using the Microbial Genome Annotation Tools and Genome Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). The median number of CDSs per strains is 2,411, and the range is from 2,259 for strain INV200 to 2,763 for strain CGSSp14BS69. The total number of CDSs in all 17 strains is 41,445 (Tables 2 and 3). Note that all 17 genomes were submitted to the NCBI Genome Annotation Pipeline (including those with previously published CDSs) to eliminate errors that may have arisen from differences in the prediction of CDSs. A multi-fasta file with all the CDSs used in this study is available in Fig. S1 in the supplemental material and can also be downloaded from SupraGen at http://centerforgenomicsciences.org/doc_frame/index-old.html.
TABLE 2.
Summary of CDSs and orthologous gene clusters for 17 S. pneumoniae strains
| Gene category | No. of orthologous clusters (% of total) | No. of CDSs (% of genes) |
|---|---|---|
| Core | 1,454 (46) | 30,070 (73) |
| Distributed | 1,140 (36) | 9,679 (23) |
| Unique | 576 (18) | 576 (1) |
| Excluded by size | 1,120 (3) | |
| Total | 3,170 (100) | 41,445 (100) |
TABLE 3.
Numbers of CDSs and orthologous clusters for individual S. pneumoniae strains
| Strain name | Genome size (kb) | No. of CDSs | No. of orthologous clusters | No. of unique clusters | % Noncore clustersa |
|---|---|---|---|---|---|
| INV200 | 2,200 | 2,259 | 1,850 | 28 | 21.4 |
| PAT6420135 | Unknown | 2,500 | 1,913 | 10 | 24 |
| R6 | 2,038 | 2,274 | 1,925 | 3 | 24.5 |
| D39 | 2,000 | 2,304 | 1,940 | 3 | 25 |
| CGSSp18BS74 | 2,033 | 2,354 | 1,955 | 13 | 25.6 |
| CGSSp3BS71 | 2,027 | 2,331 | 1,960 | 7 | 25.8 |
| CGSSp11BS70 | 2,044 | 2,336 | 1,986 | 25 | 26.8 |
| TIGR4 | 2,160 | 2,410 | 1,993 | 1 | 27 |
| INV104B | 2,200 | 2,508 | 2,012 | 74 | 27.7 |
| OXC141 | 2,200 | 2,663 | 2,014 | 67 | 27.8 |
| CGSSp9BS68 | 2,090 | 2,397 | 2,021 | 37 | 28 |
| CGSSp23BS72 | 2,053 | 2,411 | 2,022 | 41 | 28.1 |
| CGSSp19BS75 | 2,069 | 2,432 | 2,031 | 40 | 28.4 |
| CGSSp6BS73 | 2,119 | 2,434 | 2,056 | 55 | 29.3 |
| CGSSp14BS69 | 2,084 | 2,763 | 2,068 | 61 | 29.7 |
| 23F | 2,200 | 2,428 | 2,069 | 43 | 29.7 |
| TIGR670-6B | 2,100 | 2,641 | 2,157 | 68 | 32.6 |
Together, these 17 genomes contain orthologues shared among all strains (core genes), orthologues shared only between subsets of two or more strains (distributed genes), and genes unique to one strain. Genes from all strains were grouped into orthologous clusters and divided into the three categories described above (Table S2 in the supplemental material lists the CDSs organized by orthologous clusters; this table can also be downloaded from SupraGen at http://centerforgenomicsciences.org/doc_frame/index-old.html). SupraGen software was created to identify the CDSs in these orthologous clusters for any S. pneumoniae sequence.
The total number of S. pneumoniae orthologous clusters (which includes unique genes) for all 17 strains is 3,170. Forty-six percent of the total clusters (1,454 core clusters) are found in all strains and include 73% of the total number of CDSs. Eighteen percent of the total clusters (576 clusters) are unique to a single strain, and 36% of the total clusters (1,140 distributed clusters) are present in some combination of two or more strains, but not in all strains (Table 2). Figure 1 shows a histogram plotting the number of orthologous clusters against the number of genomes in which a cluster is found, where 1 and 17 genomes correspond to the unique and core sets, respectively. Table 3 gives the numbers of CDSs, orthologous clusters, and unique genes per strain. The median number of orthologous clusters per strain is 2,012, with a range from 1,850 for INV200 to 2,157 for TIGR670-6B. Note that for each strain, 21 to 32% of the orthologous gene clusters are distributed or unique genes, i.e., not common to all strains. Considering the group of 17 strains collectively, 54% of the gene clusters are noncore.
FIG. 1.

Histogram of the number of observed and predicted (by the finite supragenome model) orthologous gene clusters that are present in a given number of genomes. There were 1,454 orthologous clusters observed in all strains (core); 1,140 distributed among more than one strain, but not all; and 576 in only one strain.
In analyzing the correlation between the number of CDSs and orthologous clusters, a few factors must be noted. First, a gene may be included in a given orthologous cluster even if the CDS was not identified during annotation if a region with the required sequence similarity was found; this aspect of the analysis was designed to identify genes that were missed by the gene prediction programs. Second, when multiple highly similar copies of the same gene are present in one strain, they will all belong to the same orthologous cluster. Third, all orthologous clusters have a minimum size of 120 bp; thus, smaller genes may be included in a cluster only if they align to a longer sequence. Fourth, the same CDS may be intact in one strain and fragmented in another (this is most common in partially assembled genomes); in this case, the clustering algorithms will join the fragments by aligning them to an orthologue, thus converging multiple CDSs into one orthologous cluster. Since the comparisons are based on orthologous clusters, not on genes, they normalize for the effect of gene splitting often encountered in unfinished genomes. For these four reasons, there is not a good linear correlation between the numbers of CDSs and clusters (_R_2 = 0.439).
Phylogenetic relationships among strains.
To evaluate the relationships among strains, we constructed a dendrogram based on noncore genic differences (Fig. 2). As expected, strain D39 and its derivative strain, R6, are the most closely related pair. Interestingly, highly genetically diverse S. pneumoniae strains can be isolated from patients with the same symptoms at the same geographical location, as illustrated by the broad distribution of the eight CGS clinical isolates. Furthermore, strains OXC141 and CGSSp3BS72, which were isolated on different continents, have similar orthologous-cluster distributions. Surprisingly, the strains isolated in Norway (TIGR4) and Spain (23F) are not outliers with respect to the U.S. isolates, and the two Spanish strains (23F and TIGR670-6B) do not group together. The analysis includes two strains each of serotypes 14, 23, and 3 and three strains of serotype 6. There is great similarity between the genic contents of the two serotype 3 strains, but the same is not true for the strains with serotypes 14, 6, and 23.
FIG. 2.

Dendrogram showing the relationship among 17 S. pneumoniae strains based on orthologous cluster differences.
An exhaustive pairwise comparison was performed among all strain pairs (Fig. 3). To provide a measure of the similarities and differences between the numerous strain pairs, we created similarity, difference, and comparison scores. The similarity score corresponds to the total number of orthologous gene clusters shared; the difference score corresponds to the total number of orthologous gene clusters not shared; and the comparison score was calculated by subtracting the difference score from the similarity score to provide a genic measure of comparison between strains. We also outlined the orthologous clusters that were shared only within a given pairing; these are designated pair-unique. The average genic difference between any two strains was 407 orthologous clusters (standard deviation, 91), while the average genic similarity was 1,794 orthologous clusters (standard deviation, 50). The three strain pairs R6-D39, CGSSp3BS71-OXC141, and PAT6420135-TIGR4 have similarity scores 3 standard deviations above the mean (these are the same pairs that group in the dendrogram). Two of these pairs also have high numbers of pair-unique orthologous clusters, as might be expected. However, more-distant strains can also share unique pairs. For example, strains 23F and CGSSp18BS7 have 38 pair-unique genes, 34 of which are genes that are physically grouped on both genomes (in a colinear fashion); 21 of these are annotated as phage related, strongly suggesting that the unique similarity between these strains results from the insertion of a phage.
FIG. 3.

Global comparison of orthologous gene clusters for 17 S. pneumoniae strains. The similarity score corresponds to the total number of orthologous gene clusters shared within each strain pair; the difference score corresponds to the total number of orthologous gene clusters not shared within a strain pairing; and the comparison equals the similarity score minus the difference score. In addition, the number of clusters shared only within a strain pair is noted as pair-unique. If these values are 1, 2, or 3 standard deviations away from the mean of all pairwise comparisons, the boxes are color coded as noted in the key.
We used the sum of the difference scores as a rough measure of strain diversity to investigate whether the number of unique genes serves as an indicator for gene diversity. The plot of the total number of orthologous clusters, the number of unique orthologous clusters, and the sum of the difference scores shows a modest correlation between these values (_R_2 = 0.73 for unique orthologous clusters versus difference score) (see Fig. S2 in the supplemental material); however, the numbers of unique and pair-unique orthologous clusters are not sufficient to reveal the phylogenetic relationships among strains.
Supragenome size estimation.
These data show that S. pneumoniae has a supragenome much larger than the genome of any individual strain. Our group developed a finite supragenome model based on clustering analysis of multiple H. influenzae genomes (18). This model accounts for the numbers of core, distributed, and unique orthologous clusters and does not assume that the distributed orthologous clusters are sampled in the population with equal probability. The absence of this assumption yields a model where almost all of the supragenome can be accurately determined after the genomes of a finite number of strains are sequenced.
To visualize how the genome sequence of each additional strain adds to our understanding of the supragenome, we calculated the numbers of total and core orthologous clusters for two strains and then recalculated these parameters on an iterative basis as we added each additional strain. These exact core and supragenome values will differ depending on the order in which the strains are added to the analysis, but the trend is always the same: decay in the number of new orthologous clusters and stabilization of the number of core orthologous clusters at ∼1,400 (Fig. 4A and B). This suggests that after a finite number of genomes have been sequenced, the number of new orthologous clusters identified will be very low.
FIG. 4.

(A) Plot of the numbers of total and core observed orthologous clusters as a function of the number of strains sequenced. (B) Plot of the number of new observed orthologous clusters as a function of each genome. Numbers were calculated first for two strains and then iteratively for strains added one by one.
To address the question of the number of strains that must be sequenced before the vast majority of the orthologous clusters are identified, we fitted the clustering data from the 17 genomes to the finite supragenome model (18). This model predicts that the S. pneumoniae supragenome consists of ∼5,100 orthologous clusters, where ∼1,380 are core, ∼2,100 are unique, and the remaining are distributed. The orthologous clusters in the supragenome are represented at different frequencies within the S. pneumoniae population; a cluster that is present at a population frequency lower than 0.1 presumably does not have much impact on the population, although it may be very interesting in itself and could potentially be very important if the one strain it represents causes a pneumococcal pandemic. If all orthologous clusters are considered, the estimated supragenome size is 5,117 clusters and the core set contains 27% of the clusters. If only orthologous clusters with frequencies equal or greater then 0.1 are considered, the estimated supragenome size drops to 2,979 clusters where 46.5% are core. Accounting only for the orthologous clusters that exist in the S. pneumoniae supragenome at a frequency equal to 0.1 to greater than 95% of the supragenome is predicted to be identified after sequencing of 17 strains and 99% after sequencing of 33 strains. If orthologous clusters of all population frequencies are considered, 90% of the supragenome is predicted to be identified after 142 strains are sequenced (Table 4). The predicted and observed distributions are compared in Fig. 1, where the model fits the data well. Figure 5A shows the numbers of total and core orthologous clusters predicted for 50 or fewer strains, and Fig. 5B plots the predicted number of new orthologous clusters as a function of each genome sequenced. This prediction assumes that the 17 strains used in this study are representative of worldwide strains; if they are not, this prediction will underestimate the number of strains required for supragenome coverage.
TABLE 4.
Predicted coverage of the S. pneumoniae supragenome using the finite supragenome model
| Population frequency | Supragenome coverage (%) | No. of strains sequenced |
|---|---|---|
| ≥0.1 | 90 | 11 |
| ≥0.1 | 95 | 17 |
| ≥0.1 | 99 | 33 |
| All | 90 | 142 |
FIG. 5.

Predictions using the finite supragenome model. (A) Plot of the numbers of total and core predicted orthologous clusters as functions of the number of strains sequenced. (B) Plot of the number of new predicted orthologous clusters as a function of each genome sequenced. Numbers were calculated first for two strains and then iteratively for strains added one by one.
Unique and core orthologous clusters.
The 576 unique orthologous clusters identified in this study are distributed among all strains; the lowest number per strain is 1, for the TIGR4 strain, and the highest is 74, for INV104B (Table 3). Sixty-two percent of the unique genes are annotated as hypothetical proteins; ∼5.4% have annotations related to phage, prophage, or bacteriophage; and the remaining 33% correspond to a wide range of proteins including putative transporters, transcriptional regulators and activators, lantibiotic biosynthesis and transport proteins, macrolide efflux proteins, and many other enzymes (see Table S3 in the supplemental material).
For the most part, the genes in a given orthologous cluster have the same annotation, and somewhat surprisingly, the genes in the core orthologous clusters include a significant percentage annotated as hypothetical proteins (∼30%), perhaps suggesting that many bacterial housekeeping functions remain unknown (see Table S3 in the supplemental material). Among the annotated core orthologous clusters, no phage proteins were detected, suggesting that no single phage is conserved across all strains. Interestingly, there are a few orthologous clusters with more than 100 CDSs that are annotated as transposases, often degenerate or truncated. The role of these proteins in the S. pneumoniae genome, if any, remains to be established. A table with the annotations for genes in the core, distributed, and unique categories is provided as Table S3 in the supplemental material.
DISCUSSION
This study compared the genic contents of 17 S. pneumoniae strains. Genes from all strains were organized into orthologous clusters, and these clusters were quantified for all genomes. When the genomes are analyzed together, fewer than 50% of all the orthologous clusters (corresponding to ∼73% of the total CDSs) are conserved among all species. When the genomes of individual strains were evaluated, 21 to 32% of the orthologous clusters were noncore. Predictions using the finite supragenome model suggest that the total number of orthologous clusters in the S. pneumoniae species is around 5,100 and the total number of core orthologous clusters is around 1,380. These large strain differences illustrate the enormous genic diversity within this species, as postulated in the distributed-genome hypothesis (7, 37). The engines driving this genomic plasticity are threefold: first, it has been demonstrated that chronic infections by nasopharyngeal pathogens are generally polyclonal in nature (11, 16, 30, 31, 39-41; J. R. Gilsdorf, presented at the 9th International Symposium on Recent Advances in Otitis Media, 3 to 7 June 2007); second, the bacteria in these chronic infections adopt a biofilm mode of growth, which greatly increases the kinetics of horizontal gene transfer (8, 15, 28); and third, S. pneumoniae employs highly energetic fratricidal as well as autocompetence and autotransformation mechanisms for the release and uptake of pneumococcal DNA, respectively, from the surrounding environment (35). The pathological consequences of these phenomena, which collectively result in a continual reassortment of genic characters among strains within a polyclonal biofilm infection, are that the host's adaptive immune system continually encounters novel strains, making clearance very difficult, because the pathogen can generate diversity faster than the host can adapt to it, thus ensuring chronicity of infection.
In a previous study, we constructed individual genomic libraries from the eight CGS S. pneumoniae clinical isolates (CGSSp9BS68, CGSSp14BS69, CGSSp11BS70, CGSSp3BS71, CGSSp23BS72, CGSSp6BS73, CGSSp18BS74, and CGSSp19BS75). Of the 4,793 clones sequenced, ∼16% were not present in the TIGR4 reference strain, suggesting that many genes were not conserved across the species. In addition, the screen identified genes unrelated to any streptococcal sequences; analysis of the allocation of a subset of 58 of these found that they were not uniformly distributed across the eight strains (37). These results are in complete agreement with this study; both studies underscore the genomic plasticity of the S. pneumoniae species.
The use of the finite supragenome model suggests that 99% of orthologous clusters in the supragenome that have population frequencies equal or higher to 0.1 can be identified after sequencing of 33 strains and that the 17 available strains provide ∼95% coverage of this set. When analyzing the S. agalactiae supragenome, Tettelin and colleagues presented a different mathematical model, generated using the assumption that noncore genes are sampled in the population with equal probabilities (43). Unlike the finite supragenome model, this model predicts that a constant number of new strain-specific genes will be identified with the addition of each genome, such that sequencing a limited number of strains would not provide major coverage of the supragenome.
Our analysis includes clinical strains from multiple locations including the United States, the United Kingdom, Norway, and Spain. Diversity is generated from DNA exchange among strains; thus, it is tempting to consider that strains from the same geographical location may be more similar, since they have a higher probability of exchanging genetic information (directly or indirectly, via other strains). Interestingly, we did not observe this with our limited number of strains. While it is possible that a correlation between geographical distance and genic diversity will be observed when a larger number of strains from multiple geographic regions are sequenced and compared, we must nonetheless consider that this correlation may not exist. This result would be explained if the vast majority of the orthologous clusters in the S. pneumoniae supragenome have been in the species for a very long time, and horizontal transfer from other species and new mutations have introduced only a minority of the supragenome's orthologous clusters, or if the extent of human population migration is now so high (at least in the West) that human pathogens are essentially homogenized around the world.
This enormous genetic diversity calls attention to the need for markers of human virulence phenotypes and highlights the potential difficulty associated with this task. S. pneumoniae strains are presently categorized based on capsule type and MLST. The capsular serotype is an important virulence factor and affects the ability of pneumococci to cause invasive disease (2, 38). For example, the difference in virulence between type 2 D39, which is highly virulent in the murine model of infection, and unencapsulated R6, which is avirulent, is attributed to the loss of the capsule. However, it is critical to remember that even within the same capsular type, virulence is highly related to the genetic background of the strains (20). The virulence phenotypes displayed by the eight strains isolated in Pittsburgh differ significantly in a chinchilla model of S. pneumoniae infection; these differences may be due to distinct serotypes, genotypes, or both (M. Forbes and J. Hayes, personal communication). Our data in Fig. 2 clearly show that the serotype cannot be correlated with the genic content, since strains of serotypes 14, 6, and 23 were not grouped based on genic differences. An analysis of sequence variation of the type 6A and 6B capsular biosynthetic loci was related to the MLST profile, yet there was also ample evidence of horizontal transfer to unrelated lineages (27). Phylogenetic trees using MLST from the seven CGS clinical strains of known MLST types did not closely resemble the phylogenies created from genic differences (data not shown). Together these data suggest that in some cases, the serotype, MLST type, and/or genetic background may correlate, but in other cases, they do not, as would be expected from strains undergoing high rates of intraspecies horizontal gene transfer. Since pathogenesis is probably a consequence not only of capsular type but also of multiple other genes, MLST type and serotype alone are not ideal markers for the disease phenotype of S. pneumoniae strains.
Previous work on six strains of Streptococcus agalactiae described a supragenome with ∼80% core orthologous clusters and the remaining set consisting of partially shared and strain-specific orthologous clusters (43). In addition, these data resemble, in qualitative and quantitative terms, our comparison of 13 H. influenzae strains (18). The total number of H. influenzae orthologous clusters for the 13 strains was 2,786, of which 52% were core, 29% were distributed, and 19% were unique. Taken together, these studies suggest that a high degree of genic variation is common among multiple species. However, it may not be universal; analyses of eight Bacillus anthracis strains revealed substantially less variation among strains, with no new genes uncovered after analysis of only four genomes (43). It is possible that this degree of variation is intrinsic to naturally transforming bacteria such as H. influenzae and S. pneumoniae, which undergo extensive DNA recombination events. In addition, both of these bacteria exist exclusively in the human mucosa, where they form biofilms (15). Cells in a biofilm are embedded in an extracellular polymeric matrix that is rich in nucleic acids; thus, biofilms may provide ideal environments to foster such genomic plasticity (32). There is also quantitative similarity, since both species have core genomes that seem to stabilize around 1,400 proteins, or ∼50% of the supragenome. This similarity suggests that similar evolutionary forces may be determining the equilibrium between core genes, noncore genes, and genome size.
This diversity suggests caution in the use of model strains to test and develop vaccines and drugs, since effective targets in one strain may be missing in a significant percentage of the other strains. It is probable that these bacteria have evolved multiple and redundant mechanisms to evade immunity and adapt to variations among hosts and their commensal microbiota.
Supplementary Material
[Supplemental material]
Acknowledgments
We thank David P. Greenberg of the Children's Hospital of Pittsburgh for the eight clinical S. pneumoniae strains, as well as Susan Hollingshead, Herve Tettelin, and The Institute for Genomic Research for making strain TIGR670-6B easily accessible. We thank Mary O'Toole for help with the preparation of the manuscript and Konstantinos Liolios for creating the SupraGen website.
The sequence data for strain TIGR670-6B was produced by The Institute for Genomic Research with funds from NIAID through the University of Alabama. The sequencing of S. pneumoniae strains at The Sanger Institute was supported by the Wellcome Trust. We also acknowledge the use of the pneumococcal MLST database, which is located at Imperial College London and is funded by the Wellcome Trust. This work was supported by Allegheny General Hospital, Allegheny Singer Research Institute, and grants DC05659 (J.C.P.) DC04173 (G.D.E.), and DC02148 (G.D.E.) from NIDCD, NIH.
Footnotes
▿
Published ahead of print on 3 August 2007.
REFERENCES
- 1.Avery, O. T., C. M. MacLeod, and M. McCarty. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Inductions of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79**:**137-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brueggemann, A. B., D. T. Griffiths, E. Meats, T. Peto, D. W. Crook, and B. G. Spratt. 2003. Clonal relationships between invasive and carriage Streptococcus pneumoniae and serotype- and clone-specific differences in invasive disease potential. J. Infect. Dis. 187**:**1424-1432. [DOI] [PubMed] [Google Scholar]
- 3.Claverys, J. P., and L. S. Havarstein. 2002. Extracellular-peptide control of competence for genetic transformation in Streptococcus pneumoniae. Front. Biosci. 7**:**d1798-d1814. [DOI] [PubMed] [Google Scholar]
- 4.Daraselia, N., D. Dernovoy, Y. Tian, M. Borodovsky, R. Tatusov, and T. Tatusova. 2003. Reannotation of Shewanella oneidensis genome. OMICS 7**:**171-175. [DOI] [PubMed] [Google Scholar]
- 5.Del Beccaro, M. A., P. M. Mendelman, A. F. Inglis, M. A. Richardson, N. O. Duncan, C. R. Clausen, and T. L. Stull. 1992. Bacteriology of acute otitis media: a new perspective. J. Pediatr. 120**:**81-84. [DOI] [PubMed] [Google Scholar]
- 6.Delcher, A. L., S. Kasif, R. D. Fleischmann, J. Peterson, O. White, and S. L. Salzberg. 1999. Alignment of whole genomes. Nucleic Acids Res. 27**:**2369-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrlich, G. D., F. Z. Hu, K. Shen, P. Stoodley, and J. C. Post. 2005. Bacterial plurality as a general mechanism driving persistence in chronic infections. Clin. Orthop. Relat. Res. 2005**:**20-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrlich, G. D., R. Veeh, X. Wang, J. W. Costerton, J. D. Hayes, F. Z. Hu, B. J. Daigle, M. D. Ehrlich, and J. C. Post. 2002. Mucosal biofilm formation on middle-ear mucosa in the chinchilla model of otitis media. JAMA 287**:**1710-1715. [DOI] [PubMed] [Google Scholar]
- 9.Ewing, B., and P. Green. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8**:**186-194. [PubMed] [Google Scholar]
- 10.Ewing, B., L. Hillier, M. C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8**:**175-185. [DOI] [PubMed] [Google Scholar]
- 11.Farjo, R. S., B. Foxman, M. J. Patel, L. Zhang, M. M. Pettigrew, S. I. McCoy, C. F. Marrs, and J. R. Gilsdorf. 2004. Diversity and sharing of Haemophilus influenzae strains colonizing healthy children attending day-care centers. Pediatr. Infect. Dis. J. 23**:**41-46. [DOI] [PubMed] [Google Scholar]
- 12.Gates, G. A. 1996. Cost-effectiveness considerations in otitis media treatment. Otolaryngol. Head Neck Surg. 114**:**525-530. [DOI] [PubMed] [Google Scholar]
- 13.Gordon, D. 2004. Viewing and editing assembled sequences using Consed. John Wiley & Co., New York, NY. [DOI] [PubMed]
- 14.Gordon, D., C. Abajian, and P. Green. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8**:**195-202. [DOI] [PubMed] [Google Scholar]
- 15.Hall-Stoodley, L., F. Z. Hu, A. Gieseke, L. Nistico, D. Nguyen, J. Hayes, M. Forbes, D. P. Greenberg, B. Dice, A. Burrows, P. A. Wackym, P. Stoodley, J. C. Post, G. D. Ehrlich, and J. E. Kerschner. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296**:**202-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiltke, T. J., A. T. Schiffmacher, A. J. Dagonese, S. Sethi, and T. F. Murphy. 2003. Horizontal transfer of the gene encoding outer membrane protein P2 of nontypeable Haemophilus influenzae, in a patient with chronic obstructive pulmonary disease. J. Infect. Dis. 188**:**114-117. [DOI] [PubMed] [Google Scholar]
- 17.Hoberman, A., D. P. Greenberg, J. L. Paradise, H. E. Rockette, J. R. Lave, D. H. Kearney, D. K. Colborn, M. Kurs-Lasky, M. A. Haralam, C. J. Byers, L. M. Zoffel, I. A. Fabian, B. S. Bernard, and J. D. Kerr. 2003. Effectiveness of inactivated influenza vaccine in preventing acute otitis media in young children: a randomized controlled trial. JAMA 290**:**1608-1616. [DOI] [PubMed] [Google Scholar]
- 18.Hogg, J. S., F. Z. Hu, B. Janto, R. Boissy, J. Hayes, R. Keefe, J. C. Post, and G. D. Ehrlich. 2007. Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains. Genome Biol. 8**:**R103. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoskins, J., W. E. Alborn, Jr., J. Arnold, L. C. Blaszczak, S. Burgett, B. S. DeHoff, S. T. Estrem, L. Fritz, D. J. Fu, W. Fuller, C. Geringer, R. Gilmour, J. S. Glass, H. Khoja, A. R. Kraft, R. E. Lagace, D. J. LeBlanc, L. N. Lee, E. J. Lefkowitz, J. Lu, P. Matsushima, S. M. McAhren, M. McHenney, K. McLeaster, C. W. Mundy, T. I. Nicas, F. H. Norris, M. O'Gara, R. B. Peery, G. T. Robertson, P. Rockey, P. M. Sun, M. E. Winkler, Y. Yang, M. Young-Bellido, G. Zhao, C. A. Zook, R. H. Baltz, S. R. Jaskunas, P. R. Rosteck, Jr., P. L. Skatrud, and J. I. Glass. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183**:**5709-5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly, T., J. P. Dillard, and J. Yother. 1994. Effect of genetic switching of capsular type on virulence of Streptococcus pneumoniae. Infect. Immun. 62**:**1813-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein, J. O. 1999. Management of acute otitis media in an era of increasing antibiotic resistance. Int. J. Pediatr. Otorhinolaryngol. 49(Suppl. 1)**:**S15-S17. [DOI] [PubMed] [Google Scholar]
- 22.Kunsch, C. A., G. H. Choi, P. S. Dillon, C. A. Rosen, S. C. Barash, M. R. Fannon, and B. A. Dougherty. 16 July 2002. Streptococcus pneumoniae polynucleotides and sequences. U.S. patent 6,420,135.
- 23.Lanie, J. A., W. L. Ng, K. M. Kazmierczak, T. M. Andrzejewski, T. M. Davidsen, K. J. Wayne, H. Tettelin, J. I. Glass, and M. E. Winkler. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 189**:**38-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowe, T. M., and S. R. Eddy. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25**:**955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchler-Bauer, A., J. B. Anderson, M. K. Derbyshire, C. DeWeese-Scott, N. R. Gonzales, M. Gwadz, L. Hao, S. He, D. I. Hurwitz, J. D. Jackson, Z. Ke, D. Krylov, C. J. Lanczycki, C. A. Liebert, C. Liu, F. Lu, S. Lu, G. H. Marchler, M. Mullokandov, J. S. Song, N. Thanki, R. A. Yamashita, J. J. Yin, D. Zhang, and S. H. Bryant. 2007. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 35**:**D237-D240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margulies, M., M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. Yu, R. F. Begley, and J. M. Rothberg. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437**:**376-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mavroidi, A., D. Godoy, D. M. Aanensen, D. A. Robinson, S. K. Hollingshead, and B. G. Spratt. 2004. Evolutionary genetics of the capsular locus of serogroup 6 pneumococci. J. Bacteriol. 186**:**8181-8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molin, S., and T. Tolker-Nielsen. 2003. Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Curr. Opin. Biotechnol. 14**:**255-261. [DOI] [PubMed] [Google Scholar]
- 29.Moscoso, M., and J. P. Claverys. 2004. Release of DNA into the medium by competent Streptococcus pneumoniae: kinetics, mechanism and stability of the liberated DNA. Mol. Microbiol. 54**:**783-794. [DOI] [PubMed] [Google Scholar]
- 30.Müller-Graf, C. D., A. M. Whatmore, S. J. King, K. Trzcinski, A. P. Pickerill, N. Doherty, J. Paul, D. Griffiths, D. Crook, and C. G. Dowson. 1999. Population biology of Streptococcus pneumoniae isolated from oropharyngeal carriage and invasive disease. Microbiology 145**:**3283-3293. [DOI] [PubMed] [Google Scholar]
- 31.Murphy, T. F., S. Sethi, K. L. Klingman, A. B. Brueggemann, and G. V. Doern. 1999. Simultaneous respiratory tract colonization by multiple strains of nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease: implications for antibiotic therapy. J. Infect. Dis. 180**:**404-409. [DOI] [PubMed] [Google Scholar]
- 32.Oggioni, M. R., C. Trappetti, A. Kadioglu, M. Cassone, F. Iannelli, S. Ricci, P. W. Andrew, and G. Pozzi. 2006. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61**:**1196-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearson, W. R., and D. J. Lipman. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85**:**2444-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson, S. N., C. K. Sung, R. Cline, B. V. Desai, E. C. Snesrud, P. Luo, J. Walling, H. Li, M. Mintz, G. Tsegaye, P. C. Burr, Y. Do, S. Ahn, J. Gilbert, R. D. Fleischmann, and D. A. Morrison. 2004. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol. Microbiol. 51**:**1051-1070. [DOI] [PubMed] [Google Scholar]
- 35.Prudhomme, M., L. Attaiech, G. Sanchez, B. Martin, and J. P. Claverys. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313**:**89-92. [DOI] [PubMed] [Google Scholar]
- 36.Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132**:**365-386. [DOI] [PubMed] [Google Scholar]
- 37.Shen, K., J. Gladitz, P. Antalis, B. Dice, B. Janto, R. Keefe, J. Hayes, A. Ahmed, R. Dopico, N. Ehrlich, J. Jocz, L. Kropp, S. Yu, L. Nistico, D. P. Greenberg, K. Barbadora, R. A. Preston, J. C. Post, G. D. Ehrlich, and F. Z. Hu. 2006. Characterization, distribution, and expression of novel genes among eight clinical isolates of Streptococcus pneumoniae. Infect. Immun. 74**:**321-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, T., D. Lehmann, J. Montgomery, M. Gratten, I. D. Riley, and M. P. Alpers. 1993. Acquisition and invasiveness of different serotypes of Streptococcus pneumoniae in young children. Epidemiol. Infect. 111**:**27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith-Vaughan, H. C., A. J. Leach, T. M. Shelby-James, K. Kemp, D. J. Kemp, and J. D. Mathews. 1996. Carriage of multiple ribotypes of non-encapsulated Haemophilus influenzae in aboriginal infants with otitis media. Epidemiol. Infect. 116**:**177-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith-Vaughan, H. C., K. S. Sriprakash, J. D. Mathews, and D. J. Kemp. 1995. Long PCR-ribotyping of nontypeable Haemophilus influenzae. J. Clin. Microbiol. 33**:**1192-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith-Vaughan, H. C., K. S. Sriprakash, J. D. Mathews, and D. J. Kemp. 1997. Nonencapsulated Haemophilus influenzae in Aboriginal infants with otitis media: prolonged carriage of P2 porin variants and evidence for horizontal P2 gene transfer. Infect. Immun. 65**:**1468-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatusov, R. L., M. Y. Galperin, D. A. Natale, and E. V. Koonin. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28**:**33-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tettelin, H., V. Masignani, M. J. Cieslewicz, C. Donati, D. Medini, N. L. Ward, S. V. Angiuoli, J. Crabtree, A. L. Jones, A. S. Durkin, R. T. Deboy, T. M. Davidsen, M. Mora, M. Scarselli, I. Margarit y Ros, J. D. Peterson, C. R. Hauser, J. P. Sundaram, W. C. Nelson, R. Madupu, L. M. Brinkac, R. J. Dodson, M. J. Rosovitz, S. A. Sullivan, S. C. Daugherty, D. H. Haft, J. Selengut, M. L. Gwinn, L. Zhou, N. Zafar, H. Khouri, D. Radune, G. Dimitrov, K. Watkins, K. J. O'Connor, S. Smith, T. R. Utterback, O. White, C. E. Rubens, G. Grandi, L. C. Madoff, D. L. Kasper, J. L. Telford, M. R. Wessels, R. Rappuoli, and C. M. Fraser. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. USA 102**:**13950-13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettelin, H., K. E. Nelson, I. T. Paulsen, J. A. Eisen, T. D. Read, S. Peterson, J. Heidelberg, R. T. DeBoy, D. H. Haft, R. J. Dodson, A. S. Durkin, M. Gwinn, J. F. Kolonay, W. C. Nelson, J. D. Peterson, L. A. Umayam, O. White, S. L. Salzberg, M. R. Lewis, D. Radune, E. Holtzapple, H. Khouri, A. M. Wolf, T. R. Utterback, C. L. Hansen, L. A. McDonald, T. V. Feldblyum, S. Angiuoli, T. Dickinson, E. K. Hickey, I. E. Holt, B. J. Loftus, F. Yang, H. O. Smith, J. C. Venter, B. A. Dougherty, D. A. Morrison, S. K. Hollingshead, and C. M. Fraser. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293**:**498-506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]