Complex Formation of Yeast Rev1 with DNA Polymerase η (original) (raw)
Abstract
In Saccharomyces cerevisiae, Rev1 functions in translesion DNA synthesis (TLS) together with polymerase ζ (Polζ), comprised of the Rev3 catalytic and Rev7 accessory subunits. Rev1 plays an indispensable structural role in promoting Polζ function, and deletion of the Rev1-C terminal region that is involved in physical interactions with Rev3 inactivates Polζ function in TLS. In humans, however, Rev1 has been shown to physically interact with the Y-family polymerases Polη, Polι, and Polκ, and the Rev1 C terminus mediates these interactions. Since all the available genetic and biochemical evidence in yeast support the requirement of Rev1 as a structural element for Polζ and not for Polη, these observations have raised the possibility that in its structural role, Rev1 has diverged between yeast and humans. Here we show that although in yeast a stable Rev1-Polη complex can be formed, this complex formation involves the polymerase-associated domain of Rev1 and not the Rev1 C terminus as in humans. We also found that the DNA synthesis activity of Rev1 is enhanced in this complex. We discuss the implications of these and other observations for the possible divergence of Rev1's structural role between yeast and humans.
In Saccharomyces cerevisiae, the Rad6-Rad18 ubiquitin-conjugating enzyme complex (3, 4) promotes replication through DNA lesions via translesion synthesis (TLS) by DNA polymerase η (Polη) and Polζ and by a Rad5-Mms2-Ubc13-dependent postreplication repair pathway that repairs the discontinuities that form in the newly synthesized DNA from damaged DNA templates (13, 26, 28, 32). Polη is unique among eukaryotic TLS polymerases in its ability to promote proficient and error-free replication through UV-induced cyclobutane pyrimidine dimers (13); consequently, inactivation of Polη in both yeast and humans confers enhanced UV mutagenesis (14, 21, 30, 34) and, in humans, causes a cancer-prone syndrome, the variant form of xeroderma pigmentosum (11, 20). Although replication through cyclobutane pyrimidine dimers can be performed by Polη alone, replication through many DNA lesions requires the concerted action of two polymerases, in which one polymerase inserts the nucleotide opposite the lesion site and the other polymerase carries out the subsequent extension reaction (15, 28, 29). Polζ, comprised of the Rev3 catalytic and Rev7 accessory subunits (26), is highly specialized for conducting the extension reaction opposite a variety of DNA lesions. For example, Polζ proficiently extends from the nucleotide inserted opposite a (6-4) photoproduct or opposite an abasic site by another DNA polymerase (9, 10).
For its role in TLS, Polζ requires the Rev1 protein. Although Rev1 has a DNA polymerase activity that is specific for inserting a C opposite template G (8, 25), the Rev1 DNA polymerase activity is dispensable for Polζ's role in promoting TLS through many DNA lesions. For example, even though both Polζ and Rev1 are required for UV mutagenesis resulting from the action of Polζ at the extension step of TLS through UV lesions, the Rev1 DNA polymerase activity is not required (28). Instead, Rev1 performs a structural role in modulating Polζ function in TLS (5, 9, 24). In keeping with such a structural role for Rev1, we have shown that Rev1 forms a stable complex with Polζ and that this complex formation requires the binding of the Rev1 C terminus with the polymerase domain of Rev3 (2). Since deletion of the Rev1 C-terminal segment of 72 residues is as inhibitory to UV mutagenesis as the _rev3_Δ mutation, Rev1 binding is indispensable for Polζ function in TLS in yeast.
In mouse and human cells, Rev1 has been shown to physically interact with Polη, Polι, and Polκ and the C-terminal ∼100 residues of human Rev1 are involved in modulating these interactions (7, 27, 31). These observations suggest that Rev1 acts as a scaffold for the recruitment not only of Polζ but also of Polη, Polι, and Polκ to the replication fork stalled at a lesion site. In yeast, however, the available genetic evidence has suggested that Rev1 functions in TLS together with Polζ and not with Polη. This is exemplified by the epistasis of the _rev1_Δ mutation with the _rev3_Δ or _rev7_Δ mutations for sensitivity to DNA-damaging agents and by the enhanced sensitivity seen when the _rev1_Δ, _rev3_Δ, _or rev7_Δ mutation is combined with the _rad30_Δ mutation. Furthermore, whereas the absence of Rev1 or Polζ greatly reduces UV mutagenesis (16-19), the absence of Polη causes an increase in UV mutagenesis (14, 21, 34). Hence, the inference derived from such genetic studies and from biochemical studies of physical and functional interactions of Rev1 with Polζ (2) that in yeast Rev1 functions with Polζ would seem to be at odds with the observations of Rev1 interactions with Polη, Polι, and Polκ in mouse and human cells, since they suggest that Rev1 acts as a scaffold for these Y-family polymerases. These differences could imply that the role of Rev1 has diverged between yeast and humans such that whereas Rev1 functions only with Polζ in yeast, it additionally functions together with the various Y-family polymerases in humans. An alternative possibility to be considered is that in both yeast and humans, Rev1 is indispensable for Polζ function but that it also participates in physical interactions with Polη in yeast and with Polη, Polι, and Polκ in humans, thereby assisting in the assembly of these various polymerases at the replication fork, a role that is dispensable in both yeast and humans.
To distinguish between these possibilities and to evaluate the possible functional significance of Rev1 interactions with the Y-family polymerases, here we determined whether yeast Rev1 physically interacts with Polη and, if so, whether complex formation between these two proteins influences their respective activities. We show that a stable Rev1-Polη complex can in fact be formed from purified yeast proteins and that the DNA synthesis activity of Rev1, but not of Polη, is enhanced in the complex. We discuss the implications of these and other observations for Rev1 function in yeast and their possible bearing on Rev1 function in humans.
MATERIALS AND METHODS
Yeast strains, plasmids, and DNA substrates.
Yeast strain BJ5464 was used for protein purification. For in vitro binding assays, REV1, RAD30, and their mutants were inserted into a pBJ842 vector to produce a glutathione _S_-transferase (GST) fusion protein. DNA primer-template substrates were composed of the oligodeoxynucleotide primer (32 mer) 5′-GTTTTCCCAG TCACGACGAT GCTCCGGTAC TC-3′ annealed to a 52-mer template, 5′-TTCGTATAAT GCCTACACTX GAGTACCGGA GCATCGTCGT GACTGGGAAAAC-3′, where the X denotes either an A, a G, or an abasic site (tetrahydrofuran). For the DNA substrates shown in Fig. 4, the 75-nucleotide (nt) oligomer template 5′-AGC TAC CAT GCC TGC CTC AAG AAT TCG TAA _X_AT GCC TAC ACT GGA GTA CCG GAG CAT CGT CGT GAC TGG GAA AAC-3′ containing a G or an abasic site was annealed with the 5′-end-labeled primer N4309, 5′-GTT TTC CCA GTC ACG ACG ATG CTC CGG TAC TCC AGT GTA GGC AT-3′. DNA substrates consisted of the oligonucleotide primer, which was 5′-32P-end-labeled by using polynucleotide kinase (Roche Molecular Biochemicals) and [32P]ATP (Amersham Pharmacia Biotech), which was annealed to the template by heating a 1:1.5 molar ratio mixture of the primer-template to 95°C and allowing it to cool to room temperature overnight.
FIG. 4.

dCTP incorporation opposite template G and opposite an abasic (AP) site by Rev1 in the presence or absence of the 122-amino-acid C-terminal peptide of Polη. Rev1 (100 nM) alone or together with the Polη peptide (aa 510 to 632) (200 nM) was first preincubated overnight at 0°C. Rev1 and Rev1 plus the Polη peptide (each containing 0.2 nM Rev1) were incubated with a primer-template DNA substrate (10 nM) and increasing concentrations of dCTP for 5 min at 30°C. The nucleotide incorporation rate was plotted against the dCTP concentration, and the data were fit to the Michaelis-Menten equation describing a hyperbola. Apparent Km and kcat values were obtained from the fit and used to calculate the efficiency of deoxynucleotide incorporation (kcat /Km). The position of the abasic site in the DNA substrate is indicated by 0.
Purification of proteins.
Wild-type and mutant GST-Polη or GST-Rev1 proteins were purified on glutathione-Sepharose beads by using a protocol described earlier (12). To obtain untagged proteins, GST-fused proteins bound to glutathione-Sepharose beads were treated overnight at 4°C with PreScission protease to cleave between the GST tag and Polη or Rev1. For purification of the Rev1-Rev7 complex, the GST-Rev1-Rev7 complex was purified from yeast cells carrying the plasmids for expressing the GST-Rev1 and Rev7 proteins and the GST tag was cleaved from Rev1 as previously described (1).
In vitro interactions of Polη and Rev1 proteins.
The physical interactions between Polη and Rev1 proteins were carried out by using a protocol described before (1, 2). Briefly, GST-Rev1 or its truncated forms were incubated with Polη or its truncated derivatives and vice versa in buffer I (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM dithiothreitol [DTT], 0.01% NP-40, and 10% glycerol) in a 20-μl reaction mixture at 4°C for 30 min, followed by 10 min at 25°C. Glutathione-Sepharose beads (20 μl) were added to the mixtures, which were further incubated for 1 h with constant rocking at 4°C. The beads were spun down, and the unbound protein was collected. Then the beads were washed thoroughly three times with 10 volumes of buffer I. Finally, the bound proteins were eluted with 20 μl of sodium dodecyl sulfate (SDS) loading buffer. Various fractions were resolved on a 12% denaturing polyacrylamide gel, followed by either silver staining or Western blot analysis.
DNA polymerase reactions.
Linear DNA substrates were generated by annealing 5′ 32P-labeled 32-mer oligonucleotide primer with the template DNA substrate. The standard DNA polymerase reaction (10 μl) contained 40 mM Tris-HCl [pH 7.5], 8 mM MgCl2, 1 mM DTT, 10% glycerol, 100 μg/ml bovine serum albumin, and 10 μM each of dGTP, dATP, dTTP, and dCTP. As indicated in the legend to Fig. 3, Rev1, Polη (0.2 nM), or various Rev1-Polη complexes (0.2 nM) were incubated with linear primer-template DNA (10 nM). For assays, the reaction mixtures were assembled on ice, incubated at 30°C for 10 min, and stopped by the addition of loading buffer (30 μl) containing EDTA (20 mM), 95% formamide, 0.3% bromphenol blue, and 0.3% cyanol blue. The reaction products were resolved on 15% polyacrylamide gels containing 8 M urea.
FIG. 3.

Comparison of the DNA polymerase activity of Rev1 and Rev1-Polη complexes. (i to iii) The complete standard reaction mixture contained 0.2 nM Rev1 or Polη or the Rev1-Polη complex, 10 μM of a single dNTP, and 10 nM of a DNA substrate. Reaction mixtures were incubated at 30°C for 10 min, and the reactions were stopped by the addition of loading buffer (30 μl) containing 20 mM EDTA, 95% formamide, 0.3% bromphenol blue, and 0.3% cyanol blue. The reaction products were resolved on 15% polyacrylamide gels containing 8 M urea. Lane 1 is the buffer control. Incorporation of each of the nucleotides by Rev1 is shown in lanes 2 to 5; by Polη, in lanes 6 to 9, by the Rev1-Polη* complex, in lanes 10 to 13, and by the Rev1*-Polη complex, in lanes 14 to 17. The DNA substrates used are indicated to the right of the panels. (iii) The abasic site in the substrate is indicated by 0. Polη* and Rev1* denote the catalytically inactive forms of the polymerases.
Determination of steady-state kinetic parameters.
Steady-state kinetic analyses for deoxynucleotide incorporation opposite a nondamaged G, A, or abasic site were performed as previously described (8). Briefly, Rev1 (0.1 or 0.2 nM) or Rev1-Polη* (0.05 or 0.1 nM), where Polη* represents the catalytically inactive polymerase, or a Rev1-Polη peptide comprised of residues 510 to 632 (0.2 nM) was incubated with the DNA substrate (10 nM) and increasing concentrations of deoxynucleoside triphosphate (dNTP) for 5 or 15 min under standard reaction conditions. The intensities of the substrates and products in gel bands were quantitated by PhosphorImager, and the percentage of the primer extension was plotted as a function of dNTP concentration. The data were fit by nonlinear regression, using SigmaPlot 5.0, to the Michaelis-Menten equation describing a hyperbola, v = (_kcat_[E] × [dNTP]/(Km + [dNTP]), where [E] refers to enzyme concentration. Apparent Km and kcat steady-state parameters were obtained from the fit and used to calculate the efficiency of deoxynucleotide incorporation (kcat/Km). For comparing the steady-state kinetics of Rev1 and the Rev1-Polη peptide (amino acids [aa] 510 to 632) complex, we used either a prepurified Rev1-Polη peptide (aa 510 to 632) complex or protein samples obtained by preincubating Rev1 (100 nM) alone or Rev1 (100 nM) together with the Polη (aa 510 to 632) peptide (200 nM) in a buffer containing 25 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1 mM DTT, 10% glycerol, and 100 μg/ml bovine serum albumin at 0°C overnight.
RESULTS
Yeast Rev1 and Polη form a stable complex.
In order to investigate the physical interaction of yeast Rev1 with Polη (Rad30), we used a GST pull-down assay. A mixture of purified GST-Rev1 and Rad30 or GST-Rad30 and Rev1 protein was bound to the glutathione beads and rocked for an hour, followed by extensive washing with 150 mM NaCl-containing buffer before being eluted with SDS-containing buffer. In this type of system, the GST fusion protein binds to the beads and the interacting protein is pulled down only if the two proteins are able to form a stable complex. As shown in Fig. 1, Rad30 eluted together with Rev1, indicating the formation of a stable complex between Polη and Rev1 at a physiological salt concentration (Fig. 1, lanes 4 and 8). In control experiments, neither Rev1 or Polη showed any evidence of interaction with GST protein alone (data not shown).
FIG. 1.
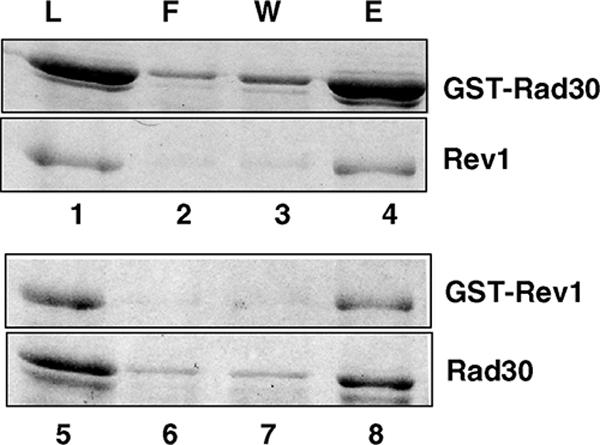
GST pull-down of Polη and Rev1 proteins. Yeast Rev1 or Rad30 (Polη) was mixed with GST-Rad30 (lanes 1 to 4) or GST-Rev1 (lanes 5 to 8), respectively. About 1 μg of each protein was used for this study. After being incubated, samples were bound to glutathione-Sepharose beads, after which they were washed and the bound proteins were eluted by SDS-sample buffer. Aliquots of each sample before being added to the beads (L) and of the flow-through fraction (F), the last washed fraction (W), and the eluted proteins (E) were analyzed on a SDS-12% polyacrylamide gel developed with silver staining.
Mapping of the regions mediating interactions between Rev1 and Polη.
To map the region of Rev1 involved in binding to Polη, the wild-type Rev1 protein and Rev1 proteins with different portions deleted were purified with the amino-terminal GST fusion from a yeast strain (Fig. 2A). The proteins were incubated with Rad30, and pull-down assays were performed on glutathione-Sepharose affinity beads (Fig. 2B). As some of the deleted Rev1 proteins are about the same molecular size as Rad30, we used Western blot analysis to detect bound Rad30 by using anti-Rad30 antibodies. As shown in Fig. 1, Rad30 bound to the wild-type Rev1 but not to GST alone (Fig. 2B, compare lanes 1 and 2). The Rev1 protein with its carboxyl-terminal 239 amino acids and amino-terminal 328 amino acids, which include the conserved BRCT domain of Rev1 (Rev1-1, aa 329 to 746), deleted (Fig. 2A) showed an interaction with Rad30 (Fig. 2B, lane 3). Further shortening of the C terminus to 640 amino acids, as in the Rev1-2 mutant, in which the polymerase-associated domain (PAD) has been truncated (Fig. 2A), abolished interaction with Rad30 (Fig. 2B, lane 4). Importantly, the Rev1 peptide which contains amino acids 567 to 767, as in Rev1-3 (Fig. 2A), was sufficient for binding Rad30 (Fig. 2B, lane 5). We verified this result from the binding of the Rev1-3 peptide to GST-Rad30 (Fig. 2C, lane 4). However, the Rev1 protein containing only the carboxyl-terminal 199 amino acids, as in Rev1-4 (Fig. 2A), did not show any binding to Rad30 (Fig. 2B, lane 6). Our results, summarized in Fig. 2A, suggest that the PAD region encompassing residues 567 to 767 of Rev1 is necessary and sufficient for interaction with Polη. Interestingly, the same region of Rev1 is also involved in binding to Rev7 (1).
FIG. 2.

Mapping of Rev1 and Polη regions involved in complex formation. (A) Schematic representation of the wild type and various mutants of Rev1 and Polη (Rad30). Based on amino acid sequence homology among the Y-family DNA polymerases, the conserved polymerase domains of yeast Rev1 (aa 1 to 985) and Rad30 (aa 1 to 632) are indicated as I and II (fingers), III and IV (palm), and V (thumb). Although not as conserved, the PAD is also shared among these polymerases. Also indicated are the carboxyl-terminal domain (CTD) and the N-terminal BRCT domain in Rev1. Following the PAD, Rad30 contains a C2H2 UBZ motif. The positions of various deletions or point mutations made in the two proteins are shown. (B) The PAD of Rev1 is necessary and sufficient for interaction with Polη. Yeast Rad30 was mixed with GST alone (lane 1), with GST-Rev1 (lane 2), or with GST-Rev1 proteins with different portions of Rev1 deleted (lanes 3 to 6). About 0.5 μg of each protein was used for this study. After being incubated, samples were bound to glutathione-Sepharose beads and extensively washed, and the bound proteins were eluted by SDS-sample buffer. Half of the eluted proteins were resolved on a SDS-12% polyacrylamide gel, and Western blot analysis was performed, using yeast anti-Polη antibodies. (C) Involvement of the Polη C terminus in mediating interactions with Rev1. Lanes 1 to 4, yeast Rev1-3 mixed with GST-Rad30; lanes 5 to 16, Rev1 mixed with GST-Rad30 with different portions of Rad30 deleted; lanes 17 to 20, Rev1 mixed with GST-Rad30 carrying the HH568,572AA mutations. After being incubated, samples were bound to glutathione-Sepharose beads and washed, and the bound proteins were eluted by SDS-sample buffer. Aliquots of each sample before being added to the beads (L) and of the flow-through fraction (F), the last washing fraction (W), and the eluted proteins (E) were analyzed on a SDS-12% polyacrylamide gel and developed with silver staining.
To map the region of Polη involved in binding to Rev1, a number of deletion and site-directed Polη mutants were generated in fusion with amino-terminal GST and pull-down assays were performed (Fig. 2A and C). Whereas the deletion of 7 amino acids from the C terminus, as in Rad30-1, had no effect on Rev1 binding, deletion of 54 amino acids from the C terminus of Polη, as in Rad30-2 (Fig. 2A), abolished the interaction with Rev1 (Fig. 2C, compare lanes 8 and 12). As the truncation in Rad30-2 is in close proximity to the C2H2 UBZ motif (6), we examined whether mutations of the two conserved histidines in this motif to alanines (HH568,572AA) affected Polη interaction with Rev1. This change in Rad30-4 (Fig. 2A), however, did not affect the Polη interaction with Rev1 (Fig. 2C, lane 20). More importantly, we found that the carboxyl-terminal peptide of 122 amino acids, as in Rad30-3, was sufficient for the interaction with Rev1 (Fig. 2B, lane 16).
Purification of various Rev1-Polη complexes.
To determine if complex formation affected the polymerase activity of either Rev1 or Polη, we generated catalytically inactive mutants of Rev1 and Polη by mutating the conserved active site residues Asp467 and Glu468 of Rev1 and Asp155 and Glu156 of Polη, present in their highly conserved motif III, to alanines (9, 14), and their N-terminal GST fusion constructs were made. To purify the different Rev1-Polη complexes, purified native Polη was added to the GST-fused catalytically inactive mutant of Rev1 (Rev1*) bound to glutathionine beads and the complex (Rev1*-Polη) was eluted by treatment with PreScission protease, which cleaved the GST attached to Rev1. Using a similar approach, the catalytically inactive Polη (Polη*) with native Rev1 was purified (Rev1-Polη* complex). A complex of Rev1 and the carboxyl-terminal polypeptide of 122 amino acids derived from Polη (Rad30-3) was also purified in a similar way.
DNA polymerase activities of Rev1 and Polη in the Rev1-Polη complexes.
To compare the DNA synthetic activity of Rev1, Polη, and Rev1-Polη complexes, we carried out DNA polymerase reactions using the same concentration of proteins (0.2 nM) and the undamaged or damaged DNA templates in the presence of individual dNTPs. The 5′, 32P-labeled, 32-nt primer was annealed to the linear 52-nt template oligomer containing an undamaged G or A residue or an abasic site at the template position, and insertion of a dGTP, dATP, dCTP, or dTTP opposite these templates was examined in a standing-start reaction (Fig. 3). Whereas opposite template G Rev1 shows incorporation of nucleotides other than a C, opposite templates A and an abasic site there is little incorporation of nucleotides other than a C (Fig. 3, compare lane 5 with lanes 2, 3, and 4 in panels i, ii, and iii). Polη incorporates a C opposite template G (Fig. 3, panel i, lane 9), a T opposite template A (Fig. 3, panel ii, lane 8), and a G and an A opposite the abasic site (Fig. 3, panel iii, lanes 6 and 7). Interestingly, the ability of Rev1 to incorporate dNTP is enhanced when it is in a complex with the catalytically inactive Polη. Thus, opposite template G, Rev1 shows an increased incorporation of each nucleotide when it is in the Rev1-Polη* complex compared to when it is used alone (Fig. 3, panel i, compare lanes 10 to 13 with lanes 2 to 5). Opposite template A, there is more incorporation of C when it is in the complex (Fig. 3, panel ii, compare lanes 13 and 5), and opposite an abasic site, incorporation of C is also increased in the complex (Fig. 3, panel iii, compare lanes 13 and 5). By contrast, we did not detect any significant change in Polη activity in the Rev1*-Polη complex (Fig. 3, compare lanes 14, 15, 16, and 17 with lanes 6, 7, 8, and 9 in panels i, ii, and iii).
Kinetic analyses of nucleotide incorporation efficiency by Rev1 in the Rev1-Polη complex.
To further analyze the effects of Rev1-Polη complex formation on Rev1 activity, we compared the steady-state kinetic parameters Km and kcat for nucleotide incorporation by Rev1 when it is alone and when it is in the Rev1-Polη* complex. The kinetics of insertion of dCTP opposite an undamaged A or G template and opposite an abasic site and of insertion of a dGTP, dATP, or dTTP opposite an undamaged G were determined in a standing-start reaction as a function of the deoxyribonucleotide concentration under steady-state conditions. From the kinetics of deoxyribonucleotide incorporation, the apparent steady-state Km and kcat values were obtained from the curve fitted to the Michaelis-Menten equation. These Km and kcat values and the efficiencies of single-nucleotide incorporation (kcat /Km) for Rev1 when it is alone and in the Rev1-Polη* complex are summarized in Table 1.
TABLE 1.
Steady-state kinetic analyses of nucleotide incorporation opposite undamaged templates A and G and opposite an abasic site by the Rev1 and Rev1-Polη* complexes
| Template nucleotide | dNTP added | Enzymeb | kcat (min−1) | Km (μM) | kcat /Km (min−1 μM−1) | Relative efficiency |
|---|---|---|---|---|---|---|
| A | dCTP | Rev1 | 1.02 ± 0.08 | 6.08 ± 1.4 | 0.17 | 1 |
| Rev1-Polη* | 4.1 ± 0.34 | 5.83 ± 1.3 | 0.7 | 4.1 | ||
| APa | dCTP | Rev1 | 1.87 ± 0.09 | 0.81 ± 0.2 | 2.3 | 1 |
| Rev1-Polη* | 8.24 ± 0.54 | 1.06 ± 0.3 | 7.77 | 3.4 | ||
| G | dGTP | Rev1 | 4.31 ± 0.4 | 12 ± 2.4 | 0.36 | 1 |
| Rev1-Polη* | 15.26 ± 0.8 | 7.6 ± 1 | 2 | 5.6 | ||
| G | dATP | Rev1 | 1.88 ± 0.3 | 192.6 ± 31 | 0.01 | 1 |
| Rev1-Polη* | 7.98 ± 0.9 | 186 ± 17.2 | 0.043 | 4.3 | ||
| G | dTTP | Rev1 | 2.45 ± 0.2 | 94.5 ± 24 | 0.026 | 1 |
| Rev1-Polη* | 8.65 ± 1.1 | 45.4 ± 11 | 0.19 | 7.3 | ||
| G | dCTP | Rev1 | 3.16 ± 0.24 | 0.045 ± 0.002 | 70.2 | 1 |
| Rev1-Polη* | 13.71 ± 0.4 | 0.032 ± 0.005 | 428 | 6.1 |
As indicated by the Km and kcat values opposite template G, the efficiency of C incorporation by Rev1 is increased approximately sixfold in the Rev1-Polη* complex compared to that by Rev1 alone, and this effect is mostly due to an increase in the kcat value. The incorporation of the incorrect nucleotide G, A, or T opposite template G is also increased approximately four- to sevenfold in the complex, and this increase, too, is primarily from the increased kcat value. The efficiency of C incorporation by Rev1 opposite template A and an abasic site are also enhanced in the complex, and this also results from an increase in the kcat value.
Stimulation of Rev1 activity by the Polη peptide containing residues 510 to 632.
Having established a stimulatory effect of full-length Polη on the enzymatic activity of Rev1, we next asked whether the 122-amino-acid-long C-terminal peptide of Polη that mediates the interaction between Rev1 and Polη is sufficient to stimulate Rev1 activity or whether other regions of Polη are also required. For these studies, we used not only the prepurified complex of Rev1-Rad30-3, but to ensure identical Rev1 concentrations, we also made the complex by adding purified Rad30-3 peptide directly to purified Rev1 in a 2:1 molar ratio. After preincubation, we compared its activity to that of Rev1 alone. The addition of the Polη peptide containing residues 510 to 632 was stimulatory to Rev1 activity opposite both the undamaged G template and the abasic site template (Fig. 4). The steady-state kinetic values measured for Rev1 alone and for Rev1 in complex with the Rad30-3 peptide are summarized in Table 2. Regardless of whether we used the preincubated mixture of Rev1 and the Rad30-3 peptide or the prepurified Rev1-Rad30-3 complex, we observed a similar stimulatory effect of the peptide on Rev1 activity. In the presence of the Rad30-3 peptide, the efficiency of C incorporation opposite both template G and the abasic site was increased approximately fourfold, and this increase in efficiency also resulted from an increase in the kcat (Table 2).
TABLE 2.
Stimulation of Rev1 activity by the Polη peptide encompassing residues 510 to 632 (Rad30-3)
| Template nucleotide | dNTP added | Enzyme | kcat (min−1) | Km (μM−1) | kca t /Km (min−1/μM−1) | Relative efficiency |
|---|---|---|---|---|---|---|
| G | dCTP | Rev1 | 1.26 ± 0.2 | 0.048 ± 0.01 | 26.3 | 1 |
| Rev1 + Rad30-3 | 3.94 ± 0.3 | 0.039 ± 0.006 | 101 | 3.84 | ||
| Apa | dCTP | Rev1 | 0.98 ± 0.05 | 1.15 ± 0.2 | 0.85 | 1 |
| Rev1 + Rad30-3 | 3.44 ± 0.3 | 0.87 ± 0.08 | 3.85 | 4.65 |
Inhibition of Rev1-Polη complex formation by Rev7.
Rev7 is an accessory but essential subunit of Polζ. Rev7 has also been shown to bind directly to Rev1, and a stable Rev1-Rev7 complex can be purified from yeast cells (1). Previously, we have shown that unlike Rev1, the Rev1-Rev7 complex does not bind Rev3; thus, the presence of Rev7 in the Rev1-Rev7 complex is inhibitory to the physical interaction of Rev1 with Rev3 in the Polζ (Rev3-Rev7) complex (2). Since the same region of the Rev1 PAD encompassing residues 567 to 767 is involved in the binding of Rev7 (1) as well as the binding of Rad30, we examined whether the binding of Rad30 by Rev1 is inhibited in the Rev1-Rev7 complex. As shown in Fig. 5, when the purified Rev1-Rev7 complex was mixed and incubated with GST-Rad30 and the proteins bound to glutathione-Sepharose beads were washed and eluted, there was no evidence of Rev1 binding to Rad30. The Rev1-Rev7 complex thus also precludes the binding of Rev1 to Polη.
FIG. 5.

Inhibition of Rev1-Polη complex formation by Rev7. A purified Rev1-Rev7 complex was mixed with GST-Rad30 (lanes 1 to 4). About 1 μg of each protein was used for the study. After being incubated, samples were bound to glutathione-Sepharose beads and washed, and the bound proteins were eluted by SDS-sample buffer. Aliquots of each sample before being added to the beads (L) and of the flow-through fraction (F), the last washing fraction (W), and the eluted proteins (E) were analyzed on an SDS-12% polyacrylamide gel developed with silver stain.
DISCUSSION
Here we provide evidence for the direct physical interaction of yeast Rev1 with Polη and show that the Rev1 PAD and the Polη C terminus mediate this interaction. In yeast, Rev1 also binds to the Rev3 subunit of Polζ and this interaction is inhibited in Rev1 with the C-terminal 72 residues deleted (2). Yeast Rev1 has also been shown to bind the Rev7 protein, and this interaction requires the same PAD region of Rev1 as is involved in Polη binding (1). The physical interaction of Rev1 with Polη does not occur in the Rev1-Rev7 complex. This observation, together with our previous result that Rev1 interaction with Rev3 is inhibited in the Rev1-Rev7 complex, implicates Rev7 binding as inhibitory to the ability of Rev1 to form a complex with Polζ or Polη.
In yeast, Rev1 plays an indispensable structural role in Polζ-dependent TLS. Also, the fact that the deletion of the C-terminal 72 residues of Rev1, which inactivates the interaction with Rev3, elicits the same degree of UV sensitivity and defectiveness in UV mutagenesis as that conferred by the _rev1_Δ or _rev3_Δ mutations indicates that Rev1 binding is essential for Polζ function (2). It is not known, however, how Rev1 binding affects the targeting of Polζ to the stalled replication fork. Rev1 could accomplish this either as a component of the multisubunit Polζ complex or as part of a different multiprotein complex that assembles with the stalled replication machinery. Polζ, as well as the other TLS Pols such as Polη, might then access the stalled replication fork through binding of Rev1 in such a multiprotein assembly. However, if Rev1 were to function as a structural element in the targeting not only of Polζ but also of Polη to the replication ensemble, the lack of Rev1's requirement for Polη function in yeast becomes rather difficult to explain.
In yeast, TLS through UV lesions requires only the structural role of Rev1: its DNA polymerase activity is not required (24, 28). For a number of other DNA lesions, however, Rev1 could function in TLS not only as a structural element but also as a DNA polymerase. Because the templating G and the incoming dCTP do not pair with each other in the Rev1 active site and the templating G is evicted from the DNA helix (23), Rev1 could proficiently incorporate a C opposite a variety of adducts attached to the _N_2 group of guanine, following which extension by Polζ (33) or another polymerase would complete the lesion bypass process. Thus, Rev1 would function in TLS not only as a structural element for Polζ but also as a DNA polymerase. In the latter role, however, Rev1 could at times function not only independently of Polζ but also in unison with Polη, where, following the dCTP insertion by Rev1 opposite a lesion site, Polη could carry out the extension reaction.
Although both yeast and human Rev1 show evidence of physical interactions with Polη and Rev7 in yeast and with Polη, Polι, Polκ, and Rev7 in humans, the region of Rev1 involved in these interactions differs between yeast and humans. Whereas in yeast the region adjoining the PAD modulates these interactions, in humans the C-terminal ∼100 residues of Rev1 are involved in these interactions. The physical interaction of yeast Rev1 with the Rev3 subunit of Polζ, however, requires the region encompassing the C-terminal ∼72 residues of Rev1. Whether human Rev1 also directly interacts with Rev3 and thereby targets Polζ to the replication ensemble remains to be determined. Nevertheless, since in yeast the Rev1 C terminus promotes its interaction with Rev3 in Polζ whereas in humans the Rev1 C terminus is involved in interactions with Polη, Polι, or Polκ, there is a possibility that in its structural role, Rev1 has diverged, at least in some ways, between yeast and humans. However, as far as the role of Rev1 as a DNA polymerase is concerned, we expect both yeast and human Rev1 to function similarly in promoting efficient and error-free TLS opposite a variety of _N_2 minor-groove adducts of guanine (23).
Interestingly, although yeast Rev7 and Polη bind the same region (PAD) of Rev1, their binding elicits very different effects on Rev1 catalytic activity. Whereas the binding of Rev7 has no effect on Rev1 activity (1), the binding of Polη stimulates Rev1 activity. Also, we found that a Polη peptide encompassing residues 510 to 632 and which can bind Rev1 is almost as stimulatory to Rev1 activity as the full-length Polη. The different effects of Rev7 and Polη on Rev1 activity suggest that they bind different regions of the Rev1 PAD. Rev7 could bind the distal portion of the Rev1 PAD (α-helices P and Q), which is away from the template-primer and the incoming nucleotide, whereas Polη could bind the more-proximal region of the Rev1 PAD that faces the template-primer and the incoming nucleotide (Fig. 6).
FIG. 6.
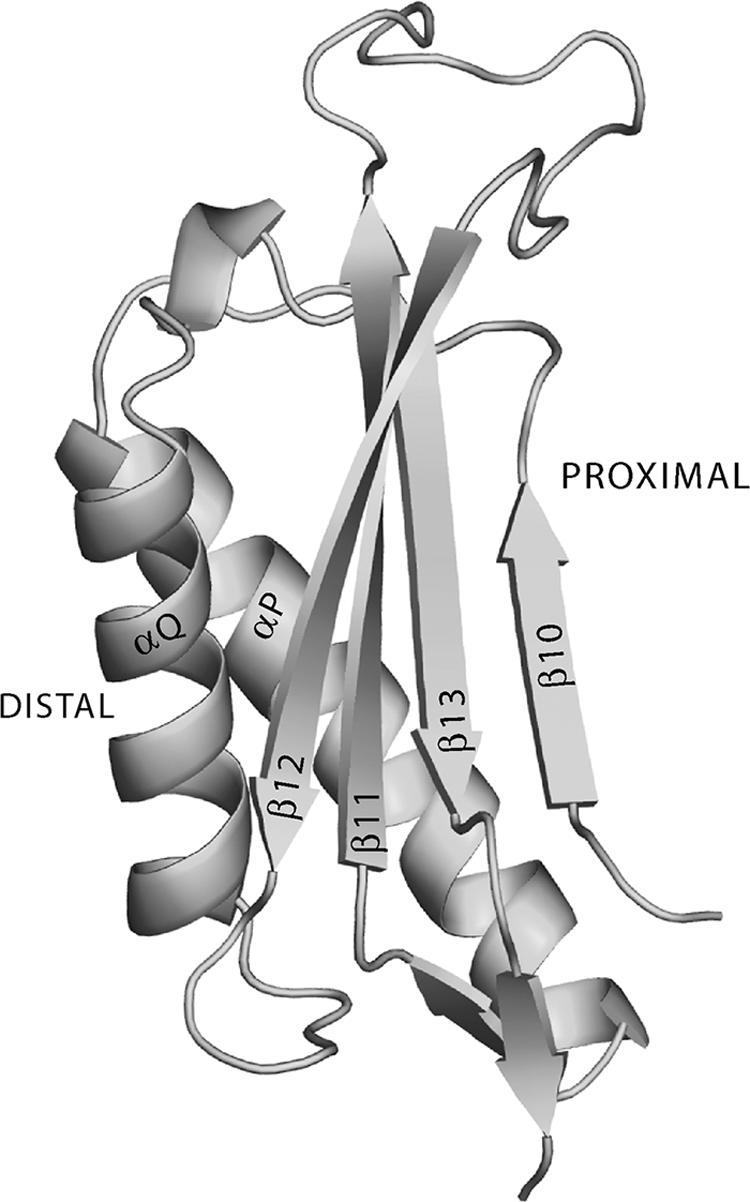
Secondary structural elements in the Rev1 PAD. The distal region of the Rev1 PAD could mediate its binding to Rev7, whereas the proximal region could be involved in interactions with Polη.
The available evidence does not allow us to unambiguously determine whether Polη's binding to the Rev1 PAD region reflects Rev1's structural role or Rev1's role in modulating TLS as a DNA polymerase. However, since Rev1 is not required for Polη function in yeast and since the Rev1 DNA synthetic activity shows an increase in the Rev1-Polη complex, this interaction is more likely to be indicative of Rev1's role as a DNA polymerase than of its role as a structural element. It is conceivable that a protein complex that combines the two TLS polymerases affords a more-efficient strategy for promoting TLS through lesions that require the consecutive action of two different polymerases. For example, the Rev1-Polη complex could be more effective in promoting lesion bypass opposite _N_2-guanine adducts, such as the _N_2-dG butadiene adducts, where, following insertion of C opposite the lesion site by Rev1, the subsequent extension could be performed by Polη (22).
Whether the complex formation of human Rev1 with Polη, Polι, or Polκ also provides for a more-efficient mode of lesion bypass, as suggested above for the yeast Rev1-Polη complex, or whether human Rev1 plays an indispensable role in the targeting of these Y-family polymerases to the replication fork remains to be established. However, the requirement of different Rev1 regions for the binding of Polη in yeast versus humans and the observation that whereas in yeast the Rev1 C terminus mediates the interaction with Polζ, in humans the Rev1 C terminus modulates interactions with Polη and other Y-family polymerases suggests that in its structural role Rev1 has diverged between yeast and humans.
Acknowledgments
This work was supported by National Institutes of Health grant CA107650.
Footnotes
▿
Published ahead of print on 17 September 2007.
REFERENCES
- 1.Acharya, N., L. Haracksa, R. E. Johnson, I. Unk, S. Prakash, and L. Prakash. 2005. Complex formation of yeast Rev1 and Rev7 proteins: a novel role for the polymerase-associated domain. Mol. Cell. Biol. 25**:**9734-9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acharya, N., R. E. Johnson, S. Prakash, and L. Prakash. 2006. Complex formation with Rev1 enhances the proficiency of yeast DNA polymerase ζ for mismatch extension and for extension opposite from DNA lesions. Mol. Cell. Biol. 26**:**9555-9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailly, V., J. Lamb, P. Sung, S. Prakash, and L. Prakash. 1994. Specific complex formation between yeast RAD6 and RAD18 proteins: a potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev. 8**:**811-820. [DOI] [PubMed] [Google Scholar]
- 4.Bailly, V., S. Lauder, S. Prakash, and L. Prakash. 1997. Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 272**:**23360-23365. [DOI] [PubMed] [Google Scholar]
- 5.Baynton, K., A. Bresson-Roy, and R. P. P. Fuchs. 1999. Distinct roles for Rev1p and Rev7p during translesion synthesis in Saccharomyces cerevisiae. Mol. Microbiol. 34**:**124-133. [DOI] [PubMed] [Google Scholar]
- 6.Bienko, M., C. M. Green, N. Crosetto, F. Rudolf, G. Zapart, B. Coull, P. Kannouche, G. Wider, M. Peter, A. R. Lehmann, K. Hofmann, and I. Dikic. 2005. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310**:**1821-1824. [DOI] [PubMed] [Google Scholar]
- 7.Guo, C., P. L. Fischhaber, M. J. Luk-Paszyc, Y. Masuda, J. Zhou, K. Kamiya, C. Kisker, and E. C. Friedberg. 2003. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 22**:**6621-6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haracska, L., S. Prakash, and L. Prakash. 2002. Yeast Rev1 protein is a G template-specific DNA polymerase. J. Biol. Chem. 277**:**15546-15551. [DOI] [PubMed] [Google Scholar]
- 9.Haracska, L., I. Unk, R. E. Johnson, E. Johansson, P. M. J. Burgers, S. Prakash, and L. Prakash. 2001. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 15**:**945-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson, R. E., L. Haracska, S. Prakash, and L. Prakash. 2001. Role of DNA polymerase η in the bypass of a (6-4) TT photoproduct. Mol. Cell. Biol. 21**:**3558-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson, R. E., C. M. Kondratick, S. Prakash, and L. Prakash. 1999. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285**:**263-265. [DOI] [PubMed] [Google Scholar]
- 12.Johnson, R. E., L. Prakash, and S. Prakash. 2006. Yeast and human translesion DNA synthesis polymerases: expression, purification, and biochemical characterization. Methods Enzymol. 408**:**390-407. [DOI] [PubMed] [Google Scholar]
- 13.Johnson, R. E., S. Prakash, and L. Prakash. 1999. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 283**:**1001-1004. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, R. E., S. Prakash, and L. Prakash. 1999. Requirement of DNA polymerase activity of yeast Rad30 protein for its biological function. J. Biol. Chem. 274**:**15975-15977. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, R. E., M. T. Washington, L. Haracska, S. Prakash, and L. Prakash. 2000. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406**:**1015-1019. [DOI] [PubMed] [Google Scholar]
- 16.Lawrence, C. W., and R. B. Christensen. 1979. Ultraviolet-induced reversion of cyc1 alleles in radiation-sensitive strains of yeast. III. rev3 mutant strains. Genetics 92**:**397-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence, C. W., and R. B. Christensen. 1978. Ultraviolet-induced reversion of cyc1 alleles in radiation-sensitive strains of yeast. I. rev1 mutant strains. J. Mol. Biol. 122**:**1-21. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence, C. W., P. E. Nisson, and R. B. Christensen. 1985. UV and chemical mutagenesis in rev7 mutants of yeast. Mol. Gen. Genet. 200**:**86-91. [DOI] [PubMed] [Google Scholar]
- 19.Lawrence, C. W., T. O'Brien, and J. Bond. 1984. UV-induced reversion of his4 frameshift mutations in r_ad6_, rev1, and rev3 mutants of yeast. Mol. Gen. Genet. 195**:**487-490. [DOI] [PubMed] [Google Scholar]
- 20.Masutani, C., R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio, and F. Hanaoka. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399**:**700-704. [DOI] [PubMed] [Google Scholar]
- 21.McDonald, J. P., A. S. Levine, and R. Woodgate. 1997. The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics 147**:**1557-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minko, I. G., M. T. Washington, L. Prakash, S. Prakash, and R. S. Lloyd. 2001. Translesion DNA synthesis by yeast DNA polymerase η on templates containing _N_2-guanine adducts of 1,3-butadiene metabolites. J. Biol. Chem. 276**:**2517-2522. [DOI] [PubMed] [Google Scholar]
- 23.Nair, D. T., R. E. Johnson, L. Prakash, S. Prakash, and A. K. Aggarwal. 2005. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309**:**2219-2222. [DOI] [PubMed] [Google Scholar]
- 24.Nelson, J. R., P. E. M. Gibbs, A. M. Nowicka, D. C. Hinkle, and C. W. Lawrence. 2000. Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol. 37**:**549-554. [DOI] [PubMed] [Google Scholar]
- 25.Nelson, J. R., C. W. Lawrence, and D. C. Hinkle. 1996. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 382**:**729-731. [DOI] [PubMed] [Google Scholar]
- 26.Nelson, J. R., C. W. Lawrence, and D. C. Hinkle. 1996. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 272**:**1646-1649. [DOI] [PubMed] [Google Scholar]
- 27.Ohashi, E., Y. Murakumo, N. Kanjo, J.-I. Akagi, C. Masutani, F. Hanaoka, and H. Ohmori. 2004. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 9**:**523-531. [DOI] [PubMed] [Google Scholar]
- 28.Prakash, S., R. E. Johnson, and L. Prakash. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74**:**317-353. [DOI] [PubMed] [Google Scholar]
- 29.Prakash, S., and L. Prakash. 2002. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 16**:**1872-1883. [DOI] [PubMed] [Google Scholar]
- 30.Stary, A., P. Kannouche, A. R. Lehmann, and A. Sarasin. 2003. Role of DNA polymerase η in the UV mutation spectrum in human cells. J. Biol. Chem. 278**:**18767-18775. [DOI] [PubMed] [Google Scholar]
- 31.Tissier, A., P. Kannouche, M.-P. Reck, A. R. Lehmann, R. P. P. Fuchs, and A. Cordonnier. 2004. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REV1 protein. DNA Repair 3**:**1503-1514. [DOI] [PubMed] [Google Scholar]
- 32.Torres-Ramos, C., S. Prakash, and L. Prakash. 2002. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 22**:**2419-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Washington, M. T., I. G. Minko, R. E. Johnson, L. Haracska, T. M. Harris, R. S. Lloyd, S. Prakash, and L. Prakash. 2004. Efficient and error-free replication past a minor-groove _N_2-guanine adduct by the sequential action of yeast Rev1 and DNA polymerase ζ. Mol. Cell. Biol. 24**:**6900-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu, S.-L., R. E. Johnson, S. Prakash, and L. Prakash. 2001. Requirement of DNA polymerase η for error-free bypass of UV-induced CC and TC photoproducts. Mol. Cell. Biol. 21**:**185-188. [DOI] [PMC free article] [PubMed] [Google Scholar]