The Function of Mitochondria in Presynaptic Development at the Neuromuscular Junction (original) (raw)
Abstract
Mitochondria with high membrane potential (ΔΨm) are enriched in the presynaptic nerve terminal at vertebrate neuromuscular junctions, but the exact function of these localized synaptic mitochondria remains unclear. Here, we investigated the correlation between mitochondrial ΔΨm and the development of synaptic specializations. Using mitochondrial ΔΨm-sensitive probe JC-1, we found that ΔΨm in Xenopus spinal neurons could be reversibly elevated by creatine and suppressed by FCCP. Along naïve neurites, preexisting synaptic vesicle (SV) clusters were positively correlated with mitochondrial ΔΨm, suggesting a potential regulatory role of mitochondrial activity in synaptogenesis. Indicating a specific role of mitochondrial activity in presynaptic development, mitochondrial ATP synthase inhibitor oligomycin, but not mitochondrial Na+/Ca2+ exchanger inhibitor CGP-37157, inhibited the clustering of SVs induced by growth factor–coated beads. Local F-actin assembly induced along spinal neurites by beads was suppressed by FCCP or oligomycin. Our results suggest that a key role of presynaptic mitochondria is to provide ATP for the assembly of actin cytoskeleton involved in the assembly of the presynaptic specialization including the clustering of SVs and mitochondria themselves.
INTRODUCTION
Mitochondria play multiple roles in cellular functions including cellular metabolism, calcium homeostasis, and programmed cell death. At both peripheral and central synapses, mitochondria are concentrated within the presynaptic nerve terminal as demonstrated by electron microscopic studies and they are presumably related to the transmitter release machinery (Herrera et al., 1985; Robitaille and Tremblay, 1987; Peters et al., 1991). Our previous study found that local synaptogenic signals induce the clustering of not only synaptic vesicles (SVs; Dai and Peng, 1995), but also mitochondria (Lee and Peng, 2006), thus suggesting that the clustering of this organelle is an integral part of presynaptic differentiation. The function of mitochondria at mature neuromuscular junctions (NMJs) has recently been explored. For example, the transmitter release under intense stimulation fails in a Drosophila NMJ mutant lacking presynaptic mitochondria (Guo et al., 2005; Verstreken et al., 2005). The calcium release from mitochondria has been shown to modulate synaptic potentiation at the vertebrate NMJ (Yang et al., 2003). However, the function of presynaptic mitochondria in the development of the NMJ is not yet known.
Within the axon, mitochondria undergo bidirectional movement and accumulate at sites of high metabolic activity (Hollenbeck, 1996). The direction of mitochondrial transport is correlated with axonal outgrowth (Morris and Hollenbeck, 1993) and its activity (Miller and Sheetz, 2004), reflected by the electrical potential (ΔΨm) across the inner membrane produced by oxidative phosphorylation-mediated electrochemical equilibrium of H+ ions. Previously, we found that mitochondria with high ΔΨm are predominantly localized at the presynaptic specialization induced by either muscle cells or growth factor–coated beads (Lee and Peng, 2006) and a recent study showed that modulation of dendritic mitochondrial ΔΨm regulates the activity-dependent synaptic plasticity in hippocampal synapses (Li et al., 2004). In this study, we examined whether mitochondrial activity as indicated by ΔΨm has a direct bearing on presynaptic development. As ΔΨm is correlated with ATP production in neuronal cells (Nguyen et al., 1997), we sought to understand the link between local ATP supply due to mitochondrial clustering and presynaptic development. Here we report an essential role of ATP production from localized synaptic mitochondria in the assembly of F-actin cytoskeleton that is involved in the clustering of synaptic vesicles (SVs) and mitochondria themselves at developing presynaptic specialization.
MATERIALS AND METHODS
Materials
MitoTracker red, JC-1, magnesium green (MgGr), jasplakinolide, and fluorescein phalloidin were obtained from Molecular Probes (Eugene, OR). Creatine, FCCP (carbonyl cyanide _p_-[trifluoromethoxy]-phenyl-hydrazone), and oligomycin were obtained from Sigma (St. Louis, MO). CGP-37157 was obtained from Calbiochem (La Jolla, CA).
Xenopus Primary Cultures and Live Cell Staining
Spinal neurons were cultured from Xenopus embryos as previously described (Peng et al., 1991). In short, neural tubes of embryos at stage 19–22 were isolated and dissociated in a Ca2+,Mg2+-free solution. Dissociated neurons were plated on glass coverslips coated with the cell attachment matrix enhanced chemiluminescence (ECL; entactin, collagen IV and laminin; Upstate Biotechnology, Waltham, MA).
To study mitochondrial ΔΨm, cultured neurons were stained with 0.25 μg/ml JC-1 for 10 min, followed by washing twice with culture medium [60 mM NaCl, 0.7 mM KCl, 0.4 mM Ca(NO3)2, 0.8 mM MgSO4, 10 mM HEPES, 10% L-15, 1% fetal bovine serum, and 0.1 mg/ml gentamicin]. The fluorescence intensity at emission wavelengths of 525 and 590 nm was measured for ratiometric analysis (Smiley et al., 1991).
To visualize the distribution of mitochondria, we used a fixable mitochondrion-selective marker MitoTracker, which is a cell-permeant probe that passively diffuses across membranes and accumulates in active mitochondria (Poot et al., 1996). Cultured neurons were stained for 5 min with 5 nM MitoTracker in culture medium. Labeled cells were washed twice with culture medium before fixation.
To study intracellular ATP level, we incubated the neurons with 10 μM MgGr-AM ester for 30 min. Emission intensity of this probe increases as a function of free intracellular Mg2+, but decreases with ATP content because of the high binding affinity of this probe to ATP than to ADP (Leyssens et al., 1996; Bernstein and Bamburg, 2003). Labeled neurons were washed and incubated in culture medium twice for 15 min each to maximize de-esterification of the probe before pharmacological treatments.
Induction of Presynaptic Specializations by Beads
To induce focal presynaptic differentiation, we stimulated cultured spinal neurons with beads coated with basic fibroblast growth factor (bFGF). Polystyrene latex beads 4 μm in diameter (Polysciences, Warrington, PA) were coated with recombinant human bFGF (R&D Systems, Minneapolis, MN) using previously described procedures (Dai and Peng, 1995). In pharmacological experiments, neurons were incubated with specific agents from 1 h before bead addition. Bead-neurite contacts were scored positive for mitochondrial and SV clustering if the mean fluorescence intensity of the corresponding marker was at least twofold over the noncontact region.
Visualization of Newly Polymerized F-Actin
The assembly of actin cytoskeleton was visualized by a previously described procedure (Dai et al., 2000). Spinal neuron cultures were pretreated with jasplakinolide at a concentration of 1 μM for 3 min and then washed with culture medium before the bead stimulation. The newly polymerized F-actin was visualized by labeling the fixed cultures with fluorescein-conjugated phalloidin for 45 min.
The sites of actin polymerization were also examined by the incorporation of rhodamine-conjugated G-actin into free barbed ends of actin filaments in saponin-permeabilized cells as described previously (Symons and Mitchison, 1991; Schafer et al., 1998). Cultured neurons were incubated with 0.45 μM rhodamine-actin (Cytoskeleton, Denver, CO) in saponin-containing buffer (20 mM HEPES, 138 mM KCl, 4 mM MgCl2, 3 mM EGTA, 40 μg/ml saponin, 1 mM ATP, and 1% bovine serum albumin [BSA]) for 45 s. The neurons were fixed immediately with 2% paraformaldehyde after labeling.
Immunocytochemical Labeling
For staining cells with antibodies or F-actin markers, neuronal cultures were fixed with 2% paraformaldehyde in PBS, followed by permeabilization with 0.5% Triton X-100. Fixed cells were blocked with phosphate-buffered saline (PBS) containing 5% BSA for 1 h and labeled with primary antibodies for 2 h followed by fluorescent secondary antibodies for 45 min (Zymed laboratories, South San Francisco, CA). To label F-actin in neurons, fixed and permeabilized cells were incubated with fluorescein-conjugated phalloidin.
Fluorescence Microscopy and Time-Lapse Imaging
All images were captured using an Olympus IX-70 inverted microscope (Melville, NY) fitted with a Hamamatsu ORCA II cooled CCD camera (Hamamatsu City, Japan). Time-lapse images were captured and processed using Sutter Lambda shutter controller (Novato, CA) and Metamorph software (Universal Imaging, West Chester, PA). The relative MgGr fluorescence intensity was measured along the entire neuritic shaft by using Metamorph software. For comparison on the fluorescence intensity between the control and the experimented groups, all of the imaging settings were kept the same, and the measured intensities were normalized against the mean of the corresponding control group. Pseudocolor images of MgGr and fluorescent phalloidin staining were processed and converted by ImageJ software (National Institutes of Health, Bethesda, MD).
Mean Squared Displacement Analysis
To quantify the movement of mitochondria, the centroid of each morphologically distinct mitochondrion was tracked by Metamorph software, and then the mean-square displacement (MSD) was calculated at each time point according to the formula (Lee et al., 1991; Dai and Peng, 1996a; Lee and Peng, 2006),
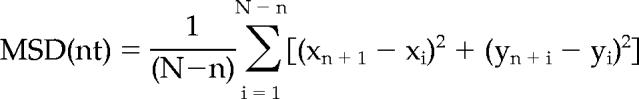
where (xn+i, yn+i) is the position of the mitochondria after a time interval of nt (t is the time interval between successive measurements) after starting at position (xi, yi). N is the total number of positions recorded; n ranges from 1 to N − 1. This analysis is typically used to describe the diffusion of particles. Although mitochondrial movement within the axon is likely mediated by transport motors along microtubules, it is usually not unidirectional and predictable at each instant. Thus, we used MSD analysis to describe this movement (Lee and Peng, 2006). Because the slope of the linear portion of the MSD plot is proportional to the particle's diffusion coefficient, it is used as a measure of the mobility of the bidirectional mitochondrial movement in this study.
RESULTS
Pharmacological Manipulation of Mitochondrial ΔΨm in Cultured Spinal Neurons
Mitochondrial ΔΨm in cultured Xenopus spinal neurons was manipulated by creatine, a known mitochondrial activity enhancer, or by FCCP, a proton ionophore that uncouples the respiratory chain from the oxidative phosphorylation (Leyssens et al., 1996). The relative ΔΨm was studied with the fluorescent probe JC-1, which exists in monomers at low ΔΨm, emitting green fluorescence at 525 nm, and in aggregates at high ΔΨm, emitting red fluorescence at 590 nm (Smiley et al., 1991; Buckman and Reynolds, 2001; Li et al., 2004). Treating 1-d-old Xenopus cultured spinal neurons with 100 μM creatine for 16 h greatly enhanced the red fluorescence of JC-1 at mitochondrial puncta along the entire neurites (Figure 1, E–H, arrows) compared with those within control neurites (Figure 1, A–D). The results were quantified by ratiometric analyses of red and green fluorescence intensity at emission wavelengths of 590 and 525 nm, respectively. As shown in Figure 1M, there was a twofold increase in this ratio after a 16-h creatine treatment. More than 80% of mitochondria reached high ΔΨm state (based on red/green fluorescence ratio larger than 1) in creatine-treated neurons, in comparison to ∼50% in the untreated neurons (Figure 1N). In contrast, a significant loss in mitochondrial ΔΨm was achieved by an acute (4 h) application of 0.5 μM FCCP (Figure 1, I–L and M). Because FCCP causes mitochondrial depolarization that releases JC-1 monomers, diffuse JC-1 green fluorescence was also observed within the axoplasm outside clusters (Figure 1K, inset). Less than 10% of mitochondria retained high ΔΨm after FCCP treatment (Figure 1N). Because long-term treatment of FCCP may initiate programmed cell death in cultured neurons (Nicholls and Budd, 2000), recovery experiments were also carried out. By washing away creatine- or FCCP-containing medium and then keeping the cultures in drug-free medium for another 4 h before JC-1 labeling, we observed that the ratiometric fluorescence intensities of JC-1 returned to a value that was not significantly different from the controls, and the ratio of mitochondrial populations with high and low ΔΨm was also restored to the control values (Supplementary Figure 1, A and B).
Figure 1.

Manipulation of mitochondrial ΔΨm by creatine and FCCP. (A–D) Cultured Xenopus spinal neurons were stained with 0.25 μg/ml JC-1 for 10 min. Live imaging of JC-1 signal showed heterogeneous populations of mitochondria with high (arrows) and low (arrowheads) ΔΨm in untreated neurons. (E–H) Treatment of 100 μM creatine for 16 h greatly enhanced the average mitochondrial ΔΨm and the population of mitochondria with higher ΔΨm (arrows). (I–L) Most of the mitochondria exhibited lower ΔΨm (arrowheads) after 0.5 μM FCCP treatment for 4 h. In addition, the signal of JC-1 monomers was less distinct and spread over the neurite (K, inset). (M) Quantification of the ratiometric analyses of JC-1 red and green fluorescence intensity in response to creatine and FCCP. Data are means ± SEM from three independent experiments; n > 80 morphologically distinct mitochondria. * p < 0.05, ** p < 0.01, t test. (N) Quantification of the populations of mitochondria with high and low ΔΨm in the cultured neurons. Mitochondria were classified as high ΔΨm, F (red/green) ≥ 1, when the fluorescence intensity at 590 nm (red) is higher than or equal to that at 525 nm (green). Mitochondria with high ΔΨm were more abundant in creatine-treated neurons, but less in FCCP-treated neurons. Data are based on 80 morphologically distinct mitochondria from three independent experiments.
Effects on Intracellular ATP Level by Mitochondrial ΔΨm Manipulation
A primary function of mitochondria is to produce ATP for various metabolic needs. Therefore, we monitored intracellular ATP content as ΔΨm was changed by creatine or FCCP with the probe magnesium green (MgGr), whose fluorescence emission correlates positively with intracellular free Mg2+ concentration. Because the affinity of Mg2+ for ATP is ∼10-fold higher than that for ADP or AMP (Leyssens et al., 1996), the fluorescence intensity emitted by MgGr is inversely proportional to the intracellular ATP content (Bernstein and Bamburg, 2003). In this set of experiments, the settings in labeling and imaging were kept constant in order to obtain semiquantitative measurements of fluorescence intensity under different conditions. As shown in Figure 2(A, B, E, and F), FCCP treatment led to a dramatic increase in MgGr fluorescence intensity along the entire length of neurite, reflecting a decrease in neuritic ATP concentration. On the other hand, creatine caused no significant change in MgGr fluorescence intensity (Figure 2, C and D). These results are quantified in Figure 2K.
Figure 2.

Influence of different mitochondrial function manipulators on intracellular ATP level. (A–J) The relative ATP content in neurons was examined by a fluorescent probe, magnesium green (MgGr), whose fluorescence intensity is inversely correlated to the intracellular ATP level. Cultured neurons were labeled with 10 μM MgGr-AM ester for 30 min, followed by fluorescence live imaging. (A, C, E, G, and I) Phase-contrast images. By comparison with the untreated neurons (B), FCCP (F), and oligomycin (H) elevated the intensity of overall MgGr fluorescence in treated neurons. In contrast, creatine (D) and CGP-37157 (J) had no significant effect on intracellular ATP level. (K) Quantification of relative MgGr fluorescence intensity along neurites (±SD from two independent experiments, n > 20 neurites. **p < 0.01, t test).
ΔΨm-dependent Mitochondrial Movement and SV Clustering
Mitochondria exhibit anterograde and retrograde movements within Xenopus neurite (Lee and Peng, 2006). To understand whether their movements are dependent on ΔΨm, creatine, or FCCP was applied to neurons under time-lapse recording after labeling with MitoTracker. The movement was quantified by calculating the mean speed of translocation and MSD analyses. As the rate of mitochondrial movement is correlated with the neurite outgrowth (Morris and Hollenbeck, 1993), only neurites with no obvious extension during the 100-s recording period were chosen for observation by fluorescence microscopy. An example of the control neurite is shown in Figure 3, A–C, and in Supplementary Video 1. We observed one “fast moving” mitochondrion with a translocation rate of >0.15 μm/s (Figure 3, A–C; Supplementary Video 1, d), six “slow-moving” mitochondria at a speed of 0.05–0.15 μm/s (c, e, j, k, l, and m) and six “stationary” mitochondria which were essentially immobile at <0.05 μm/s (a, b, f, g, h, and i). The mean diffusion coefficient calculated from the slope of the linear portion of MSD plots, as shown in Figure 3C, was 1.01 μm2/s (range: 8 × 10−4 to 4.02). In neurons treated with 100 μM creatine for 16 h, there were more mitochondria in fast- and slow-moving categories (Figure 3, D–F; Supplementary Video 2; two “fast moving”: h and k; eight “slow moving”: a, c, d, e, f, i, j, and m; and three “stationary”: b, g, and l). The mean diffusion coefficient of mitochondria in creatine-treated cultures was 2.728 μm2/s (range: 0.02–16.12). Interestingly, mitochondrial movement was largely reduced in the presence of FCCP and all mitochondria were in “stationary” category (Figure 2, G–I; Supplementary Video 3, a–k). The mean diffusion coefficient of mitochondria in FCCP-treated cultures was reduced to 0.014 μm2/s (range: 3 × 10−4 to 0.13). These results suggest that mitochondrial translocation within these neurites is ΔΨm-dependent. As directional transport of mitochondria may correlate to their ΔΨm (Miller and Sheetz, 2004), we also tested if creatine and FCCP affected anterograde and/or retrograde transport of mitochondria. Based on the above classification, anterograde- and retrograde-moving mitochondria were classified as “fast,” “slow,” or “stationary” (Table 1). Creatine treatment increased the percentage of mitochondria in fast and slow categories, but the most obvious effect was on the retrograde movement and largely reduced the percentage of mitochondria in stationary category. On the contrary, FCCP arrested the bidirectional movement of >90% mitochondria.
Figure 3.

Regulation of mitochondrial movement by their ΔΨm. Cultured neurons were treated with creatine (D–F) and FCCP (G–I) for 16 and 4 h, respectively. Mitochondria were labeled with MitoTracker (B, E, and H). (A, D, and G) The corresponding phase-contrast images. The mobility of mitochondria along the naïve neurites in the presence of different mitochondrial ΔΨm manipulators was followed by time-lapse recording for 100 s. The corresponding mitochondria, marked with a–m (A–F) or a–k (G–I), were analyzed by MSD in which the diffusion coefficients of the mitochondria were reflected by their slope. The directionality of each mitochondrion's movement is indicated along its MSD plot. (C, F, and I), *, anterograde; #, retrograde; and unmarked for stationary. Time-lapse video clips with a 5-s interval between adjacent frames are available in Supplementary Videos 1–3.
Table 1.
Effects of mitochondrial ΔΨm manipulators on bidirectional mitochondrial movements
| Treatment | Anterograde | Retrograde | Stationary | ||
|---|---|---|---|---|---|
| Fast | Slow | Fast | Slow | ||
| Control | 13.3 | 23.3 | 5 | 5 | 53.3 |
| Creatine | 15 | 28.3 | 11.7 | 15 | 30 |
| FCCP | 0 | 1.7 | 3.3 | 1.7 | 93.3 |
| Oligomycin | 10 | 10 | 2.5 | 17.5 | 60 |
Apart from the effects on mitochondrial movement, we also investigated whether ΔΨm affected presynaptic development by examining SV clustering in naïve neurite. Previous studies have shown that SVs form clusters that are capable of activity-induced transmitter release along the axon before target contact (Bixby and Reichardt, 1985; Dai and Peng, 1996b). As shown in Figure 4A, discrete SV clusters were observed by immunolabeling with an antibody against the SV-associated protein synapsin-1 (Benfenati et al., 1991). Creatine treatment (at 100 μM for 16 h) significantly increased the number of SV clusters along the axon, whereas FCCP (at 0.5 μM for 4 h) gave the opposite effect (Figures 4, B and C). These observations were quantified by measuring the area occupied by SV clusters per unit axonal length as shown in Figure 4D. Not only did creatine increase the number and area of SV clustering, it also enhanced the synapsin labeling intensity (Figure 4E), suggesting an increase in SV density within these clusters. In contrast, FCCP caused a decrease in both the extent and intensity of SV clustering (Figure 4, D and E). After treated cultures were returned to normal medium for 4 h, the extent of SV clustering became the same as that seen in untreated cultures (Supplementary Figure 1, C and D). Thus, SV cluster formation could be reversibly regulated by ΔΨm in naïve neurites.
Figure 4.

Mitochondrial ΔΨm-dependent clustering of SVs along the naïve neurites. (A–C) The cultured neurons were treated with creatine or FCCP for 4 or 16 h. They were then fixed and labeled with a SV marker, synapsin-1. More SV clusters were found along neurite in cultures treated with 100 μM creatine for 16 h (arrows in B) than in untreated cultures (arrows in A). In 4- and 16-h time points, fewer SV clusters were detected in 0.5 μM FCCP-treated cultures (arrow in C). (D and E) Area and intensity of SV clusters were measured after background subtraction by ImageJ software. Quantification of the area of SV clusters per unit length of neurite (D) and normalized fluorescence intensity of SV clusters (E). Means from three independent experiments are shown; n >30 neurites. *p < 0.05, t test.
Correlation of Mitochondrial ΔΨm and Bead-induced Presynaptic Differentiation
Our previous study showed that the formation of mitochondrial and SV clusters can be focally induced by growth factor–coated beads (Lee and Peng, 2006). In this study, we used this model to understand whether these specializations associated with presynaptic development are dependent on mitochondrial function. After a short synaptic induction by bFGF beads for 30 min, an increase in MgGr fluorescence intensity was locally detected at bead-neurite contacts and this was associated with mitochondrial clusters near these sites (Figure 5). This local reduction in ATP may reflect its great demand due to processes underlying presynaptic development. Next, we tested if mitochondrial ΔΨm manipulation affects presynaptic development induced by bFGF beads. Enhancing ΔΨm by creatine had no effect on SV or mitochondrial clustering induced by beads (Figure 6, D–F compared with control A–C). However, abolishing ΔΨm by FCCP greatly suppressed the formation of bead-induced mitochondrial clusters (Figure 6I), consistent with the inability of this organelle to move along the neurite as described above. Local SV clustering at the bead-neurite contact was also inhibited by FCCP (Figure 6, G and H).
Figure 5.

Local ATP depletion at bFGF bead-induced presynaptic specializations. Cultured spinal neurons were labeled sequentially with MitoTracker and MgGr and then were stimulated with bFGF-coated beads for 30 min. To clearly show the intensity difference, original fluorescence image of MgGr staining (B) was converted to pseudocolor image by ImageJ software (C). bFGF beads (asterisks in A) caused a dramatic decrease in ATP level at bead contact sites where presynaptic specializations developed, as evidenced by an increase in MgGr intensity (arrows in C). These sites were also enriched in mitochondria (arrows in D).
Figure 6.

Mitochondrial ΔΨm and presynaptic differentiation induced by beads. (A–I) MitoTracker-loaded neurons were treated with either 100 μM creatine or 0.5 μM FCCP at 1 h before bead stimulation and being kept in the medium throughout the experiment. After bead stimulation for 4 h, the neurons were fixed and labeled with synapsin-1 antibody. bFGF beads induced the clustering of SVs and mitochondria in both control (arrows in A–C) and creatine-treated cultures (arrows in D–F). However, in FCCP-treated cultures, neither SVs nor mitochondria were clustered at bead-neurite contacts (arrowheads in G–I). (J) Quantification of the presynaptic differentiation. Bead-neurite contacts were scored positive for mitochondrial and SV clustering if the mean fluorescence intensity of the corresponding marker was at least twofold over the noncontact region. Data are means ± SEM from three independent experiments; n > 150 bead-neurite contacts. * p < 0.05, ** p < 0.01, t test.
To further understand the role of ATP production on presynaptic development, we examined the effect of the ATP synthesis inhibitor oligomycin that blocks mitochondrial ATP synthase (Bertina et al., 1974) on presynaptic development. Unlike FCCP, oligomycin did not arrest mitochondrial movement along neurites (Supplementary Video 4), but caused increases in retrograde transport of slow-moving mitochondria (Table 1). Consistent with its established effect, it caused a significant elevation of MgGr fluorescence along the neurite at concentrations as low as 1 nM (Figure 2, G, H, and K). In the presence of oligomycin, bFGF bead-induced SV clustering (Figures 6J) was significantly inhibited. Mitochondrial clustering was, however, much less affected (Figures 6J). These results further suggest that ATP production from synaptic mitochondria plays an importance role in the development of presynaptic specializations.
In addition to ATP production, mitochondria also serve to buffer intracellular Ca2+ during neurotransmission within the nerve terminal (Alnaes and Rahamimoff, 1975; Yang et al., 2003). We thus also tested whether their function in calcium homeostasis plays a role in their involvement of presynaptic development through the use of a benzothiazepine compound CGP-37157, which selectively inhibits the mitochondrial Na+/Ca2+ exchanger (Cox et al., 1993; Baron and Thayer, 1997; Scanlon et al., 2000). In contrast to metabolic inhibitors, CGP-37157 did not affect bead-induced presynaptic differentiation (Figure 6J) and intracellular ATP level (Figure 2, I–K). Taken together, the ATP production, not the calcium sequestration function, of mitochondria is essential for the translocation and docking of mitochondria to sites of presynaptic development, which is a prerequisite for the assembly of SV clusters.
Regulation of F-Actin Assembly by Mitochondrial ΔΨm
To understand the dependence of SV clustering on ΔΨm, its effect on actin polymerization was examined. Previous studies have shown that presynaptic F-actin cytoskeleton plays an important role in the formation and maintenance of SV clusters (Hirokawa et al., 1989; Dai and Peng, 1996a; Peng et al., 1997; Richards et al., 2004). F-actin assembly is a major cellular process that consumes metabolic energy in neurons (Bernstein and Bamburg, 2003). Thus, mitochondrial activity as reflected by ΔΨm should have a significant bearing on F-actin assembly. Using the bead model, we found that F-actin, visualized by fluorescent phalloidin staining, was highly localized at bead-neurite contacts where mitochondrial clusters were also detected (Figure 7, A–D, arrows). In contrast, other mitochondrial clusters within varicosities that were not induced by beads were not associated with F-actin concentration (Figure 7, B–D, arrowheads). To visualize the newly polymerized F-actin, we used our previously established method to detect de novo actin polymerization associated with acetylcholine receptor clustering in muscle (Dai et al., 2000). This method involved first treating live neuronal cultures with another F-actin–binding agent, jasplakinolide which competes with phalloidin for F-actin binding in order to mask all pre-existing F-actin (Bubb et al., 1994). After presynaptic stimulation with beads, the newly assembled cytoskeleton was visualized by postlabeling with fluorescent phalloidin in fixed cultures. The complete masking of F-actin with jasplakinolide was confirmed by examining growth cones where dynamic F-actin turnover took place. In untreated neurons, F-actin enrichment was detected within the periphery domain of the growth cone (Figure 7, E and F). The phalloidin labeling was abolished after 1 μM jasplakinolide masking (Figure 7, G and H). This low concentration of jasplakinolide was sufficient to mask all the existing F-actin at the growth cone and along the neuritic shaft (Figure 7H) without a dramatic change in the growth cone morphology (Figure 7G). Jasplakinolide-treated neurons were then stimulated by beads, and new F-actin was probed by phalloidin after fixation. As shown in Figure 7, I and J, newly polymerized F-actin was mainly observed at the bead-neurite contact after 2 h of bead stimulation. This new F-actin polymerization induced by beads was largely inhibited by FCCP (Figure 7, K and L) and oligomycin, but not CGP-37157 (Figure 7Q). Although jasplakinolide was used at a low concentration in this study, it nevertheless may alter the rate of actin dynamics as a previous study suggests (Bubb et al., 1994). We thus also used another method to visualize actin assembly through the incorporation of rhodamine-conjugated G-actin (Rh-actin) into barbed ends of polymerizing actin filaments in saponin-permeabilized cultures. Rh-actin signals thus represent sites of new F-actin assembly. In untreated cultures, an elevated level of Rh-actin signal was detected at bead-neurite contacts (Figure 7, M and N). Suppression of either mitochondrial ΔΨm with FCCP or mitochondrial ATP production with oligomycin also abolished the incorporation of Rh-actin into F-actin structures at the contact site (Figure 7, O, P, and R).
Figure 7.

Mitochondrial ΔΨm-dependent assembly of F-actin by beads. (A–D) MitoTracker-labeled neurons were induced by bFGF beads for 2 h and then fixed for F-actin staining. The bead-induced mitochondrial cluster (arrow in C) was enriched with F-actin cytoskeleton as visualized by FITC-phalloidin (arrow in B). On the other hand, mitochondrial clusters away from beads were not associated with F-actin concentration (arrowheads in B–D). (E–H) To examine the role of newly polymerized F-actin, cultured neurons were incubated with 1 μM jasplakinolide (Jasp) for 3 min to mask the pre-existing F-actin before bead addition. In control neurons, F-actin, labeled by phalloidin, was highly enriched at the peripheral domain of the growth cone (arrow in F), but this was not seen in neurons pretreated with Jasp (arrowhead in H). (I–L) After masking the pre-existing F-actin with Jasp, the treated neurons were stimulated by bFGF beads for 2 h. Assembly of F-actin was locally induced at the bead-neurite contact (asterisk in I), which was prominently displayed in pseudocolor image (arrow in J). FCCP suppressed the local F-actin assembly induced by bead (arrowhead in L). (M–P) The effect of mitochondrial manipulators on bead-induced actin polymerization was also examined by the incorporation of rhodamine-conjugated G-actin (Rh-actin) into the barbed ends of actin filaments. Cultured neurons were stimulated by beads for 2 h and then incubated with 0.45 μM Rh-actin in saponin-containing buffer for 45 s. Rh-actin signal at the bead contact sites was significantly reduced in FCCP-treated neurons (arrowhead in P) when compared with the control (arrow in N). (Q and R) Quantifications of relative fluorescence intensity of rhodamine-conjugated phalloidin (after Jasp treatment; Q) and Rh-actin (R) at bead-contacts versus bead-free areas. Data are means ± SEM from three independent experiments; n > 30 bead-neurite contacts. **p < 0.01, t test.
DISCUSSION
The aggregation of mitochondria is a prominent feature of the nerve terminal. Previous studies have revealed the role of synaptic mitochondria in modulating the efficacy and plasticity of mature synapses (Alnaes and Rahamimoff, 1975; Herrera et al., 1985; Nguyen et al., 1997; Li et al., 2004). At the developing NMJ, our recent study has shown that mitochondria become coclustered with SVs at developing presynaptic specializations within minutes upon presentation of synaptogenic stimulus to spinal neurons. The results of the present study have indicated an essential role of localized mitochondrial activity in presynaptic differentiation.
The Regulation of Presynaptic Development by Mitochondrial ΔΨm Manipulators
JC-1 provided us with a simple probe for monitoring ΔΨm in live neurons. This dye has been extensively used as a semiquantitative indicator of neuronal mitochondrial ΔΨm (Ankarcrona et al., 1995; White and Reynolds, 1996; Buckman and Reynolds, 2001). We provided evidence that ΔΨm in cultured neurons could be reversibly modulated by creatine or FCCP and such manipulation affected mitochondria's axonal mobility and SV clustering. Creatine phosphate modulates ATP production by donating its high-energy phosphate in converting ADP to ATP (Ishida et al., 1994). Exogenous creatine therefore acts as a temporal and spatial buffer of energy which enhances the conversion rate of ATP from ADP. On the other hand, FCCP, an ionophore for proton, increases the permeabilization of inner mitochondrial membrane to protons and consequently depolarizes this membrane, therefore reducing ATP production (Nicholls and Budd, 2000). The relatively long latency in the effect of creatine on ΔΨm is likely due to the fact that its entry into the cell is rate-limited by a transporter-mediated process compared with the direct passage of FCCP (Moller and Hamprecht, 1989). Moreover, there is a possibility that the increase in SV clusters by creatine treatment involves transcriptional regulation of SV proteins.
In cultured neurons, axonal transport of mitochondria is bidirectional along microtubules or actin filaments by different motor proteins (Morris and Hollenbeck, 1993, 1995). Labeling the nerve-muscle or bead-nerve cocultures with JC-1 revealed that mitochondria with higher ΔΨm are preferentially located at the synaptic sites along the axon (Lee and Peng, 2006). This suggests that even during development, there is a requirement for high, local ATP production to support presynaptic development. Consistent with this, the present study showed that mitochondria were localized near the sites of local ATP consumption as a result of presynaptic differentiation induced by bFGF beads (Figure 5). The mechanism for targeting mitochondria to the presynaptic nerve terminal is likely mediated by the protein milton and syntabulin in Drosophila NMJs and in rat hippocampal synapses, respectively (Stowers et al., 2002; Cai et al., 2005). A previous study showed that a mitochondrial uncoupler CCCP blocks the bidirectional mitochondrial movements, whereas an inhibitor of the electron transport chain antimycin causes higher retrograde mitochondrial movement in neurons (Miller and Sheetz, 2004). In agreement with that study, our present results showed that depolarization by FCCP completely abolished mitochondrial translocation in both anterograde and retrograde directions along the axon, presumably due to the fact that ATP hydrolysis powers the microtubule motor proteins, such as kinesin and dynein, for anterograde and retrograde translocation, respectively. Like antimycin, inhibition of mitochondrial ATP production by oligomycin caused an increase in retrograde (slow) and a decrease in anterograde (slow) movements of mitochondria. An increase in retrograde mitochondrial transport may drive mitochondria with low ATP production away from the axon toward the soma. Because mitochondrial clustering induced by beads was not affected by oligomycin treatment, the docking of mitochondria to the synaptic sites is likely independent on the direction of mitochondrial movement.
The Role of F-Actin in the Formation of SV Clusters
The clustering, mobilization, fusion and recycling of SVs are vital to neurotransmitter release at the nerve terminal. The F-actin cytoskeleton is involved in regulating each one of these events (Dillon and Goda, 2005). At the mature nerve terminal, SVs are organized into two functional pools, the readily releasable pool (RRP) and the reserve pool (RP; Rizzoli et al., 2003; Rizzoli and Betz, 2005). RRP is a population of vesicles docked at the active zone and primed for release, whereas RP is a cluster of vesicles residing distally from the active zone. Structural studies with electron microscopy and immunolabeling have shown that F-actin is associated with SV clusters within the nerve terminal (Hirokawa et al., 1989; Dai and Peng, 1996a; Bloom et al., 2003). At Drosophila NMJs, F-actin disruption by cytochalasin D disrupts RP while leaving the RRP intact (Kuromi and Kidokoro, 1998). It is suggested that F-actin provides cytoskeletal tracks for SV replenishment as shown by the translocation of SVs from RP to RRP via an actin-based motor protein myosin V (Evans et al., 1998). In this study, polymerization of F-actin is locally induced at sites of synaptogenic induction in close association with mitochondrial clusters. These results further support that local actin polymerization is involved, at least in part, in the formation of presynaptic specialization.
The Importance of ATP Production in Actin-mediated Presynaptic Differentiation
The assembly of actin filaments requires ATP hydrolysis. In neurons, ∼50% of cellular ATP supply is utilized in cytoskeletal assembly (Bernstein and Bamburg, 2003). Thus, mitochondria that become localized at the nascent presynaptic region may serve as a local powerhouse in the assembly of F-actin and other presynaptic components, like SV clusters. With magnesium green as an ATP probe, our experiment has shown that depolarization of mitochondrial ΔΨm by FCCP significantly depleted the intracellular ATP content and this may account for the observed inhibition of presynaptic differentiation. ATP depletion by oligomycin similarly inhibited actin polymerization and SV clustering. Interestingly, although FCCP inhibited both SV and mitochondrial clustering, oligomycin only affected the former. A relatively low level of intracellular ATP may be sufficient for the function of motor proteins responsible for the synaptic targeting of mitochondria, a notion in agreement with our finding that mitochondrial translocation was not affected by oligomycin treatment (Supplementary Video 4). In agreement with our findings, mutant Drosophila NMJs lacking presynaptic mitochondria show a diffused, but not clustered, pattern of SV distribution in the nerve terminal, and this results in a reduction in the size of synaptic boutons (Guo et al., 2005). On the other hand, creatine, which enhanced presynaptic differentiation, did not cause an increase in intracellular ATP level. This may be due to the fact that creatine enhances local ATP resynthesis at sites of high metabolic activity but does not increase the overall ATP content in the neuron.
Because mitochondria also contribute to Ca2+ buffering in cultured neurons, we also tested whether this function is involved in presynaptic differentiation with a Na+/Ca2+ exchanger inhibitor CGP-37157. Our results showed that all three presynaptic events, SV and mitochondrial clustering as well as F-actin polymerization, were not affected by this compound. Thus, ATP production is specifically required for presynaptic differentiation. This conclusion is consistent with a study on Drosophila NMJs with a dynamin-related protein (Drp1) mutation that the mobilization of RP vesicles is inhibited in the absence of presynaptic mitochondria and this can be partially rescued by exogenous ATP (Verstreken et al., 2005). It remains unclear whether local ATP production by presynaptic mitochondria could regulate activity-dependent mobilization and release of SVs.
Taken together, this study shows that mitochondrial ATP production regulates presynaptic differentiation in an actin-dependent mechanism. The F-actin–based cytoskeleton may form the scaffold for the assembly of presynaptic organelles including SVs and mitochondria themselves.
Supplementary Material
[Supplemental Materials]
ACKNOWLEDGMENTS
We are very grateful for helpful comments from anonymous reviewers in improving the article. This work was supported by Research Grants Council Grants HKUST6107/01M and AoE/B-15/01 and by National Institutes of Health Grant NS36754. C.W.L. is supported by the Croucher Foundation Fellowship.
Footnotes
REFERENCES
- Alnaes E., Rahamimoff R. On the role of mitochondria in transmitter release from motor nerve terminals. J. Physiol. 1975;248:285–306. doi: 10.1113/jphysiol.1975.sp010974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankarcrona M., Dypbukt J. M., Bonfoco E., Zhivotovsky B., Orrenius S., Lipton S. A., Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Baron K. T., Thayer S. A. CGP37157 modulates mitochondrial Ca2+ homeostasis in cultured rat dorsal root ganglion neurons. Eur. J. Pharmacol. 1997;340:295–300. doi: 10.1016/s0014-2999(97)01433-7. [DOI] [PubMed] [Google Scholar]
- Benfenati F., Valtorta F., Greengard P. Computer modeling of synapsin I binding to synaptic vesicles and F-actin: implications for regulation of neurotransmitter release. Proc. Natl. Acad. Sci. USA. 1991;88:575–579. doi: 10.1073/pnas.88.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B. W., Bamburg J. R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003;23:1–6. doi: 10.1523/JNEUROSCI.23-01-00002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertina R. M., Steenstra J. A., Slater E. C. The mechanism of inhibition by oligomycin of oxidative phosphorylation in mitochondria. Biochim. Biophys. Acta. 1974;368:279–297. doi: 10.1016/0005-2728(74)90175-3. [DOI] [PubMed] [Google Scholar]
- Bixby J. L., Reichardt L. F. The expression and localization of synaptic vesicle antigens at neuromuscular junctions in vitro. J. Neurosci. 1985;5:3070–3080. doi: 10.1523/JNEUROSCI.05-11-03070.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom O., Evergren E., Tomilin N., Kjaerulff O., Low P., Brodin L., Pieribone V. A., Greengard P., Shupliakov O. Colocalization of synapsin and actin during synaptic vesicle recycling. J. Cell Biol. 2003;161:737–747. doi: 10.1083/jcb.200212140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb M. R., Senderowicz A. M., Sausville E. A., Duncan K. L., Korn E. D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Buckman J. F., Reynolds I. J. Spontaneous changes in mitochondrial membrane potential in cultured neurons. J. Neurosci. 2001;21:5054–5065. doi: 10.1523/JNEUROSCI.21-14-05054.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q., Gerwin C., Sheng Z. H. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 2005;170:959–969. doi: 10.1083/jcb.200506042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. A., Conforti L., Sperelakis N., Matlib M. A. Selectivity of inhibition of Na(+)-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- Dai Z., Luo X., Xie H., Peng H. B. The actin-driven movement and formation of acetylcholine receptor clusters. J. Cell Biol. 2000;150:1321–1334. doi: 10.1083/jcb.150.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z., Peng H. B. Presynaptic differentiation induced in cultured neurons by local application of basic fibroblast growth factor. J. Neurosci. 1995;15:5466–5475. doi: 10.1523/JNEUROSCI.15-08-05466.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z., Peng H. B. Dynamics of synaptic vesicles in cultured spinal cord neurons in relationship to synaptogenesis. Mol. Cell Neurosci. 1996a;7:443–452. doi: 10.1006/mcne.1996.0032. [DOI] [PubMed] [Google Scholar]
- Dai Z., Peng H. B. From neurite to nerve terminal: induction of presynaptic differentiation by target-derived signals. Semin. Neurosci. 1996b;8:97–106. [Google Scholar]
- Dillon C., Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu. Rev. Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Evans L. L., Lee A. J., Bridgman P. C., Mooseker M. S. Vesicle-associated brain myosin-V can be activated to catalyze actin-based transport. J. Cell Sci. 1998;111(Pt 14):2055–2066. doi: 10.1242/jcs.111.14.2055. [DOI] [PubMed] [Google Scholar]
- Guo X., Macleod G. T., Wellington A., Hu F., Panchumarthi S., Schoenfield M., Marin L., Charlton M. P., Atwood H. L., Zinsmaier K. E. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Herrera A. A., Grinnell A. D., Wolowske B. Ultrastructural correlates of naturally occurring differences in transmitter release efficacy in frog motor nerve terminals. J. Neurocytol. 1985;14:193–202. doi: 10.1007/BF01258447. [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Sobue K., Kanda K., Harada A., Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J. Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck P. J. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Ishida Y., Riesinger I., Wallimann T., Paul R. J. Compartmentation of ATP synthesis and utilization in smooth muscle: roles of aerobic glycolysis and creatine kinase. Mol. Cell Biochem. 1994;133-134:39–50. doi: 10.1007/BF01267946. [DOI] [PubMed] [Google Scholar]
- Kuromi H., Kidokoro Y. Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron. 1998;20:917–925. doi: 10.1016/s0896-6273(00)80473-0. [DOI] [PubMed] [Google Scholar]
- Lee C. W., Peng H. B. Mitochondrial clustering at the vertebrate neuromuscular junction during presynaptic differentiation. J. Neurobiol. 2006;66:522–536. doi: 10.1002/neu.20245. [DOI] [PubMed] [Google Scholar]
- Lee G. M., Ishihara A., Jacobson K. A. Direct observation of brownian motion of lipids in a membrane. Proc. Natl. Acad. Sci. USA. 1991;88:6274–6278. doi: 10.1073/pnas.88.14.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssens A., Nowicky A. V., Patterson L., Crompton M., Duchen M. R. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J. Physiol. 1996;496(Pt 1):111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Miller K. E., Sheetz M. P. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Moller A., Hamprecht B. Creatine transport in cultured cells of rat and mouse brain. J. Neurochem. 1989;52:544–550. doi: 10.1111/j.1471-4159.1989.tb09154.x. [DOI] [PubMed] [Google Scholar]
- Morris R. L., Hollenbeck P. J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993;104(Pt 3):917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Morris R. L., Hollenbeck P. J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. V., Marin L., Atwood H. L. Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J. Neurophysiol. 1997;78:281–294. doi: 10.1152/jn.1997.78.1.281. [DOI] [PubMed] [Google Scholar]
- Nicholls D. G., Budd S. L. Mitochondria and neuronal survival. Physiol. Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Peng H. B., Baker L. P., Chen Q. Tissue culture of Xenopus neurons and muscle cells as a model for studying synaptic induction. Methods Cell Biol. 1991;36:511–526. doi: 10.1016/s0091-679x(08)60294-0. [DOI] [PubMed] [Google Scholar]
- Peng H. B., Xie H., Dai Z. Association of cortactin with developing neuromuscular specializations. J. Neurocytol. 1997;26:637–650. doi: 10.1023/a:1018545827369. [DOI] [PubMed] [Google Scholar]
- Peters A., Palay S. L., Webster H. d. The Fine Structure of the Nervous System: Neurons and Their Supporting Cells. New York: Oxford University Press; 1991. [Google Scholar]
- Poot M., Zhang Y. Z., Kramer J. A., Wells K. S., Jones L. J., Hanzel D. K., Lugade A. G., Singer V. L., Haugland R. P. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 1996;44:1363–1372. doi: 10.1177/44.12.8985128. [DOI] [PubMed] [Google Scholar]
- Richards D. A., Rizzoli S. O., Betz W. J. Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J. Physiol. 2004;557:77–91. doi: 10.1113/jphysiol.2004.062158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli S. O., Betz W. J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rizzoli S. O., Richards D. A., Betz W. J. Monitoring synaptic vesicle recycling in frog motor nerve terminals with FM dyes. J. Neurocytol. 2003;32:539–549. doi: 10.1023/B:NEUR.0000020609.19873.e8. [DOI] [PubMed] [Google Scholar]
- Robitaille R., Tremblay J. P. Non-uniform release at the frog neuromuscular junction: evidence of morphological and physiological plasticity. Brain Res. 1987;434:95–116. doi: 10.1016/0165-0173(87)90019-1. [DOI] [PubMed] [Google Scholar]
- Scanlon J. M., Brocard J. B., Stout A. K., Reynolds I. J. Pharmacological investigation of mitochondrial Ca(2+) transport in central neurons: studies with CGP-37157, an inhibitor of the mitochondrial Na(+)-Ca(2+) exchanger. Cell Calcium. 2000;28:317–327. doi: 10.1054/ceca.2000.0171. [DOI] [PubMed] [Google Scholar]
- Schafer D. A., Welch M. D., Machesky L. M., Bridgman P. C., Meyer S. M., Cooper J. A. Visualization and molecular analysis of actin assembly in living cells. J. Cell Biol. 1998;143:1919–1930. doi: 10.1083/jcb.143.7.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley S. T., Reers M., Mottola-Hartshorn C., Lin M., Chen A., Smith T. W., Steele G. D., Jr, Chen L. B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers R. S., Megeath L. J., Gorska-Andrzejak J., Meinertzhagen I. A., Schwarz T. L. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Symons M. H., Mitchison T. J. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P., Ly C. V., Venken K. J., Koh T. W., Zhou Y., Bellen H. J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- White R. J., Reynolds I. J. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J. Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., He X. P., Russell J., Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J. Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Materials]