Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation (original) (raw)
Abstract
PD-1 is an immunoinhibitory receptor expressed by activated T cells, B cells, and myeloid cells. Mice deficient in PD-1 exhibit a breakdown of peripheral tolerance and demonstrate multiple autoimmune features. We report here that the ligand of PD-1 (PD-L1) is a member of the B7 gene family. Engagement of PD-1 by PD-L1 leads to the inhibition of T cell receptor–mediated lymphocyte proliferation and cytokine secretion. In addition, PD-1 signaling can inhibit at least suboptimal levels of CD28-mediated costimulation. PD-L1 is expressed by antigen-presenting cells, including human peripheral blood monocytes stimulated with interferon γ, and activated human and murine dendritic cells. In addition, PD-L1 is expressed in nonlymphoid tissues such as heart and lung. The relative levels of inhibitory PD-L1 and costimulatory B7-1/B7-2 signals on antigen-presenting cells may determine the extent of T cell activation and consequently the threshold between tolerance and autoimmunity. PD-L1 expression on nonlymphoid tissues and its potential interaction with PD-1 may subsequently determine the extent of immune responses at sites of inflammation.
Keywords: binding, proliferation inhibition, cytokine secretion inhibition, tissue expression, peripheral tolerance
Introduction
The balance of positive and negative signals is of central importance in maximizing the ability of the adaptive immune response to defend the host while maintaining immunologic tolerance and preventing autoimmunity. PD-1 is a type I transmembrane protein that is transcriptionally induced in activated T cells, B cells 1 2 3, and myeloid cells (Nishimura, H., and T. Honjo, unpublished data). The extracellular region of PD-1 consists of a single Ig-like variable (IgV) domain 4, and the cytoplasmic region contains an immunoreceptor tyrosine-based inhibitory motif (for a review, see reference 5). Recent work has revealed that the PD-1 receptor acts to downregulate immune responses and its loss leads to a breakdown of peripheral tolerance. Mice deficient in PD-1 develop lupus-like proliferative arthritis and glomerulonephritis with predominant IgG3 deposition as they age 6. Additionally, in a manner similar to that reported in the lupus-prone MRL mice, introduction of the lpr mutation leads to accelerated disease onset and severity of symptoms. Furthermore, 2C-TCR (anti–H-2Ld) transgenic mice homozygous for the PD-1 null mutation in the autoreactive H-2b/d background develop a chronic graft-versus-host–like disease 6. In this model, peripheral 2C T cells show a memory rather than a naive phenotype and infiltrate into tissues including the epidermis, despite efficient negative selection in the thymus. However, it remains to be determined whether this regulation is achieved at the activation phase (lymph nodes), at the effector phase (tissues), or both. Direct approaches to this question as well as further characterization of this inhibitory pathway have been hampered by a lack of knowledge of the identity of the ligand for PD-1.
PD-1 is structurally similar to CTL-associated antigen 4 (CTLA-4), which binds B7-1 and B7-2 and plays a crucial role for the maintenance of T cell homeostasis (for reviews, see references 7 and 8). Although PD-1 does not have the MYPPPY motif that is critical for B7-1 and B7-2 binding, the extracellular regions of PD-1 and CTLA-4 each consist of a single IgV domain, with 23% identity to each other. This is in comparison to the ∼30% identity between CTLA-4 and CD28 9. Thus, we reasoned that the ligand of PD-1 might be structurally related to B7-1 and B7-2. We have pursued this hypothesis by searching for B7-like molecules and testing their binding to PD-1. In this work, we identify a ligand of PD-1 and demonstrate that this receptor–ligand interaction leads to the inhibition of lymphocyte proliferation.
Materials and Methods
Molecular Cloning.
A BLAST search of the human expressed sequence tag (EST) database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) identified two overlapping ESTs with homology to B7-1 and B7-2 (AA292201 and AA399416). A 389-bp portion of the cDNA was amplified by PCR using as primers 5′-dCAGCTATGGTGGTGCCGACTACAA-3′ and 5′-dAGGTGCTAGGGGACAGTGTTAGACA-3′. The PCR product was biotin labeled and used to isolate a full-length cDNA by Cloncapture (CLONTECH Laboratories, Inc.) from a human placenta cDNA library in the pAXEF vector 10. A search of the National Center for Biotechnology Information murine EST database identified overlapping sequences with homology to human PD-1 ligand 1 (PD-L1; AA896104 and AA823166). A 409-bp portion was amplified by PCR using as primers 5′-dgagagcctcgcgtccaaag-3′ and 5′-dGTGGTTTTGCCCTGGCTGTGATCT-3′. The PCR product was biotin labeled and used to isolate a full-length cDNA by Cloncapture (CLONTECH Laboratories, Inc.) from a murine-activated T cell cDNA library in the pAXEF vector.
Fusion Proteins and Cell Transfections.
The Ig fusion proteins used consist of the complete extracellular region of a receptor linked to the hinge-CH2-CH3 domains of either human Igγ1 or the same domains of murine Igγ2a (with four point mutations blocking Fc receptor and complement binding) to give Ig(γ1) and Ig(γ2a) fusions, respectively 11. These recombinant proteins were produced in COS cells transiently transfected with LipofectAMINE (GIBCO BRL) or stably transfected Chinese hamster ovary (CHO) cell lines and purified from conditioned media using protein A–Sepharose.
The human PD-L1 cDNA was subcloned into pEF6 (Invitrogen) and linearized with ScaI. This DNA was coelectroporated into CHO-K1 cells with a plasmid construct containing a hygromycin B resistance gene under the control of a phosphoglycerate kinase gene promoter. Cells were selected in 800 μg/ml hygromycin B and cloned by limiting dilution. The murine PD-L1 cDNA in pAXEF was linearized with ApaLI and coelectroporated into CHO-K1 cells with a puromycin resistance gene under the control of a phosphoglycerate kinase gene promoter. Cells were selected in 10 μg/ml puromycin and cloned by limiting dilution.
Murine T Cell Assays.
Splenic T cells were collected by anti-IgM panning from pools of splenocytes from two to three age-matched C57BL/6 and C57BL/6–PD-1 −/− mice, resulting in 80% purity. 4 × 104 cells/well in 96 U-bottomed plates (Iwaki) were incubated with the indicated concentrations of reagents. Purified anti–mouse CD3 (145-2C11) alone or in combination with hPD-L.Ig(γ2a) or mouse IgG2a were precoated overnight at 4°C. Cells were cultured for 72 h, then pulsed with 0.5 μCi of [3H]thymidine (Amersham Pharmacia Biotech) per well for the last 18 h, and incorporated label was measured.
Human T Cell Proliferation and Cytokine Assays.
PBMCs were isolated by Ficoll-Hypaque gradient centrifugation. CD4+ T cell populations (85–90% purity) were purified by negative selection using a cocktail of mAbs and immunomagnetic beads (PerSeptive Biosystems). Anti-CD3, control IgG, and fusion protein were covalently attached to polyurethane-coated tosyl-activated Dynabeads (Dynal) according to the manufacturer's instructions and as described previously 12. Anti-CD3 Ab (UCHT1; BD PharMingen) at the indicated concentration was added to 107 beads/ml in 0.1 M phosphate buffer, pH 7.4. Control IgG was added to the bead suspension in order to maintain a constant total Ig concentration of 5 μg/ml during binding. Similarly, anti–CD3/hPD-L.Ig(γ2a) beads were prepared with the indicated anti-CD3 Ab concentration, a constant concentration of either hPD-L1.Ig representing 40% of the total bound protein (2 μg/107 beads), or control IgG to make up the remaining total bound protein. 105 T cells were cultured in 96-well flat-bottomed plates, and beads were added at a 1:1 bead/cell ratio in the presence or absence of the indicated concentrations of anti-CD28 Ab (CD28.2; BD PharMingen). Proliferation was determined by labeling cultures for the last 6 h of a 4-d assay with 1 μCi [3H]thymidine/well. For analysis by cytokine ELISAs, cultures were set up as described above and supernatants were harvested at the indicated times. IFN-γ, IL-10, and IL-2 concentrations were determined using commercially available ELISA kits (Genzyme).
Dendritic Cell Isolation.
Human dendritic cells were derived from peripheral blood. Mononuclear cells were isolated after fractionation on a Ficoll gradient. Nonadherent cells were removed and the remaining cells were cultured in 150 ng/ml human GM-CSF (R & D Systems) and 100 ng/ml human IL-4 (R & D Systems) for 2 d. The nonadherent dendritic cells were isolated (CD80+CD86+HLA-DR+CD54+CD58+CD1a+) and cultured in GM-CSF alone or activated with GM-CSF, 2.5 μg/ml LPS (Sigma-Aldrich), and 10 ng/ml human IFN-γ. At 4 and 20 h after activation, cells were harvested and RNA was isolated using the RNeasy kit (QIAGEN).
Murine bone marrow mononuclear cells were immunodepleted of granulocytes, lymphocytes, and Ia+ cells by magnetic-activated cell sorting and cultured in petri dishes with GM-CSF and IL-4. Dendritic cells were harvested as the nonadherent population after 7 d of culture, and demonstrated to be 75–80% CD11c+, high Ia+ cells. Cells were activated with LPS and human IFN-γ.
RNA Expression Analysis.
For quantitative PCR analysis, cellular RNA was deoxyribonuclease treated, reextracted, and converted to first strand cDNA. 6-carboxyfluorescein (FAM)-labeled hPD-L1, hB7-1, hB7-2, and human glyceraldehyde 3-phosphate dehydrogenase (hGAPDH) probes were purchased from PE Biosystems (hPD-L1: 5′-dGCCGAAGTCATCTGGACAAG-3′ and 5′-dTCTCAGTGTGCTGGTCACAT-3′, probe 5′-FAM-dCACCACCACCAATTCCAAGA-3′; hB7-1: 5′-dACGTGACCAAGGAAGTGAAAGAA-3′ and 5′-dTGCCAGCTCTTCAACAGAAACAT-3′, probe 5′-FAM-dTGGCAACGCTGTCCTGTGGTCAC-3′; and hB7-2: 5′-dGGGCCGCACA-AGTTTTGAT-3′ and 5′-dGCCCTTGTCCTTGATCTGA-AGA-3′, probe 5′-FAM-dCGGACAGTTGGACCCTGAGA-CTTCACA-3′.
PCR reactions were set up in 96-well plates using reagents from the PerkinElmer TaqMan™ EZ kit, according to the manufacturer's instructions. Standard curves were set up for each of the four genes analyzed. 40 cycles of PCR were run in an ABI Prism 7700 Sequence Detector (PerkinElmer) and GAPDH was used to normalize the PD-L1, B7-1, and B7-2 results.
The Mu19KsubA chip (Affymetrix) was used for GeneChip® hybridization analysis. The sequence of a portion of murine PD-L1 is represented by the EST TC17781 of The Institute for Genomic Research (http://www.tigr.org) on this chip. RNA isolation, chip hybridization, and scanning were performed as described 13.
For RNA blot hybridization, the 1.6-kb human and 3.6-kb murine PD-L1 cDNAs were excised by digestion with XbaI and labeled by random priming with [γ-32P]ATP and the Klenow fragment of DNA polymerase I. RNA blots were hybridized as described 14.
Chromosomal Analysis.
The chromosomal location of the human PD-L1 gene was identified using monochromosomal somatic cell hybrid DNA templates (Quantum Technologies) for PCR amplification using _PD-L1_–specific primers. The oligonucleotide primers used were: 5′-dCCCAGGTAATATTCTGAATGTGTC-3′ and 5′-dATTCCATAAATATCTGCTGAATGT-3′. Each PCR used 100 ng of template DNA, with 30 cycles of 94°C, 30 s; 60°C, 30 s; and 68°C, 60 s. The PCR products were analyzed by agarose gel electrophoresis and ethidium bromide staining.
Results
Identification of a Novel B7 Homologue That Binds PD-1.
The human and murine PD-L1 cDNAs were identified in a B7 homology–based search of EST databases. A BLAST search of the National Center for Biotechnology Information database revealed two human overlapping ESTs with similarity to B7-1 and B7-2 molecules (accession nos. AA292201 and AA399416). Using these sequences, a full-length cDNA was isolated from a human placenta cDNA library. In parallel, two murine ESTs were found that correspond to the murine orthologue of PD-L1 (accession nos. AA896104 and AA823166). Using these sequences, a full-length cDNA was isolated from a murine activated T cell cDNA library. Human and mouse PD-L1 molecules are members of the B7 gene family and share a common structural organization consisting of an IgV and an IgC domain in the extracellular region 15, with a hydrophobic transmembrane domain followed by a short, charged intracellular region (Fig. 1). Human PD-L1 with 290 residues is identical to B7-H1, which was reported to have T cell stimulation activities 16. Murine PD-L1 cDNA encodes a polypeptide with 70% amino acid identity to its human orthologue (Fig. 1). PD-L1 has amino acid identities of 21, 20, and 23% to B7-1, B7-2, and the ligand of inducible costimulator (ICOS), respectively 11 15 17 18.
Figure 1.

Amino acid sequence alignment of murine and human B7 gene family members. Identical amino acids are boxed in black and conservative substitutions in gray. Predicted signal and transmembrane domains of PD-L1 are underlined. Amino acids comprising the binding site of human B7-1 are indicated with a dot (reference 26). The sequences of the human (reference 11) and murine (references 17 and 18) ligands of ICOS (ICOS-L) have been reported previously. The full-length cDNA sequences of the human and murine PD-L1 cDNAs have been deposited with EMBL/GenBank/DDBS under accession nos. AF233516 and AF233517, respectively.
The ability of PD-L1 to bind PD-1 has been determined by flow cytometry and BIAcore binding assays. Human and murine PD-1.Ig fusion proteins bound to both human and murine PD-L1 expressed on CHO cells as detected by flow cytometry (Fig. 2 A). However, neither human CTLA-4.Ig, human CD28.Ig, or human ICOS.Ig bound to either PD-L1–expressing line. The PD-1 fusion proteins did not bind CHO cells transfected with vector alone. Further confirmation of the PD-1–PD-L1 interaction was obtained using surface plasmon resonance with a BIAcore instrument. The human and murine PD-1.Ig proteins and human CTLA-4.Ig were immobilized on the flow cell surfaces of a dextran chip, and tested for binding to soluble human PD-L1.Ig. PD-L1.Ig bound to both human and murine PD-1.Ig but not to human CTLA-4.Ig (Fig. 2 B). This binding was blocked by competition with coinjected soluble PD-1.Ig but not CTLA-4.Ig. Soluble forms of human B7-1 and B7-2 did not bind immobilized human PD-1.Ig (data not shown).
Figure 2.


The binding of PD-1 to PD-L1. (A) CHO cells stably transfected with human or murine PD-L1 or vector alone were stained with hPD-1.Ig(γ2a), mPD-1.Ig(γ1), hCTLA-4.Ig(γ2a), hCD28.Ig(γ1), or hICOS.Ig(γ2a) (species matched), and developed with goat anti–murine IgG2a-PE or anti–human IgG-FITC antisera. (B) PD-L1.Ig was tested for binding to immobilized mPD-1.Ig(γ1) (gray bars), hPD-1.Ig(γ1) (black bars), and hCTLA-4.Ig(γ1) (white bars) using surface plasmon resonance on a BIAcore 2000 instrument. Receptor-Fc fusion proteins were immobilized on a CM5 dextran chip by amine coupling with normal human serum/_N_-ethyl-_N_′-(dimethylamino)propyl I carbodiimide hydrochloride (EDC) in 10 mM sodium acetate, pH 4.0, as described (reference 27). The amounts of protein immobilized were 5,383 response units (RU) for mPD-1.Ig(γ1), 5,416 RU for hPD-1.Ig(γ1), and 11,493 RU for hCTLA-4.Ig(γ1). Concentrated COS-conditioned medium from hPD-L1.Ig(γ2a)–transfected cells was analyzed with (+) or without (−) coinjection of 100 μg/ml of soluble mPD-1.Ig, hPD-1.Ig, or hCTLA-4.Ig for competition. Binding was quantified as an increase in RU at 60 s after the end of injection compared with a baseline established 20 s before injection.
PD-L1, B7-1, and B7-2 Are Coexpressed on Antigen-presenting Cells.
We next examined the pattern of PD-L1 expression on antigen-presenting cells and compared it with that of B7-1 and B7-2 expression. First, we analyzed expression in human blood monocytes by RNA blot hybridization and found that PD-L1 is not expressed by unstimulated monocytes, but is rapidly upregulated upon IFN-γ treatment (Fig. 3 A). Treatment of monocytes with another proinflammatory cytokine, TNF-α, led to a low level induction similar to that found with medium alone, presumably as a result of activation by adherence to plastic (Fig. 3 A). In addition to the major 4.2-kb PD-L1 mRNA, we also observed a minor 1.8-kb PD-L1 mRNA species in IFN-γ–treated monocytes. The major 4.2-kb mRNA species has also been reported by Dong et al. 16. We also detected expression of PD-L1 by human B cells activated by cell surface Ig cross-linking, but not by the Raji cell line (Fig. 3 A). Similarly, B7-1 is not expressed by unstimulated monocytes but is upregulated in response to IFN-γ with similar kinetics to PD-L1 expression. In contrast, B7-2 mRNA is constitutively expressed in monocytes and levels are unaffected by IFN-γ or TNF-α.
Figure 3.
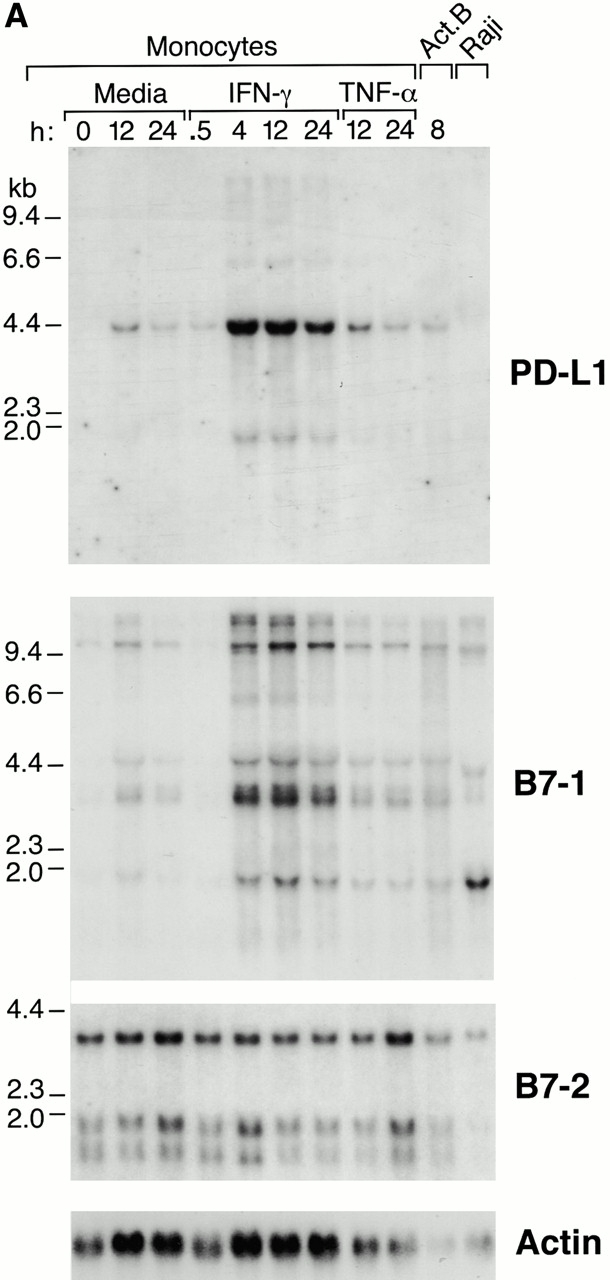




Expression of PD-L1 in antigen-presenting cells and murine tissues. (A) Northern blot analysis of total RNA of human peripheral blood monocytes stimulated with 500 U/ml human IFN-γ, 100 U/ml human TNF-α, or media alone, and anti-Ig–activated human B cells, with human PD-L1, B7-1, B7-2, and β-actin cDNA probes. Cells were prepared and stimulated as described (reference 28). Act.B, actinomycin B. (B) Isolated human peripheral blood dendritic cells (DC) were cultured in either human GM-CSF alone (gray bars) or in GM-CSF, LPS, and IFN-γ (black bars) for 4 or 20 h, after which RNA was isolated for quantitative (real time) PCR analysis. Fluorescence is plotted as a ratio of PD-L1, B7-1, or B7-2 signal to the GAPDH signal. (C) Northern blot analysis of human keratinocyte total RNA with a human PD-L1 cDNA probe. Keratinocytes were isolated and activated with PMA and IFN-γ, as described previously (reference 10). (D) Northern blot analysis of murine tissue polyA+ RNAs (Ambion) with a murine PD-L1 cDNA probe. (E) The human PD-L1 gene was amplified by PCR from monochromosomal somatic cell hybrid genomic DNAs containing the indicated human chromosome as well as hamster, murine, and human genomic DNAs.
Second, PD-L1, B7-1, and B7-2 mRNA expression by human dendritic cells was examined by quantitative PCR. Human peripheral blood–derived dendritic cells were treated with GM-CSF alone or activated with GM-CSF, LPS, and IFN-γ. As a result of activation by LPS and IFN-γ, PD-L1 mRNA was rapidly induced with a 16-fold increase at 4 h and a 34-fold increase at 20 h relative to noninduced cells (Fig. 3 B). B7-1 and B7-2 mRNAs were also induced upon activation: B7-1 was induced 21-fold at 4 h and 22-fold at 20 h. B7-2 showed little induction at 4 h; however, expression was induced fivefold at 20 h (Fig. 3 B). Expression of PD-L1 by murine bone marrow–derived dendritic cells treated with LPS and IFN-γ was examined using GeneChip® hybridization. PD-L1 expression in these cells follows a pattern similar to that observed on human dendritic cells: a fivefold induction of the PD-L1 mRNA relative to the uninduced cells at 6 and 20 h after induction (data not shown). These data demonstrate that PD-L1 is expressed by antigen-presenting cells and lymphocytes, and it is induced on dendritic cells in a manner similar to B7-1 and B7-2. We also demonstrated an induction of PD-L1 by treatment of human keratinocytes with phorbol ester and IFN-γ (Fig. 3 C).
In murine tissues, we found an ∼3.7-kb PD-L1 mRNA transcript by Northern blot hybridization. Dong et al. 16 have reported a 4.2-kb PD-L1 (B7-H1) mRNA species in normal human tissue RNA. The distribution of the murine PD-L1 mRNA closely resembled that of the human with high levels in the heart, thymus, and lung, and low levels in the kidney, spleen, and liver (Fig. 3 D).
By PCR of monochromosomal human somatic cell hybrids, we demonstrated that the human PD-L1 gene is located on chromosome 9 (Fig. 3 E), unlike the B7-1 and B7-2 genes, which are clustered on chromosome 3 19. Thus, the parallel upregulation we observe for these genes on activated dendritic cells is not the result of clustering at a single chromosomal site. In addition, this also distinguishes PD-L1 from the B7-like butyrophilins, which are in the MHC gene complex of human chromosome 6 20.
PD-1–PD-L1 Interaction Inhibits CD3-mediated T Cell Proliferation.
To examine the functional significance of the PD-1–PD-L1 interaction, we compared the effect of PD-L1.Ig on the proliferation of murine T cells derived from wild-type and _PD-1_–deficient mice 21. We first examined the proliferation of purified splenic T cells from wild-type and PD-1 −/− mice to anti-CD3 mAb and found they were essentially identical (Fig. 4 A). Thus, the loss of PD-1 did not impair the ability of T cells to respond to stimulation through the TCR, nor did it confer a hyperproliferative phenotype in a primary stimulation. To examine the effects of PD-L1, an optimal concentration of anti-CD3 mAb and varying concentrations of human PD-L1.Ig or an IgG control were precoated on plastic plates. Proliferation of purified splenic T cells was measured 3 d after stimulation by incorporation of [3H]thymidine. As shown in Fig. 4 B, human PD-L1.Ig inhibited proliferation of PD-1 +/+ T cells in a dose-dependent fashion relative to the IgG control (Fig. 4 B). In contrast, human PD-L1.Ig had no effect on the proliferation of PD-1 −/− T cells under the same conditions. These results show that PD-L1 can attenuate TCR-mediated T cell proliferation. Furthermore, the failure of PD-L1 to inhibit _PD-1_–deficient T cells indicates that PD-L1 conveys this signal via its interaction with PD-1.
Figure 4.


TCR activation of murine and human T cells in the presence of PD-L1 results in inhibition of T cell proliferation and cytokine production. (A) Splenic T cells from wild-type (open squares) and PD-1 −/− (filled circles) C57BL/6 mice were stimulated for 72 h with varying concentrations of precoated anti-CD3 Ab. (B) Splenic T cells from wild-type (squares) and PD-1 −/− (circles) C57BL/6 mice were stimulated for 72 h with 10 μg/ml of anti-CD3 mAb in combination with varying concentrations of hPD-L.Ig(γ2a) (filled symbols) or control mIgG2a (open symbols). [3H]Thymidine incorporation was measured in triplicate. These data are representative of four separate experiments. (C) Purified human CD4+ T cells were stimulated for 4 d with anti-CD3 mAb/control IgG–coated beads (stippled bars), anti-CD3 mAb/hPD-L1.Ig(γ2a)–coated beads (hatched bars), or medium alone (black bars). The bead/cell ratio was 1:1. Proliferation was determined by [3H]thymidine incorporation in triplicate wells. Supernatants were collected at 96 h after activation, and cytokine concentrations were measured using commercially available ELISA kits. The data are representative of two separate experiments.
The functional consequences of PD-L1 interaction with its receptor were also examined using human T cells. Purified CD4+ T cells obtained from PBMCs were activated with beads coated with anti-CD3 mAb and either human PD-L1.Ig or a control Ig. Proliferation and cytokine production were assessed 96 h after stimulation. As shown in Fig. 4 C, cells activated with anti-CD3 mAb/PD-L1.Ig–coated beads showed a 69% decrease in proliferation relative to anti-CD3 mAb/control Ig–activated cells. Furthermore, activation of cells in the presence of PD-L1 also impairs cytokine secretion. In the presence of PD-L1, the secretion of IFN-γ and IL-10 is decreased by ∼80 and 60%, respectively (Fig. 4 C). IL-2 production was below detection under these activation conditions at both 24 and 96 h. However, under conditions in which costimulation in the form of soluble anti-CD28 is provided, activation of cells in the presence of PD-L1 also leads to a decrease in IL-2 production (data not shown). Thus, activation of murine and human T cells in the presence of PD-L1 leads to inhibition of both proliferation and cytokine secretion.
The Outcome of PD-1–PD-L1 Interaction Depends on the Strength of TCR and CD28 Signals.
To examine the relationship between TCR-, CD28-, and PD-1–mediated signals, human CD4+ T cells were stimulated with suboptimal or optimal concentrations of anti-CD3 mAb, a fixed concentration of PD-L1.Ig, and increasing concentrations of soluble anti-CD28 mAb. Using anti-CD3 mAb–coated beads, the concentrations required for suboptimal and optimal T cell stimulation were established (data not shown). Under conditions of suboptimal TCR engagement (anti-CD3 mAb at 1 μg/ml), minimal proliferation is observed in the absence of costimulation (Fig. 5 A). The addition of increasing concentrations of soluble anti-CD28 mAb leads to an up to 30-fold increase in proliferation. Under these conditions, activation of T cells in the presence of PD-L1 results in an 80% reduction in proliferation (Fig. 5 A). A maximal level of costimulation (anti-CD28 at 250 ng/ml) was required to rescue the inhibition of proliferation mediated by PD-L1 stimulation. In contrast, under saturating conditions of TCR activation (anti-CD3 mAb at 2 μg/ml), PD-L1–mediated inhibition of T cell proliferation is only observed in the absence of CD28 costimulation (Fig. 5 B).
Figure 5.

TCR/PD-L1 activation in the presence of CD28 costimulation results in inhibition of T cell proliferation. Purified human CD4+ cells were stimulated for 4 d with anti-CD3 mAb/control mIgG–coated beads (gray bars), or anti-CD3 mAb/hPD-L1.Ig(γ2a)–coated beads (black bars) in the presence of various concentrations of soluble anti-CD28 mAb. Stimulations were performed at (A) suboptimal (1 μg/ml) and (B) optimal (2 μg/ml) concentrations of anti-CD3 Ab. The bead/cell ratio was 1:1. Proliferation was determined by [3H]thymidine incorporation in triplicate wells. The data are representative of two separate experiments.
Discussion
Recent studies of _PD-1_–deficient mice have revealed a critical immunoregulatory role for this receptor, as its absence leads to autoimmunity. As PD-1 is structurally similar to CTLA-4, we hypothesized that the ligand of PD-1 might be a member of the B7 gene family and searched for novel B7 homologues. One of these, later termed PD-L1, was found to bind PD-1 specifically. The functional significance of this interaction has been demonstrated in T cell assays, in which engagement of PD-1 by PD-L1 leads to the inhibition of TCR-mediated lymphocyte proliferation and cytokine secretion. PD-1 is also expressed by B cells and myeloid cells, and the significance of PD-1 in these cell types is evidenced by the autoantibody generation and myeloid hyperplasia of _PD-1_–deficient mice 6 21. Thus, PD-L1 is expected to act on a wider range of cell types than we have demonstrated in this report.
We have demonstrated that PD-L1 signaling can inhibit the TCR-mediated proliferation of T cells. The capacity of the PD-1–PD-L1 interaction to downregulate a CD3/CD28-stimulated response shows that PD-1 engagement results in the delivery of a strong inhibitory signal. However, the functional consequences of the PD-1–PD-L1 interaction are dependent on the relative strength of signals delivered via the TCR and CD28. Increasing levels of TCR or CD28 signaling can circumvent the inhibitory effects of PD-1 ligation at the activation stage. In the absence of the PD-1–PD-L1 interaction, the threshold of TCR signal needed for T cell activation will be lowered, consistent with the autoimmune reactions seen in _PD-1_–deficient mice 6.
In our experiments, inhibition was observed when PD-L1 was coimmobilized with anti-CD3. This would allow for coligation of the TCR and PD-1, consistent with the immunoreceptor tyrosine-based inhibitory motif of PD-1 delivering a negative signal by recruitment of an Src homology 2 (SH2) domain–containing tyrosine phosphatase. Indeed, using the A20II1.6 B lymphoma line, we have demonstrated that cocross-linking of the B cell receptor and PD-1 reduces the calcium influx observed by crosslinking of the B cell receptor alone, and leads to interaction of the cytoplasmic region of PD-1 with SHP-2 (our unpublished data). In addition, there is inhibition of the tyrosine phosphorylation of a variety of signaling molecules leading to suppression of the proliferative response.
Dong et al. 16 have shown an increase in T cell proliferation and IL-10 secretion by T cells stimulated with PD-L1/B7-H1 and low levels of anti-CD3. Previous work has demonstrated that PD-1 expression is not constitutive but induced by antigen receptor cross-linking 2 3. Thus, PD-1 would be expected to be expressed and capable of interacting with PD-L1 in our assays in which the T cells are stimulated with higher levels of anti-CD3. It is unclear whether the PD-L1/B7-H1 costimulation reported by Dong et al. 16 is PD-1 dependent or whether it could be mediated by an alternative receptor for PD-L1. If a second, costimulatory receptor exists for PD-L1, this situation would parallel that of the costimulatory/inhibitory CD28/CTLA-4 receptor–B7-1/B7-2 ligand pairs. We do not find that PD-L1 induces IL-10 secretion in our assays. It should be noted that Dong et al. find that neutralizing Ab against IL-2 blocks the B7-H1–mediated increase in IL-10 production, suggesting that the IL-10 production is IL-2 dependent 16. Thus, the increase in IL-10 secretion may be due to the amplification of a population of cells rather than a direct induction.
The PD-1 gene resembles that of CTLA-4 in that it is expressed only after T cell activation. Thus, we expect that the PD-1–PD-L1 interaction may be most important in setting the TCR threshold requirements at sites of restimulation. Previously activated T cells that migrate to the periphery may reencounter antigen on cells expressing PD-L1 but little or no B7-1, B7-2, or other costimulatory molecules. Such an interaction would fail to result in T cell expansion, except under conditions of strong TCR ligation. In the absence of CTLA-4, mice develop a rapid and fatal T cell proliferative disease that results in damage to multiple organs 22 23. If PD-1 ligation provides a checkpoint for T cell activation later in an immune response than CTLA-4 ligation, this could account for the milder phenotype of _PD-1_–deficient mice compared with _CTLA-4_–deficient mice. Alternatively, the differences in phenotype between mice with these null mutations may reflect differences in the expression patterns of their respective ligands.
The expression patterns of B7-1, B7-2, and PD-L1 are distinct. B7-2, one of the ligands of CD28 and CTLA-4, is constitutively expressed on monocytes; however, constitutive expression of B7-1 and B7-2 is not seen in any organ. B7-1 and B7-2 expression can be induced in dendritic cells, macrophages, and B cells 15, as well as some types of fibroblast and epithelial cell. In contrast, PD-L1 is expressed constitutively by nonlymphoid, parenchymal organs such as the heart, placenta, skeletal muscle, and lung, but not the small intestine (16; Fig. 3 D). PD-L1 is also expressed in some cancers, as three ESTs are from human ovarian tumors. This raises the possibility that some tumors may use PD-L1 to inhibit an antitumor immune response. We found that PD-L1 expression by monocytes and keratinocytes was enhanced by stimulation with IFN-γ. Upon activation, keratinocytes express MHC class II, but not B7-1 or B7-2 24. Induction of PD-L1 expression by cytokines such as IFN-γ or other inflammatory stimuli could result in attenuation of TCR/CD28–mediated T cell activation. This may be of particular significance in the effector sites of inflammatory or autoimmune responses. As PD-1 is expressed on activated T as well as B cells 2, the expression of PD-L1 in nonlymphoid tissues as well as on dendritic cells may allow for regulation of potentially autoreactive lymphocytes at sites of immune activation as well as at effector sites**.** This may be a particularly important mechanism in limiting activities of both T and B cells in the heart, lung, kidney, and placenta where PD-L1 is highly expressed. In this context, Fas-L, a potent immune negative regulator, is also expressed on nonlymphoid tissues including the small intestine and testis 25. Interestingly, the tissue expression patterns of PD-L1 and Fas-L appear to be in general mutually exclusive, suggesting nonredundant regulation of peripheral tolerance by the Fas–Fas-L and PD-1–PD-L1 pathways.
Acknowledgments
We thank Ms. M. Yamamoto and Y. Tabuchi for their technical assistance and Ms. K. Saito for her secretarial help. We thank Dr. N. Minato for helpful discussions.
This work was supported by a Center of Excellence grant from the Ministry of Education, Science, Sports, and Culture of Japan, and by National Institutes of Health grants AI39671, AI41584, and CA84500 (to G.J. Freeman).
Footnotes
Abbreviations used in this paper: CHO, Chinese hamster ovary; CTLA-4, CTL-associated antigen 4; EST, expressed sequence tag; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ICOS, inducible costimulator; IgV, Ig-like variable; PD-L1, PD-1 ligand 1.
G.J. Freeman, A.J. Long, and Y. Iwai contributed equally to this work. C.R. Wood and T. Honjo share senior authorship.
References
- Ishida Y., Agata Y., Shibahara K., Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- Vibhakar R., Juan G., Traganos F., Darzynkiewicz Z., Finger L.R. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp. Cell Res. 1997;232:25–28. doi: 10.1006/excr.1997.3493. [DOI] [PubMed] [Google Scholar]
- Shinohara T., Taniwaki M., Ishida Y., Kawaichi M., Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1) Genomics. 1994;23:704–706. doi: 10.1006/geno.1994.1562. [DOI] [PubMed] [Google Scholar]
- Vivier E., Daeron M. Immunoreceptor tyrosine-based inhibition motifs. Immunol. Today. 1997;18:286–291. doi: 10.1016/s0167-5699(97)80025-4. [DOI] [PubMed] [Google Scholar]
- Nishimura H., Nose M., Hiai H., Minato N., Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- Sperling A.I., Bluestone J.A. The complexities of T-cell costimulationCD28 and beyond. Immunol. Rev. 1996;153:155–182. doi: 10.1111/j.1600-065x.1996.tb00924.x. [DOI] [PubMed] [Google Scholar]
- Thompson C.B., Allison J.P. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7:445–450. doi: 10.1016/s1074-7613(00)80366-0. [DOI] [PubMed] [Google Scholar]
- Harper K., Balzano C., Rouvier E., Mattei M.-G., Luciani M.F., Golstein P. CTLA-4 and CD28 activated lymphocyte molecules are closely related in both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J. Immunol. 1991;147:1037–1044. [PubMed] [Google Scholar]
- Freeman G.J., Cardoso A.A., Boussiotis V.A., Anumanthan A., Groves R.W., Kupper T.S., Clark E.A., Nadler L.M. The BB1 monoclonal antibody recognizes both cell surface CD74 (MHC class II-associated invariant chain) as well as B7-1 (CD80), resolving the question regarding a third CD28/CTLA-4 counter receptor. J. Immunol. 1998;161:2708–2715. [PubMed] [Google Scholar]
- Ling V., Wu P.W., Finnerty H.F., Bean K.M., Spaulding V., Fouser L.A., Leonard J.P., Hunter S.E., Zollner R., Thomas J.L. Identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J. Immunol. 2000;164:1653–1657. doi: 10.4049/jimmunol.164.4.1653. [DOI] [PubMed] [Google Scholar]
- Blair P.J., Riley J.L., Levine B.L., Lee K.P., Craighead N., Francomano T., Perfetto S.J., Gray G.S., Carreno B.M., June C.H. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-XL induction. J. Immunol. 1998;160:12–15. [PubMed] [Google Scholar]
- Byrne M.C., Whitley M.Z., Follettie M.T. Preparation of mRNA for expression monitoring Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology Supplement. Vol. 49 2000. 22 John Wiley & Sons, Inc; New York: 2.1–22.2.13. [Google Scholar]
- Freeman G.J., Lombard D.B., Gimmi C.D., Brod S.A., Lee K., Laning J.C., Hafler D.A., Dorf M.E., Gray G.S., Reiser H. CTLA-4 and CD28 are coexpressed in most T-cells after activation. J. Immunol. 1992;149:3795–3801. [PubMed] [Google Scholar]
- Boussiotis V.A., Freeman G.J., Gribben J.G., Nadler L.M. The role of B7-1/B7-2:CD28/CTLA-4 pathways in the prevention of anergy, induction of productive immunity and down-regulation of the immune response. Immunol. Rev. 1996;153:5–26. doi: 10.1111/j.1600-065x.1996.tb00918.x. [DOI] [PubMed] [Google Scholar]
- Dong H., Zhu G., Tamada K., Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Swallow M.M., Wallin J.J., Sha W.C. B7h, a novel costimulatory homolog of B7-1 and B7-2, is induced by TNFα. Immunity. 1999;11:423–432. doi: 10.1016/s1074-7613(00)80117-x. [DOI] [PubMed] [Google Scholar]
- Yoshinaga S.K., Whoriskey J.S., Khare S.D., Sarmiento U., Guo J., Horan T., Shih G., Zhang M., Coccia M.A., Kohno T. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 1999;402:827–832. doi: 10.1038/45582. [DOI] [PubMed] [Google Scholar]
- Reeves R.H., Patch D., Sharpe A.H., Borriello F., Freeman G.J., Edelhoff S., Disteche C. The costimulatory genes CD80 and CD86 are linked on mouse chromosome 16 and human chromosome 3. Mamm. Genome. 1997;8:581–582. doi: 10.1007/s003359900508. [DOI] [PubMed] [Google Scholar]
- Henry J., Miller M.M., Pontarotti P. Structure and evolution of the extended B7 family. Immunol. Today. 1999;20:285–288. doi: 10.1016/s0167-5699(98)01418-2. [DOI] [PubMed] [Google Scholar]
- Nishimura H., Nakano T., Minato N., Honjo T. Immunological studies on PD-1 deficient miceimplication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998;10:1563–1572. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- Tivol E.A., Borriello F., Schweitzer A.N., Lynch W.P., Bluestone J.A., Sharpe A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Waterhouse P., Penninger J.M., Timms E., Wakeham A., Shahinian A., Lee K.P., Thompson C.B., Griesser H., Mak T.W. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Nickloff B.J., Mitra R.S., Lee K., Turka L.A., Green J., Thompson C., Shimizu Y. Discordant expression of CD28 ligands, BB-1, and B7-1 on keratinocytes in vitro and psoriatic cells in vivo. Am. J. Pathology. 1993;142:1029–1040. [PMC free article] [PubMed] [Google Scholar]
- Suda T., Takahashi T., Golstein P., Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Ikemizu S., Gilbert R.J.C., Fennelly J.A., Collins A.V., Harlos K., Jones E.Y., Stuart D.I., Davis S.J. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12:51–60. doi: 10.1016/s1074-7613(00)80158-2. [DOI] [PubMed] [Google Scholar]
- Fitz L.J., Morris J.C., Towler P., Long A., Burgess P., Greco R., Wang J., Gassaway R., Nickbarg E., Kovacic S. Characterization of murine Flt4 ligand/VEGF-C. Oncogene. 1997;15:613–618. doi: 10.1038/sj.onc.1201191. [DOI] [PubMed] [Google Scholar]
- Freedman A.S., Freeman G.J., Rhynhart K., Nadler L.M. Selective induction of B7/BB-1 on interferon-gamma stimulated monocytesa potential mechanism for amplification of T cell activation through the CD28 pathway. Cell. Immunol. 1991;137:429–437. doi: 10.1016/0008-8749(91)90091-o. [DOI] [PubMed] [Google Scholar]