Intrinsic Defect in T Cell Production of Interleukin (IL)-13 in the Absence of Both IL-5 and Eotaxin Precludes the Development of Eosinophilia and Airways Hyperreactivity in Experimental Asthma (original) (raw)
Abstract
Interleukin (IL)-5 and IL-13 are thought to play key roles in the pathogenesis of asthma. Although both cytokines use eotaxin to regulate eosinophilia, IL-13 is thought to operate a separate pathway to IL-5 to induce airways hyperreactivity (AHR) in the allergic lung. However, identification of the key pathway(s) used by IL-5 and IL-13 in the disease process is confounded by the failure of anti–IL-5 or anti–IL-13 treatments to completely inhibit the accumulation of eosinophils in lung tissue. By using mice deficient in both IL-5 and eotaxin (IL-5/eotaxin−/−) we have abolished tissue eosinophilia and the induction of AHR in the allergic lung. Notably, in mice deficient in IL-5/eotaxin the ability of CD4+ T helper cell (Th)2 lymphocytes to produce IL-13, a critical regulator of airways smooth muscle constriction and obstruction, was significantly impaired. Moreover, the transfer of eosinophils to IL-5/eotaxin−/− mice overcame the intrinsic defect in T cell IL-13 production. Thus, factors produced by eosinophils may either directly or indirectly modulate the production of IL-13 during Th2 cell development. Our data show that IL-5 and eotaxin intrinsically modulate IL-13 production from Th2 cells and that these signaling systems are not necessarily independent effector pathways and may also be integrated to regulate aspects of allergic disease.
Keywords: allergy, cytokines, eosinophils, lung, inflammation
Introduction
Allergic asthma is recognized as a chronic inflammatory disease of the airways that is characterized by reversible airways obstruction in association with aberrant CD4+ Th 2 lymphocyte responses to common environmental stimuli (1, 2). The hallmark features of allergic asthma, elevated serum IgE, mucus hypersecretion, eosinophilia, and enhanced bronchial reactivity, or airways hyperreactivity (AHR),*to nonspecific spasmogenic stimuli, have all been linked to the effector functions of Th2 cytokines (e.g., IL-4, IL-5, IL-9, IL-10, and IL-13) and Th2 cells are obligatory for the development and expression of disease (2). Importantly, it is these pathogenic processes that are thought to promote airways obstruction in asthma, which predisposes to wheezing, shortness of breath, and life-threatening limitations in airflow.
Although the etiology and pathophysiology of asthma are complex, two independent models have been proposed as possible key mechanisms whereby Th2 cells regulate disease processes that predispose to structural and functional alterations of the airways. The first model identifies IL-5–regulated eosinophilia as a central pathogenic pathway. IL-5 regulates eosinophil function (development, activation, migration, and survival) and is a critical molecular switch for the induction of blood and tissue eosinophilia. This model is based on extensive clinical and experimental investigations that show a strong correlation between IL-5, eosinophils, and their secreted products with the severity and exacerbation of disease (3–12). The second model provides evidence that IL-13 underscores the development of mucus hypersecretion and AHR in the allergic lung, and implies dissociation between eosinophilia and these phenomena (13, 14). Although in comparison to IL-5 the data on the role of IL-13 in asthma are limited, the potency of this molecule in inducing AHR and mucus hypersecretion in experimental models has provided compelling evidence for a key pathogenic role in the induction of airways obstruction (13–15). Furthermore, production or expression of IL-13 and its receptor subunits have been directly linked to asthma and cells that play key roles in pathogenesis (16–18). Although these two models have been viewed as independent effector pathways, IL-13 has been shown to promote eosinophilia by IL-5– and eotaxin- (a CC chemokine) regulated mechanisms, which suggests that these molecules may cooperate to regulate certain aspects of the asthma phenotype (19–21).
Although extensive investigations have implicated IL-5–regulated eosinophilia as a central effector mechanism in asthma and an important clinical target for the resolution of this disease, the role of this pathway in the development and exacerbation of pathogenesis remains highly controversial. In experimental models, the inhibition of the actions of IL-5 consistently suppress pulmonary eosinophilia in response to antigen inhalation, however, this effect does not always correlate with a reduction of AHR (9, 10, 22–25). Indeed, this dichotomy is highlighted by findings from our laboratory in which allergic IL-5–deficient (IL-5−/−) mice of the C57BL/6 strain (10) do not develop antigen-induced AHR, whereas BALB/c mice develop enhanced reactivity independently of this factor (23). Recent clinical trials with humanized monoclonal antibodies raised against IL-5 limited eosinophil migration into the lung, but also failed to resolve AHR (26). In a small number of studies, eosinophils have also been dissociated from the induction of AHR in asthma (27, 28).
These anti–IL-5 studies indicate that mechanisms may operate in the allergic lung independently of this cytokine and eosinophils to induce disease, however, they do not unequivocally dissociate this cell from pathological processes. Although we and others have observed that eosinophil trafficking to the allergic lung is profoundly attenuated in IL-5−/− mice or those treated with anti–IL-5 antibodies by comparison to wild-type (WT) responses, a significant residual tissue eosinophilia can persist in these mice after allergen inhalation (9, 10, 22, 23, 25). Often, eosinophils in IL-5–depleted mice are present in the tissue even though they are almost absent from bronchalveolar lavage fluid. Furthermore, the degree of residual tissue eosinophilia directly correlates with the development of AHR and is under genetic regulation. In allergic IL-5−/− mice, tissue eosinophilia is 10–100-fold greater in the BALB/c strain where AHR persists in comparison to the C57BL/6 strain where airways reactivity is abolished (10, 23). Thus, it is important to recognize that anti–IL-5 treatment may not completely inhibit the accumulation of eosinophils in the allergic lung and that the residual cells may still contribute to pathogenesis through other regulatory processes.
One possibility is that local chemokine systems can operate independently of IL-5 to recruit eosinophils into the allergic lung. Of particular interest in allergic inflammation is the role of the eosinophil chemokine, eotaxin, because of its demonstrated potency and selectivity for eosinophil recruitment in experimental models and its strong clinical association with disease in humans (18, 29–33). Eotaxin production is also regulated by IL-13. Furthermore, although IL-5 is a cofactor for eotaxin-induced eosinophilia, we have also observed that this chemokine can regulate eosinophil migration independently of IL-5 (29). Similarly, we have shown that eotaxin plays a more pronounced role in contrast to IL-5 in regulating eosinophil recruitment to sites of allergic inflammation of the gastrointestinal tract (34). Thus, the secretion of IL-13 from activated Th2 cells may promote eotaxin production and eosinophil accumulation, and this pathway may contribute to disease processes independently of IL-5.
To investigate the possibility that pathways operated by eotaxin in the allergic lung can contribute to eosinophil accumulation and disease progression in the absence of IL-5, we generated BALB/c mice deficient in both IL-5 and eotaxin (IL-5/eotaxin−/−). Our data show that eotaxin plays a critical role in regulating eosinophil accumulation in the allergic lung independently of IL-5. Eotaxin and IL-5 deficiency not only abolished tissue accumulation of eosinophils, but also impaired the ability of antigen-specific CD4+ T cells to produce IL-13 and precluded the development of AHR. Thus, IL-5 and eotaxin not only regulate eosinophil migration, they also supply intrinsic signals that either directly or indirectly modulate Th2 cell production of IL-13 and subsequently, bronchoconstriction.
Materials and Methods
Genetically Manipulated Mice.
6–12-wk-old BALB/c mice were used in all experiments and obtained from the Special Pathogen Free Facility or the Gene Targeting Facility of the John Curtin School of Medical Research (Australian National University, Canberra, Australia). IL-5_−_ / −, eotaxin_−_ / −, and IL-5/eotaxin_−_ / _−_were each backcrossed for 12 generations with the BALB/c strain. BALB/c IL-5 transgenic (Tg) mice (∼49 transgene copies, male, 6–8-wk-old, and backcrossed to the 12th generation [35]) were obtained from the University of Adelaide (South Australia, Australia). Mice were treated according to the Australian National University Animal Welfare guidelines and were housed in an approved containment facility.
Induction of Allergic Airways Disease by OVA Sensitization.
6–8-wk-old mice were sensitized by intraperitoneal injection of 50 μg OVA/1 mg alhydrogel (Commonwealth Serum Laboratories) in 0.9% sterile saline. Nonsensitized mice received 1 mg alhydrogel in 0.9% saline. On days 12, 14, 16, and 18, all groups of mice were aeroallergen challenged with OVA (antigen) as previously described (10). 24 h after the last challenge, AHR was measured, the mice were killed by cervical dislocation, and T cell responses and inflammation and morphological changes to the airways were characterized.
Isolation of Donor Eosinophils.
Donor eosinophils for in vivo transfer and in vitro stimulation were derived from the peritoneal cavity of naive IL-5 Tg BALB/c mice and isolated using a FACStar™ Plus flow cytometer (Becton Dickinson) based on the forward, side, and light scatter properties of these cells as previously described (29). The purity of the eosinophils was measured on cytospin preparations and was ≥98%. The contaminating population consisted of macrophages and neutrophils and was devoid of lymphocytes (unpublished data).
Generation of Allergen-specific CD4+ T Cells.
OVA-specific CD4+ T cells were derived from mice that had been sensitized with OVA as previously described. In some experiments, mice received 2 × 106 eosinophils by intraperitoneal injection on the day of sensitization and again 3 d later. 6 d after sensitization, recipient mice were killed by cervical dislocation, the spleens were excised, and the splenocytes were disaggregated. Erythrocytes were lysed and the washed splenocytes were resuspended at 5 × 106 cells/ml in MLC with 10% heat-inactivated FCS, l-glutamine (2 mM), and neomycin sulfate (50 mg/liter). Splenocytes were then cultured for 4 d at 37°C in the presence of 200 μg/ml OVA to generate OVA-specific T cells. CD4+ T cells were then isolated using high gradient magnetic MiniMACS separation columns (Miltenyi Biotec) as previously described (15). 2 × 106 purified CD4+ T cells were then washed and resuspended in PBS and transferred to naive recipients. Purified CD4+ T cell populations were also analyzed for OVA-specific cytokine production. The purity of the enriched CD4+ cell fraction was uniformly >96% as determined by flow cytometry (unpublished data) and was devoid of granulocytes.
Antigen-specific Cytokine Production from Purified CD4+ T Cell Populations.
T cells (5 × 105 cells/well) were incubated with 25 μg/ml freshly isolated mitomycin C–treated splenocytes (5 × 105 cells/well) or purified eosinophils (2.5 × 105 cells/well) in complete medium in the presence of 200 μg/ml OVA in 96-well plates (250 μl/well) for 96 h to determine cytokine production. Cell-free culture supernatants were then collected and stored in aliquots at −70°C until analysis.
The Role of CD4+ T Cell–derived IL-13 in the Induction of AHR.
2 × 106 CD4+ T cells (WT or IL-13–deficient) were injected intravenously to naive recipient BALB/c WT or IL-5/eotaxin_−_ / −_mice. Control mice received PBS vehicle intravenously. 24 h after the adoptive transfer, mice were exposed to an aerosol of 10 mg/ml OVA in 0.9% saline for 30 min and then every day for 6 d. AHR to methacholine was measured 24 h after the last aeroallergen challenge and lung inflammation and morphology were characterized as previously described (10, 15, 36). In some experiments, recombinant murine IL-13 or PBS was instilled to the trachea of IL-5/eotaxin_− / _−_mice as previously described (20) and AHR was measured 48 h later.
Characterization of Lung Morphology.
Lung tissue representing the central (bronchi-bronchiole) and peripheral (alveoli) airways were fixed in 10% phosphate-buffered formalin, sectioned, and stained with Carbol's chromotrope-hematoxylin for the identification of eosinophils. The number of eosinophils in the central bronchi–bronchiole area were identified by morphological criteria and quantified as previously described (10, 36).
Stimulation of Peribronchial Lymph Nodes (PBLNs).
PBLNs were excised and filtered through nylon mesh (70 μm). The filtrate was then centrifuged at 500 g for 5 min at 4°C and the cell pellet was resuspended in red blood cell lysis solution and centrifuged at 500 g for 5 min at 4°C. The resulting pellet was cultured (5 × 105 cells/well) in complete medium in the presence of 200 μg/ml OVA in 96-well plates (250 μl/well) for 96 h. Cell-free culture supernatants were then collected and stored in aliquots at −70°C until cytokine levels were determined.
Analysis of Cytokines by ELISA.
IL-13 (R&D Systems), IL-4, and IL-5 (both from BD PharMingen) concentrations were determined in culture supernatants from OVA-stimulated CD4+ T cells and OVA-stimulated PBLN homogenates by ELISA according to the manufacturer's protocol.
Measurement of AHR.
Responsiveness to β-methacholine (methacholine) was assessed in conscious, unrestrained mice by barometric plethysmography, using apparatus and software supplied by Buxco Electronics. This system yields a dimensionless parameter known as enhanced pause (Penh) that reflects changes in wave form of the pressure signal from the plethysmography chamber combined with a timing comparison of early and late expiration. Measurement was performed as previously described (36). Notably, we have confirmed in the BALB/c strain that changes in Penh in response to methacholine directly correlate with changes in airway resistance to this spasmogen (20). Thus, measuring changes in Pehn reflects alterations in resistance and is indicative of enhanced airways responsiveness.
Stimulation of Purified Eosinophils.
2 × 106 FACS®-purified eosinophils were incubated with 10 μg/ml anti–mouse CD28 for 18 h in complete medium in 24-well culture plates in the presence or absence of recombinant IL-5 (20 ng/ml). For stimulation with IgA/anti-IgA, highly purified eosinophils were first incubated with secretory IgA at a final concentration of 5 μg/ml. After 1 h of incubation at 37°C, cells were transferred into 24-well plates and stimulated with 10 μg/ml anti-IgA monoclonal antibody for 18 h in the presence of recombinant IL-5 (20 ng/ml). After an 18-h culture, cell viability was determined by trypan blue exclusion (>90% viable cells) and RNA was extracted from ∼2 × 106 eosinophils.
Reverse Transcription (RT)-PCR Analysis.
Total RNA was isolated from PBLN cells and eosinophils by standard methods with RNAzol B (Biotech Laboratories). An RT-PCR procedure was performed as previously described (37) to determine the relative quantities of mRNA for various cytokines. The primers and probes for all genes were purchased from GIBCO BRL. Primer and probe sequences for HPRT have been described elsewhere (38). Primer and probe sequences for IL-4, IL-5, IL-13, IL-12, CCR3, IL-18, and eotaxin are as follows: IL-4: sense, GAATGTACCAGGAGCCATATC, antisense, CTCAGTACTACGAGTAATCCA, probe, AGGGCTTCCAAGGTGCTTCGCA; IL-5: sense, GACAAGCAATGAGACACGATGAGG, antisense, GAACTCTGCAGGTAATCCAGG, probe, GGGGGTACTGTGGAAATGCTTAT; IL-13: sense, CTCCCTCTGACCCTTAAGGAG, antisense, GAAGGGGCCGTGGCGAAA-CAG, probe, TCCAATTGCAATGCCATCTAC; IL-12(p40): sense, CGTGCTCATGGCTGGTGCAAAG, antisense, CTTCATCTGCAAGTTCTTGGGC, probe, TCTGTCTGCAGAGAAGGTCACA; CCR3: sense, AAG TAC AGG AAG CTA CAA ATT ATG, antisense, AGC AGA GTT TTA ATG ATT CCT GAG, probe, GGCCTTGCAGGACTGGCAGC; IL-18: sense, ACT GTACAACCGCAGTAATACGG, antisense, AGTGAACATTACAGATTTATCCC, probe, GAACAAGATCATTTCCTTTGAGG; and eotaxin: sense, TCCACCATGCAGAGCTCCACAG, antisense, CCCACATCTCCTTTCAT-GCCCC, probe, GGAACACAATGGGACGAGTTAGG. After the appropriate number of PCR cycles, the amplified DNA was analyzed by gel electrophoresis and Southern blotting, and detected using the enhanced chemiluminescence detection system as recommended by the manufacturer (Amersham Pharmacia Biotech). PCR amplification with the HPRT reference gene was performed to assess variations in cDNA or total RNA loading between samples.
Generation of Th2 Cells from Naive CD4+ T Cells.
CD4+ T cells from naive mice were isolated from splenocytes with magnetic-activated cell sorting and cultured at 5 × 106 cells/ml in complete medium in the presence of anti-CD3 (50 ng/ml; clone 2C11), recombinant murine IL-4 (20 ng/ml), and anti–IFN-γ (40 μg/ml; clone R46A2) for 4 d to generate Th2 cells (15). In some experiments, recombinant IL-18 (20 or 200 ng/ml; R&D Systems) was also added to the cultures. Th2 cells (1.25 × 106 cells/well) were then restimulated in the presence of anti-CD3 (50 ng/ml) and anti-CD28 (1 μg/ml) in 96-well plates (250 μl/well) for 72 h to determine cytokine release. Cell-free culture supernatants were collected and stored in aliquots at −70°C until analysis by ELISA.
Collection of Sputum Samples.
Children with moderate, persistent asthma were recruited from the outpatient department at the University Children's Hospital of Freiburg (Freiburg, Germany). The diagnosis of asthma was based according to the criteria of the American Thoracic Society (39). The patients were in a stable condition and had been free of respiratory infections. All patients were under continuous inhalation treatment with 400–800 μg budesonide per day. Sputum induction was performed as previously described (40). In brief, 10 min after the inhalation of 200 μg salbutamol, subjects inhaled hypertonic saline (3, 4, and 5%) via an ultrasonic nebulizer (Ultraneb 2000; De Villbiss) with the output set at maximum (4.5 ml/min) for three consecutive periods of 10 min for each concentration. Lung function was recorded before the procedure and every 5 min for safety by using a Masterscope 4.0 (Jaeger). To collect sputum, subjects were asked to rinse their mouth, blow their nose, swallow water, and then expectorate the sputum onto a plastic Petri dish after the first 10-min period of inhalation and every 5 min thereafter. Adequate plugs of sputum were selected to reduce contamination with saliva and were processed immediately. Sputum processing was performed as previously described (40). In brief, the weight of selected plugs was determined and twice their volume of dithiothreitol 0.1% (sputalysin; Calbiochem) was added. Samples were placed in a water bath at 37°C for 15 min to ensure the complete dissolution and PBS was added to achieve a 25-fold diluted final concentration. Cell-free supernatant was stored at −70°C until additional analysis was performed. Eosinophil cationic protein (ECP; detection limit: 4 ng/ml, diluted sample: 100 ng/ml; Pharmacia & Upjohn) and IL-18 (detection limit: 12 pg/ml, diluted sample: 300 pg/ml; Medical & Biological Laboratories Co Ltd.) were measured in the supernatants of the selected plugs. The study was approved by the ethics committee of the Albert-Ludwigs University of Freiburg (Freiburg, Germany) and written informed consent from parents and patients was obtained in advance.
Statistical Analysis.
The significance of the differences between experimental groups was analyzed using Student's unpaired t test. Values were reported as the mean ± SEM. Differences in mean values were considered significant if P < 0.05. The relation between variables was calculated using Spearmann's rank correlation coefficients.
Results
Cooperative Signaling Events between IL-5 and Eotaxin Regulate AHR and Tissue Levels of Eosinophils.
Baseline levels of airways reactivity were not significantly different between nonallergic WT, eotaxin−/−, IL-5−/−, or IL-5/eotaxin−/− BALB/c mice (Fig. 1 a). However, antigen (OVA) inhalation induced a marked AHR in allergic WT, eotaxin−/−, and IL-5−/− mice (Fig. 1 a). In contrast, in the absence of both IL-5 and eotaxin, AHR was completely abrogated (Fig. 1 a), suggesting that these molecules act in concert (potentially through eosinophils) to regulate enhanced reactivity (data is only shown for 25 mg methacholine, but is representative of the full concentration effect curve to methacholine). To determine the impact of IL-5 and eotaxin deficiency on eosinophil expansion and migration in response to antigen inhalation in the lung, numbers of this leukocyte were quantified in the bone marrow, blood, and pulmonary compartments of allergic mice deficient in these factors. Eosinophils expanded in the bone marrow (unpublished data) and blood (Fig. 1 b) compartments of allergic WT and eotaxin−/− mice. However, in the absence of IL-5, eosinophilia in these compartments was not observed. Although blood and bone marrow eosinophilia was critically regulated by IL-5, the accumulation of eosinophils in the pulmonary compartment was modulated by both IL-5 and eotaxin (Fig. 1 c). Eosinophil numbers observed in the airway wall of allergic IL-5−/− or eotaxin−/− mice were markedly decreased in comparison to those observed in WT mice, whereas in the absence of both IL-5 and eotaxin, eosinophil accumulation was totally abrogated (Fig. 1 c). Only occasionally were eosinophils identified in the perivascular region of the allergic IL-5/eotaxin−/− lung at numbers similar to those seen in nonallergic mice (unpublished data). Notably, although slightly attenuated, mucus hypersecretion was still a marked feature in the allergic lung of IL-5/eotaxin−/− mice (unpublished data).
Figure 1.
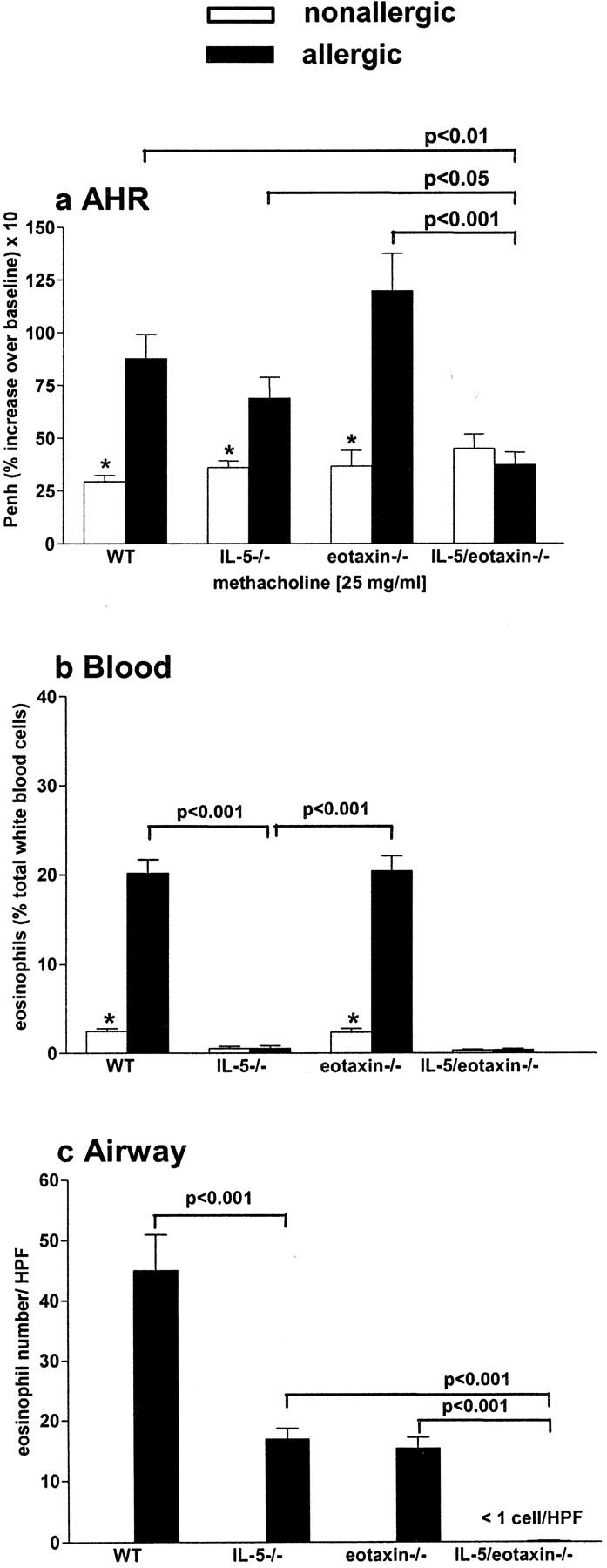
IL-5 and eotaxin deficiency abrogates AHR. Groups of mice (WT, IL-5 −/−, eotaxin−/−, or IL-5/eotaxin−/−) were sensitized with saline (nonallergic) or OVA (allergic) and aeroallergen challenged with OVA. (a) AHR data represent the percentage increase in Penh at 25 mg/ml methacholine over baseline reactivity in the absence of cholinergic stimuli. The maximal response to methacholine (25 mg/ml) is shown, but these results are representative of the full dose response curve. Data is the mean of six to eight mice ± SEM per group. (b) The percentages of blood eosinophils. Data is the mean ± SEM of four mice per group. (c) Mean number of eosinophils residing in the airway wall per 10 similar HPFs (×1,000) for each group. Data is the mean ± SEM of four mice per group). *, significant differences (P < 0.05) between respective nonallergic and allergic groups. Levels of significant differences are also indicated for other groups.
These data indicate that both IL-5 and eotaxin can independently recruit eosinophils into the lung, but collectively they provide the primary signals for the homing of this cell to allergic airways. Importantly, in the absence of IL-5 and a pronounced blood eosinophilia, mechanisms still exist to recruit eosinophils into the allergic lung from the basal eosinophil population. These data also suggest that it is the residual eosinophil population, which is very pronounced by comparison with the airways of nonallergic mice, in the lung of allergic IL-5−/− and eotaxin−/− mice that may promote AHR.
IL-13 Production by Antigen-specific CD4+ T Cells and in PBLNs Is Impaired in the Absence of IL-5 and Eotaxin.
As signals elicited by CD4+ T cells have been shown to play an obligatory role in the induction of AHR (2), the impact of IL-5/eotaxin deficiency on CD4+ T cell production of IL-13, IL-4, and IL-5 was examined after their isolation from allergic WT, IL-5_−_ / −, eotaxin_−_ / −, and IL-5/eotaxin_−_ / −_mice. After in vitro antigen stimulation of whole splenocyte populations taken from allergic mice, CD4+ T cells were isolated and restimulated at equivalent numbers (5 × 105 cells/well = 2 × 106 cells/ml) with antigen-loaded mitomycin C–treated splenocytes (Fig. 2 , a–c). Stimulation of IL-5_− / −, eotaxin_−_ / −, IL-5/eotaxin_−_ / −, and WT CD4+ T cells under identical conditions promoted the production of equivalent levels of IL-4 but not IL-13 (Fig. 2, a and b). The level of IL-13 produced by IL-5_−_ / −, eotaxin_−_ / −, or IL-5/eotaxin_−_ / −_CD4+ T cells was significantly reduced when compared with those derived from WT mice (Fig. 2 a). Notably, the levels of IL-13 in IL-5/eotaxin_− / −_CD4+ T cell cultures were 10-fold less compared with WT cultures and were further reduced in comparison to the levels observed with IL-5_− / −_or eotaxin_− / −_mice. The reduced levels of IL-13 did not correlate with an overall suppression in Th2-type cytokine production, as IL-5 levels produced by eotaxin_− / _−_CD4+ T cells and IL-4 levels in all cultures were similar to those observed in WT CD4+ T cell cultures (Fig. 2, b and c).
Figure 2.
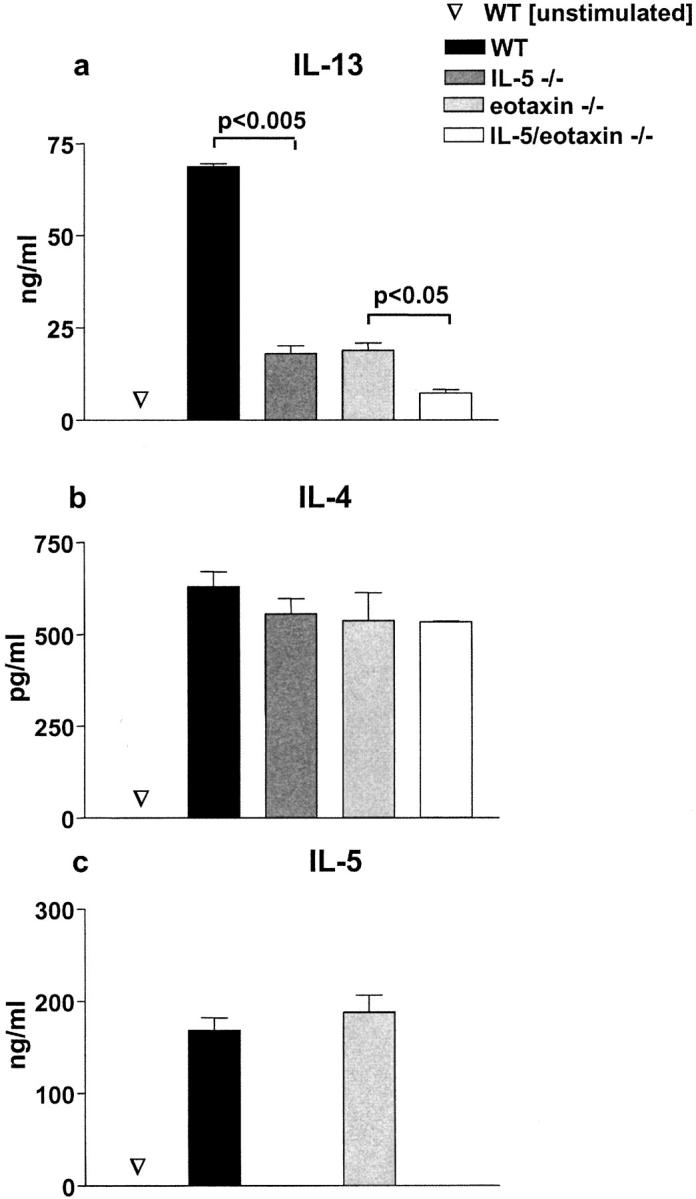
Intrinsic defect in IL-13 production by antigen-specific CD4+ T cells in the absence of IL-5 and eotaxin. Production of (a) IL-13, (b) IL-4, and (c) IL-5 by purified OVA-specific CD4+ T cells generated from WT, IL-5 −/−, eotaxin−/−, or IL-5/eotaxin−/− mice. Cultures were performed in triplicate. CD4+ T cells were derived from splenocytes because these T cell cytokine profiles were directly reflective of those observed after the stimulation of PBLN cultures. Data represent the mean ± SEM of n = 4 cultures per group. Significant differences (P < 0.05) between respective groups are shown.
The limited ability of CD4+ T cells isolated from allergic IL-5/eotaxin−/− mice to produce IL-13 also correlated with the reduced levels of IL-13 and the number of eosinophils found in the PBLN. This correlation was also observed in IL-5−/− mice and eotaxin−/− mice (unpublished data). Antigen provocation of WT mice induced a significant increase in the number of eosinophils in the PBLN (50-fold; 7.2 ± 0.5 eosinophils/high power field [HPF]) as compared with nonallergic mice (0.14 ± 0.06 eosinophils/HPF). Accumulation of eosinophils in PBLN directly correlated with increased levels of IL-13 production within the lymph nodes (48.5 ± 6.1 ng/ml in allergic WT and not detectable in nonallergic WT in PBLN homogenates). In contrast, in allergic IL-5/eotaxin−/− mice, eosinophil numbers (0.73 ± 0.18 eosinophils/HPF) in the PBLN after antigen inhalation were similar to those observed in WT nonallergic mice and IL-13 levels were markedly diminished (<7.0 ± 0.4 ng/ml in PBLN homogenates). Thus, eosinophil deficiency in IL-5/eotaxin−/− mice (and in IL-5−/− and eotaxin −/− mice, unpublished data) directly correlated with impaired CD4+ T cell production of IL-13 and with the levels of cytokine generated in the allergic lung. Furthermore, the complete removal of this granulocyte from pulmonary tissues in IL-5/eotaxin−/− mice, which is where IL-13 levels were the lowest, correlated with the abolition of airways reactivity to cholinergic stimuli (Fig. 1, a and c). Collectively, these data show that the abolition of IL-5 or eotaxin signaling predisposes (potentially due to eosinophil deficiency) to an intrinsic defect in T cell production of IL-13 that precludes the development of AHR.
Transfer of IL-13–producing Antigen-specific CD4+ T Cells to Naive IL-5/Eotaxin−/− Mice Reconstitutes Normal IL-13 Levels in the Allergic Lung and Induces AHR and Eosinophilia.
The observations in allergic IL-5/eotaxin−/− mice suggested that the absence of these molecules resulted in the abolition of AHR by limiting the ability of CD4+ T cells to produce IL-13. Therefore, we adoptively transferred WT antigen-specific CD4+ T cells that are competent in their ability to produce IL-4, IL-5, and IL-13 to naive WT and IL-5/eotaxin−/− mice to directly determine the contribution of T cell–derived IL-13 (in association with IL-5) to the induction of disease. Transfer of these CD4+ T cells (2 × 106) into naive WT mice followed by the subsequent delivery of antigen (OVA) to the airways induced the hallmark features of allergic airways inflammation, which included AHR, mucus hypersecretion (unpublished data), and peripheral blood and pulmonary eosinophilia (Fig. 3 , a–c). Adoptive transfer of this CD4+ T cell population into naive IL-5/eotaxin−/− mice also induced AHR to levels observed in WT mice (Fig. 3 a). However, in the absence of endogenous IL-5 and eotaxin, although peripheral blood and pulmonary eosinophilia were induced, responses were attenuated in comparison to the WT (Fig. 3, b and c). Concomitant with the transfer of CD4+ T cells, the levels of IL-4, IL-5, and IL-13 increased in the lung (PBLN; Fig. 3, d–f). Stimulation of PBLN cultures from IL-5/eotaxin−/−– and WT-recipient mice with antigen promoted the production of IL-4, IL-13, and IL-5 (Fig. 3, d–f). The levels of IL-13 and IL-4 produced in PBLN cultures from IL-5/eotaxin−/− mice were similar to those observed in WT mice (Fig. 3, d and e). In contrast, the level of IL-5 produced from IL-5/eotaxin−/− cultures was significantly lower than that observed in WT mice (Fig. 3 f). This latter finding can be explained by the inability of the recipient's endogenous PBLN cells to produce IL-5 and is consistent with the observed reduction in peripheral blood and pulmonary eosinophilia in IL-5/eotaxin−/− mice receiving CD4+ T cells in comparison with WT responses (Fig. 3, b and c). Thus, by overcoming the functional defect in the production of IL-13 and IL-5 within the CD4+ T cell compartment, the hallmarks of allergic asthma could be induced in IL-5/eotaxin−/− mice. These data also show that T cell activation processes by antigen are functional in IL-5/eotaxin−/− mice.
Figure 3.

Adoptive transfer of IL-13–producing CD4+ T cells to naive IL-5/eotaxin−/− mice reconstitutes normal IL-13 levels in the allergic lung and induces AHR and eosinophilia. CD4+ T cells (2 × 106 per mouse) were adoptively transferred into IL-5/eotaxin−/− or WT mice. Control groups received PBS. (a) AHR. Data represent the percentage increase in Penh at 25 mg/ml methacholine over baseline reactivity in the absence of cholinergic stimuli. The maximal response to methacholine (25 mg/ml) is shown, but these results are representative of the full dose response curve. Data is the mean of six to eight mice ± SEM per group. (b) The percentage of blood eosinophils. Data represent the mean ± SEM of three or four mice per group. (c) Mean number of airway wall/smooth muscle eosinophils per 10 similar HPF (×1,000) for each group. Data mean ± SEM of two or three mice per group. (d–f) Production of (d) IL-13, (e) IL-4, and (f) IL-5 from PBLN cells after OVA restimulation. T cell cytokine levels were determined in PBLN culture because these directly reflect the trafficking of the transferred cells to the lung and the subsequent release of cytokines in the pulmonary compartment. *, significant differences (P < 0.05) between control and T cell transferred groups. Levels of significant differences are also indicated for other groups.
To further confirm the critical requirement of IL-13 for the induction of AHR, antigen-specific IL-13–deficient CD4+ T cells, which are only defective in their ability to produce IL-13, were adoptively transferred to naive IL-5/eotaxin−/− mice (Fig. 4 a). Recombinant mIL-13 was also instilled into the trachea of mice deficient in both IL-5 and eotaxin (Fig. 4 b). In contrast to WT CD4+ T cells, IL-13–deficient T cells failed to induce AHR. However, when only IL-13 was delivered to the pulmonary microenvironment, AHR was induced. These data confirm the essential requirement of IL-13 in the induction of AHR.
Figure 4.
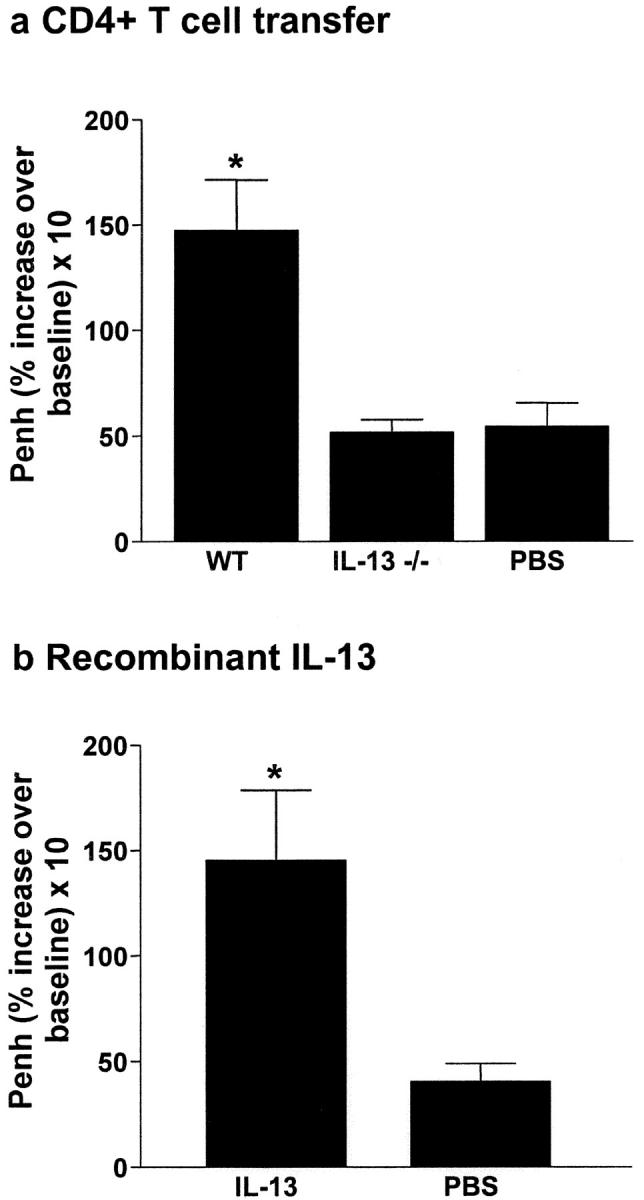
IL-13 plays a critical role in the generation of AHR. (a) Adoptive transfer of IL-13–deficient CD4+ T cells (2 × 106 per mouse) to naive IL-5/eotaxin−/− mice failed to induce AHR. Control groups received PBS. (b) In contrast, instillation of mIL-13 in the lungs of naive IL-5/eotaxin−/− mice induced AHR. Data is the mean of four to eight mice ± SEM per group. *, significant differences (P < 0.05) between groups is shown.
Adoptive Transfer of Eosinophils to IL-5/Eotaxin−/− Mice Reestablishes IL-13 Production from CD4+ T Cells.
Collectively, the above findings in allergic mice deficient in IL-5 and/or eotaxin lead us to speculate that limitations in the ability of their CD4+ T cells to produce IL-13 may be a direct result of eosinophil deficiency, rather than the absence of the cytokines themselves. To examine the role of eosinophils in regulating IL-13 secretion from CD4+ T cells in vivo, we purified and adoptively transferred eosinophils (4 × 106) into IL-5/eotaxin−/− mice and then sensitized these mice with antigen (OVA). The transferred eosinophil population was highly enriched and did not contain T lymphocytes as determined by FACS® analysis and morphological criteria after cytocentrifugation (unpublished data). 6 d after sensitization and intraperitoneal injection of eosinophils, whole splenocyte populations were removed and cultured with antigen for 4 d. After primary stimulation, CD4+ T cells (2 × 106 cells/ml) were purified and restimulated with antigen-loaded mitomycin C–treated splenocytes or eosinophils (Fig. 5 , a and b). Similarly, CD4+ T cells were also prepared from sensitized WT and IL-5/eotaxin−/− mice that did not receive exogenous eosinophils. Secondary stimulation of WT CD4+ T cells with antigen-loaded splenocytes induced a significant increase in the levels of IL-13 compared with IL-5/eotaxin−/− CD4+ T cells (Fig. 5 a). However, when eosinophils were transferred to IL-5/eotaxin−/− mice before sensitization, the levels of IL-13 produced by CD4+ T cells after secondary stimulation with antigen-loaded splenocytes was equivalent to that observed in WT cultures (Fig. 5 a). To ascertain whether the eosinophil was promoting IL-13 production from T cells indirectly or had the ability to directly engage these cells through antigen presentation, CD4+ T cells from all groups of sensitized mice were subjected to secondary stimulation in the presence of antigen-loaded eosinophils (Fig. 5 b). Secondary stimulation of CD4+ T cells from WT mice in the presence of eosinophils produced significantly higher levels of IL-13 compared with IL-5/eotaxin−/− CD4+ T cells (Fig. 5 b), and these levels were similar to those observed in cells stimulated with splenocytes (Fig. 5 a). Moreover, secondary stimulation of CD4+ T cells from IL-5/eotaxin−/− mice, which received eosinophils before sensitization, produced similar levels of IL-13 to those observed in T cell cultures from WT mice (Fig. 5 b). These data support the concept that eosinophil deficiency induced in the absence of IL-5 and eotaxin predisposes to an intrinsic defect in T cells that subsequently impairs the ability of antigen-specific CD4+ T cells to produce IL-13.
Figure 5.
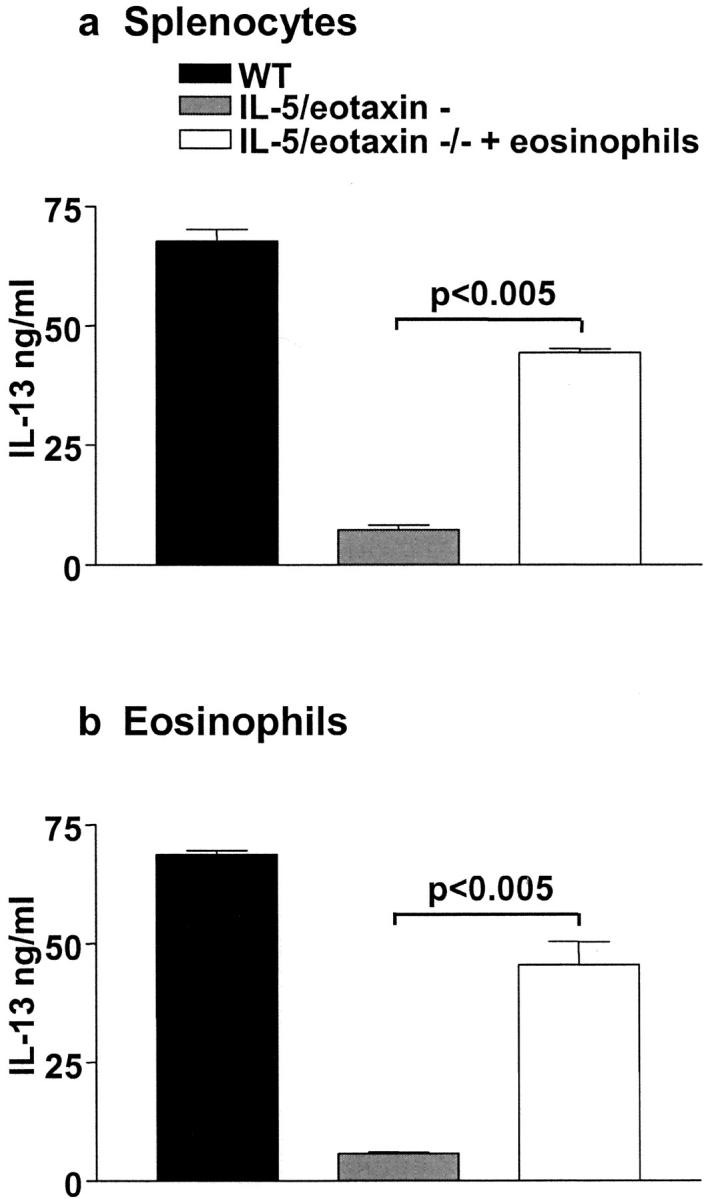
Adoptive transfer of eosinophils to IL-5/eotaxin−/− mice reestablishes normal production of IL-13 from antigen-specific CD4+ T cells. IL-13 production from purified CD4+ T cells derived from allergic WT and IL-5/eotaxin−/− mice or allergic IL-5/eotaxin−/− mice that had been injected with eosinophils intraperitoneally before sensitization. CD4+ T cells stimulated in the presence of OVA with (a) freshly isolated and mitomycin C–treated splenocytes or (b) with purified eosinophils. Cultures were performed in triplicate. Data represent the mean ± SEM of n = 4 cultures per group. Significant differences (P < 0.05) between respective groups are shown. Cytokine production was analyzed from T cells derived from splenocytes after eosinophil transfer experiments. Eosinophils injected intraperitoneally migrate to the spleen and colocalize with T cells in this compartment. Thus, by taking T cells from the spleen we know that they have been exposed to the transferred eosinophil population.
IL-18 Expression Is Decreased in PBLN of Allergic IL-5/Eotaxin−/− Mice and Up-regulated in IL-5–primed Eosinophils.
As IL-18 has been identified as an important modulator of Th2 cell immunity and, in particular, a key regulator of IL-13 production from this cell (41), we investigated the levels of expression of IL-18 in allergic PBLN isolated from WT, IL-5−/−, and IL-5/eotaxin−/− mice. RT-PCR for IL-5, eotaxin, IL-13, and IL-18 was performed and specificity of PCR products was confirmed by Southern blot analysis using 30-mer oligonucleotide probes for HPRT, IL-5, eotaxin, IL-13, and IL-18 (Fig. 6 a). In allergic WT mice all factors were abundantly expressed. In contrast, the levels of expression of these factors, in particular IL-13 and IL-18, were attenuated in the absence of IL-5. Moreover, in allergic IL-5/eotaxin−/− mice, the levels of IL-13 and IL-18 expression were low or barely detectable (Fig. 6 a), which also correlates with the low levels of eosinophils in PBLN that were previously described. Thus, eosinophil deficiency correlated with the decreased expression of IL-13 and IL-18 in allergic lymph nodes. To determine if eosinophils could express IL-18, we cultured purified eosinophils with IL-5 and stimulated these cells with anti-CD28 or IgA–anti-IgA complex to activate cytokine gene expression (42). CCR3, the receptor for eotaxin, and IL-4 were expressed in unstimulated eosinophils (Fig. 6 b). Stimulation of eosinophils with IL-5 up-regulated the expression of IL-18. Notably, IL-18 expression was additionally enhanced by the stimulation of IL-5–primed eosinophils with anti-CD28 or IgA–anti-IgA complex. Although IL-12 expression was also induced under these conditions, we could not detect transcripts for IL-13. These data indicate that eosinophils potentially have the ability to synthesize IL-18, which could subsequently influence T cell function when released into the same microenvironment.
Figure 6.

IL-18 expression is decreased in PBLN of allergic IL-5/eotaxin−/− mice and up-regulated in IL-5–primed eosinophils. (a) PBLN cells from WT, IL-5−/−, and IL-5/eotaxin−/− were isolated and RT-PCR was performed for HPRT, IL-5, eotaxin, IL-13, and IL-18. To confirm specificity, PCR products were probed by Southern blot analysis using 30-mer oligonucleotide probes for these factors. The mean fold decreases in cytokine levels in comparison to WT are as follows: eotaxin in IL-5−/− (10-fold reduction); IL-13 in IL-5−/− (15-fold reduction) and in IL-5/eotaxin−/− (40-fold reduction); and IL-18 in IL-5−/− (15-fold reduction) and IL-5/eotaxin−/− (30-fold reduction). PBLN were analyzed to directly reflect the production of factors within the allergic lung. (b) Highly purified eosinophils were stimulated with IL-5/anti-CD28 or IgA–anti-IgA complex for 18 h to activate cytokine gene expression. RT-PCR was performed for HPRT, CCR3, IL-18, IL-13, IL-4, and IL-12 (p40). PCR products were probed by Southern blot analysis using 30-mer oligonucleotide probes for these factors.
IL-18 Restores IL-13 Production in Polarized Th2 Cells from IL-5/Eotaxin−/− Mice.
To define at what stage IL-5 and eotaxin deficiency may impair IL-13 production from T cells, and to identify the potential of IL-18 to overcome the intrinsic defect, we isolated naive CD4+ T cells from the spleens of WT, IL-5/eotaxin−/−, and IL-5 Tg mice. Eosinophils, which localize with T cells in the spleens of WT mice, are absent in spleens from IL-5/eotaxin−/− mice and overly abundant in this compartment in IL-5 Tg mice (unpublished data). Naive CD4+ T cells were then polarized to the Th2 phenotype under standard conditions in the absence of antigen and antigen-presenting cells and restimulated with anti-CD28/anti-CD3. Like antigen-specific CD4+ T cells isolated from allergic mice, polarized Th2 cells from IL-5/eotaxin−/− mice were limited in their ability to produce IL-13 (Fig. 7 a) but not IL-4 (Fig. 7 b) when compared with WT Th2 cells. In contrast, Th2 cells derived from naive IL-5 Tg mice produced exaggerated levels of IL-13 in response to stimulation (Fig. 7 a, right). Incubation of naive CD4+ T cells isolated from IL-5/eotaxin−/− mice with IL-18 during polarization and subsequent secondary stimulation restored the ability of these T cells to produce IL-13 (Fig. 7 c). Stimulation of Th2 cells from IL-5/eotaxin−/− mice with IL-18 (20 ng) produced similar levels of IL-13 compared to WT cells stimulated with anti-CD28 and anti-CD3 (Fig. 7 a). These data suggest that there is an intrinsic and selective dysfunction in IL-13 production during CD4+ Th2 cell differentiation in the absence of IL-5/eotaxin. Moreover, the defect in IL-13 production is proportionally linked to eosinophil deficiency and is overcome by signals elicited by IL-18. Although not conclusive, these data (Figs. 6 and 7) are suggestive that eosinophils may regulate IL-13 production from CD4+ T cells through IL-18–mediated mechanisms.
Figure 7.
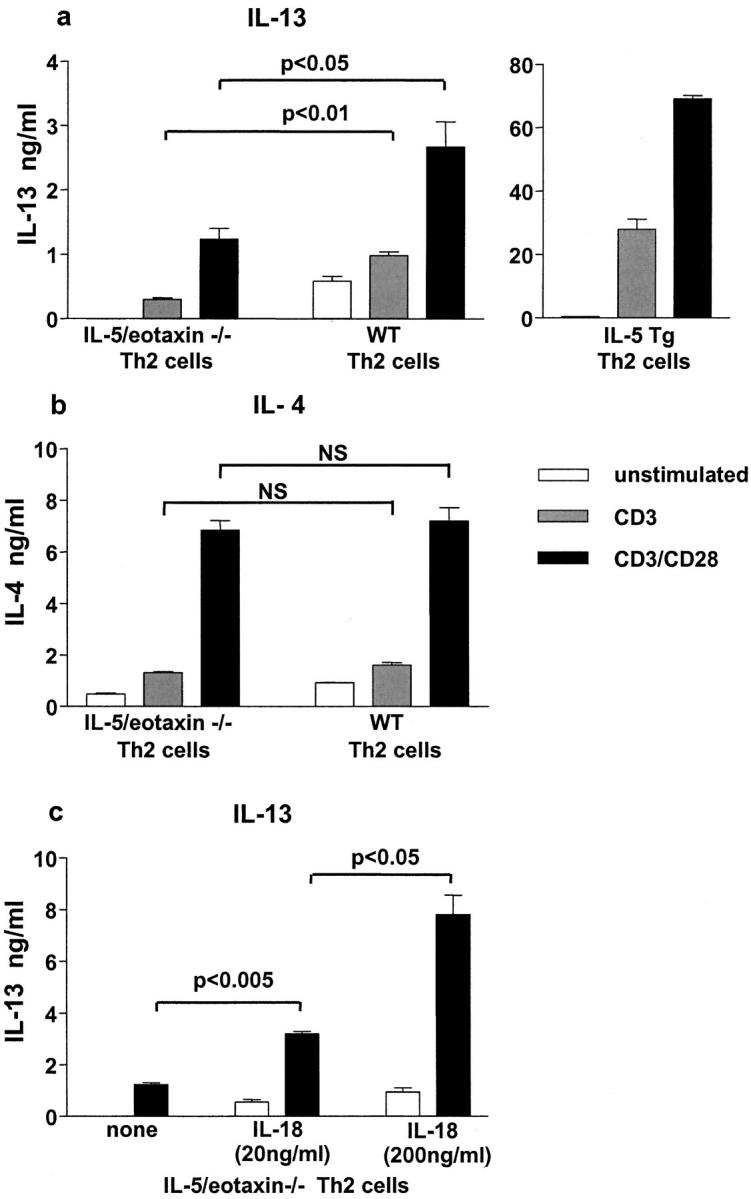
IL-13 production is impaired in polarized Th2 cells derived from IL-5/eotaxin−/− mice, but the defect is overcome by addition of IL-18 to cultures. Production of (a) IL-13 and (b) IL-4 by CD4+ Th2 cells after polyclonal stimulation. Th2 cells were generated from purified naive CD4+ T cells isolated from the spleens of WT, IL-5/eotaxin−/−, or IL-5 Tg mice. (c) Release of IL-13 by CD4+ Th2 cells after polyclonal (solid bar) in the absence or presence of IL-18 (20 and 200 ng/ml, respectively). Cultures were performed in triplicate. Data represent the mean ± SEM of n = 4 cultures per group. The spleen was used as a source of naive T cells to enable the generation of sufficient Th2 cells.
IL-18 Correlates with Eosinophilic Airway Inflammation in Asthma.
To determine the potential relevance of our observations to human disease, we measured the levels of IL-18 and ECP in the induced sputum of children suffering from allergic asthma (40). Analysis of IL-18 (mean [min/max]: 2,385 [300–8,025] pg/ml) and ECP (mean: 1,509 [<100/4,625] ng/ml) in 23 paired samples showed a highly significant correlation (rs = 0.70, P = 0.0002) between this cytokine and the eosinophil-derived product in the airways. Thus, we could also demonstrate a strong correlation between IL-18, eosinophils, and allergic inflammation in the human lung.
Discussion
In this investigation we show that deficiency in IL-5, eotaxin, or both molecules predisposes to an intrinsic defect in T cells that subsequently impairs the ability of antigen-specific CD4+ T cells from allergic mice or in vitro–polarized Th2 cells to produce IL-13. Furthermore, this defect in T cell production of IL-13 (in IL-5/eotaxin−/− mice) correlates with the abrogation of AHR. Although we cannot unequivocally exclude the possibility that IL-5/eotaxin regulate T cell production of IL-13 independently of eosinophils, our data indicate that eosinophil deficiency caused by the absence of these cytokines predisposes to this intrinsic defect in T cells. The number of eosinophils present in tissues where T cells colocalize at baseline or during allergic inflammation directly correlates with the subsequent potential of CD4+ T cells to produce IL-13. Eosinophils transferred to IL-5/eotaxin−/− mice before sensitization also restored the ability of antigen-specific CD4+ T cells to produce IL-13. Although the IL-5R α chain, which is essential for IL-5 signal transduction, is expressed in B lymphocytes and in limited studies has been implicated in the generation of cytotoxic T lymphocytes, its expression in CD4+ Th cells has not been observed (43, 44). A direct effect of IL-5 on this subclass of lymphocyte is therefore unlikely. Furthermore, CD4+ T cell production of IL-5 was not impaired in allergic eotaxin−/− mice (Fig. 2), which suggests that the T cell defect was not due solely to the absence of IL-5, but to involved processes downstream of these molecules. Notably, in allergic IL-5−/− C57BL/6 mice, IL-13 levels (unpublished data) in PBLN were lower than those reported here in IL-5−/− BALB/c mice, further linking total tissue eosinophil numbers with IL-13 production and the development of AHR.
Our data support recent investigations that show eosinophils can modulate CD4+ T cell function. Eosinophils in the allergic lung can present antigen and traffic to local lymph nodes where they colocalize with T cells (45) and this granulocyte can induce proliferation and cytokine secretion from Th2 cells (46). Eosinophils can also secrete a wide range of T cell growth and chemotactic factors (47). Thus, evidence is emerging that eosinophils may not only act as terminal effector cells, but can also actively modulate allergic inflammation by amplifying type 2 cytokine responses.
Notably, eosinophil deficiency did not predispose to a generalized deficiency in CD4+ T cell cytokine production, nor in the ability of the immune system to activate this T cell. IL-4 secretion was not significantly altered in CD4+ T cells derived from mice deficient in IL-5, eotaxin, or IL-5 and eotaxin. Furthermore, the transfer of WT CD4+ T cells to eotaxin/IL-5−/− mice not only highlighted the importance of T cells and presumably T cell–derived IL-13 for the onset of AHR and pathophysiology, but also demonstrated that normal activation mechanisms for T cells can operate in the absence of eosinophils. The specific role for IL-13 in the mechanism of AHR was demonstrated by the transfer of IL-13–deficient T cells to eotaxin/IL-5−/− mice where AHR did not develop, and the delivery of IL-13 alone to the airway microenvironment, inducing AHR, of these factor-deficient mice. Thus, these data suggest that the CD4+ T cell defect may relate specifically to impaired production of IL-13 and that eosinophils can provide a factor that either directly or indirectly regulates IL-13 production. The limitation in T cell IL-13 production predisposes to the abrogation of AHR.
The molecular mechanisms that regulate IL-13 production in T cells are largely unknown. However, we were interested in the role of IL-18 as this cytokine has been identified not only as a modulator of both Th1 and Th2 development (41, 48), but more importantly as a potent cofactor for the regulation of the expression of IL-13 from T cells (41). Notably, in the allergic lymph nodes from WT, IL-5−/−, or IL-5/eotaxin−/− mice, the levels of expression of IL-18 directly correlated with the levels of IL-13, the production of IL-13 mRNA, and the number of eosinophils localized to this compartment. Furthermore, eosinophils primed with IL-5 and stimulated with molecules that promote cytokine production in this cell showed that IL-18 expression was up-regulated. Thus, eosinophils, through the secretion of IL-18, may potentially regulate IL-13 production from CD4+ T cells located within the same microenvironment. Indeed, we were able to show a direct correlation between the number of eosinophils within the microenvironment of naive CD4+ T cells and their subsequent ability to produce IL-13 after clonal expansion to the Th2 phenotype. Th2 cells derived from the spleens of IL-5/eotaxin−/− mice where eosinophils are absent were limited in their ability to produce IL-13, whereas Th2 cells generated from the spleens of IL-5 Tg mice where eosinophils are overly abundant produced exaggerated levels of this cytokine in comparison to WT Th2 cells. Furthermore, the addition of IL-18 to cultures during polarization restored the ability of Th2 cells derived from eosinophil-deficient mice to produce normal levels of IL-13. Current data on the role of IL-18 in the regulation of Th2 immune responses are paradoxical (41, 48) as it has been shown to both suppress and amplify allergic responses. However, this cytokine has been shown to regulate IL-13 production from Th2 cells and promote eotaxin production and eosinophilia (49, 50). Recently, higher serum levels of IL-18 have been observed in patients with acute asthma exacerbation. Our data in stable asthmatics indicate a close correlation between IL-18 and ECP in induced sputum. This result, derived directly from the bronchial system, further supports a link between IL-18, eosinophils, and the pathogenesis of allergic airways inflammation.
The synonymous association between IL-5 and eosinophilia in conjunction with clinical trial data has propagated the concept that eosinophils are not central mediators of asthma but rather bystander cells recruited to the airways in response to aberrant Th2 cell activation. This concept is also supported by some studies in animal models. However, these investigations have failed to consider the relevance of pathways that may recruit eosinophils into tissues independently of IL-5 to disease processes. Here we demonstrate that eosinophils might be recruited to the allergic lung independently of IL-5 by eotaxin-dependent mechanisms and that this eosinophilia is directly linked to the development of disease. Eosinophilia, albeit reduced, is also a predominant feature in the lung of IL-5−/− mice infected with Toxocara canis (51). Thus, although eotaxin and IL-5 cooperate to regulate eosinophil recruitment to the allergic lung, they can also operate independently of one another to induce eosinophil accumulation. However, in the absence of both of these molecules, eosinophil accumulation in the allergic lung was ablated and AHR did not develop. These data indicate that although IL-5 can amplify eosinophil recruitment to the allergic lung, the primary role of this cytokine appears to be in the promotion of eosinophilia in the blood and bone marrow compartments in response to antigen provocation. Notably, in the absence of IL-5, eotaxin induced the recruitment of eosinophils to the lung without significant blood eosinophilia or expansion of the eosinophil pool in the bone marrow. Apparently, eosinophils produced by steady-state hematopoiesis and/or residing in tissues can be efficiently recruited by eotaxin to the allergic lung in the absence of IL-5. In previous investigations eotaxin has been shown not only to promote eosinophil accumulation in tissues, but also induce the release of this cell and its progenitors from the bone marrow (52). The lower abundance of eosinophils in the blood of allergic IL-5−/− mice may mask the development of eosinophilia in this compartment in response to eotaxin. Further, it is likely that once eosinophils and/or progenitors enter the circulation in IL-5−/− mice in response to eotaxin they are rapidly sequestered into the allergic lung. Indeed, evidence is accumulating for a local pulmonary role for eosinophil progenitors in the pathogenesis of allergic disease (53). Potentially, in the absence of IL-5, eotaxin may regulate the recruitment of eosinophils and progenitors to the allergic lung that then undergo maturation in the Th2 immune environment.
In summary, our data demonstrate that eosinophils are able to accumulate in allergic lungs of BALB/c mice in the absence of IL-5 and promote disease. A key role for eosinophils appears to be in the modulation of IL-13 production from CD4+ T cells. It is tempting to speculate that within the allergic lung, eosinophils may sequester antigen and localize to regional lymph nodes where they modulate IL-13 production from T cells during expansion by the secretion of IL-18. This mechanism may have evolved to promote the expulsion of parasites from the intestinal mucosa. Eosinophils loaded with parasitic antigens may enhance the production of IL-13 by T cells in gut-associated lymphoid tissue, which subsequently promotes the expulsion of the pathogen by increasing gastrointestinal motility by amplifying cholinergic responsiveness and enhancing mucus secretion. Collectively, our findings indicate that IL-5 and IL-13 signaling systems are not necessarily mutually exclusive effector mechanisms, and may also be integrated through eosinophils to regulate certain aspects of allergic disease. The observation that eosinophils may regulate disease processes in the absence of IL-5 has important implications for therapeutic approaches to allergic disorders. It is potentially dangerous to conclude that eosinophils do not play a role in generating AHR or other pathologies by using data from treatments that attenuate, but do not critically reduce eosinophil numbers in the allergic airways. Finally, our investigations are the first to identify a fundamental link between the innate (eosinophils) and adaptive (T cells) immune responses for the regulation of IL-13 production.
Acknowledgments
We thank Elke Strauch, Olaf Moske, and Sandra Thomas for the collection and processing of the sputum samples.
This work was supported by a Human Frontiers grant (R90262/1999-M 102) to P.S. Foster and M.E. Rothenberg, and a grant by the German Research Association (MA2241/1) to J. Mattes.
Footnotes
*
Abbreviations used in this paper: AHR, airways hyperreactivity; ECP, eosinophil cationic protein; HPF, high power field; PBLN, peribronchial lymph node; Penh, enhanced pause; RT, reverse transcription; Tg, transgenic; WT, wild-type.
References
- 1.Bochner, B.S., B.J. Undem, and L.M. Lichtenstein. 1994. Immunological aspects of allergic asthma. Annu. Rev. Immunol. 12:295–335. [DOI] [PubMed] [Google Scholar]
- 2.Wills-Karp, M. 1999. Immunological basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 17:255–281. [DOI] [PubMed] [Google Scholar]
- 3.Gleich, G.J., and C. Adolphson. 1993. Bronchial hyperreactivity and eosinophil granule proteins. Agents Actions Suppl. 43:223–230. [DOI] [PubMed] [Google Scholar]
- 4.Azzawi, M., B. Bradley, P.K. Jeffery, A.J. Frew, A.J. Wardlaw, G. Knowles, B. Assoufi, J.V. Collins, S. Durham, and A.B. Kay. 1990. Identification of activated T lymphocytes and eosinophils in bronchial biopsies in stable atopic asthma. Am. Rev. Respir. Dis. 142:1407–1413. [DOI] [PubMed] [Google Scholar]
- 5.Bentley, A.M., Q. Meng, D.S. Robinson, Q. Hamid, A.B. Kay, and S.R. Durham. 1993. Increases in activated T lymphocytes, eosinophils, and cytokine mRNA expression for interleukin-5 and granulocyte/macrophage colony-stimulating factor in bronchial biopsies after allergen inhalation challenge in atopic asthmatics. Am. J. Respir. Cell Mol. Biol. 8:35–42. [DOI] [PubMed] [Google Scholar]
- 6.Sur, S., G.J. Gleich, M.C. Swanson, K.R. Bartemes, and D.H. Broide. 1995. Eosinophilic inflammation is associated with elevation of interleukin-5 in the airways of patients with spontaneous symptomatic asthma. J. Allergy Clin. Immunol. 96:661–668. [DOI] [PubMed] [Google Scholar]
- 7.Jarjour, N.N., W.J. Calhoun, E.A. Kelly, G.J. Gleich, L.B. Schwartz, and W.W. Busse. 1997. The immediate and late allergic response to segmental bronchopulmonary provocation in asthma. Am. J. Respir. Crit. Care Med. 155:1515–1521. [DOI] [PubMed] [Google Scholar]
- 8.Hamid, Q., M. Azzawi, S. Ying, R. Moqbel, A.J. Wardlaw, C.J. Corrigan, B. Bradley, S.R. Durham, J.V. Collins, P.K. Jeffery, et al. 1991. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J. Clin. Invest. 87:1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamelmann, E., A. Oshiba, J. Loader, G.L. Larsen, G. Gleich, J. Lee, and E.W. Gelfand. 1997. Antiinterleukin-5 antibody prevents airway hyperresponsiveness in a murine model of airway sensitization. Am. J. Respir. Crit. Care Med. 155:819–825. [DOI] [PubMed] [Google Scholar]
- 10.Foster, P.S., S.P. Hogan, A.J. Ramsay, K.I. Matthaei, and I.G. Young. 1996. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 183:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gleich, G.J., N.A. Flavahan, T. Fujisawa, and P.M. Vanhoutte. 1988. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J. Allergy Clin. Immunol. 81:776–781. [DOI] [PubMed] [Google Scholar]
- 12.Robinson, D., Q. Hamid, A. Bentley, S. Ying, A.B. Kay, and S.R. Durham. 1993. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J. Allergy Clin. Immunol. 92:313–324. [DOI] [PubMed] [Google Scholar]
- 13.Grunig, G., M. Warnock, A.E. Wakil, R. Venkayya, F. Brombacher, D.M. Rennick, D. Sheppard, M. Mohrs, D.D. Donaldson, R.M. Locksley, et al. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 282:2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wills-Karp, M., J. Luyimbazi, X. Xu, B. Schofield, T.Y. Neben, C.L. Karp, and D.D. Donaldson. 1998. Interleukin-13: central mediator of allergic asthma. Science. 282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 15.Mattes, J., M. Yang, A. Siqueira, K. Clark, J. MacKenzie, A.N. McKenzie, D.C. Webb, K.I. Matthaei, and P.S. Foster. 2001. IL-13 induces airways hyperreactivity independently of the IL-4R alpha chain in the allergic lung. J. Immunol. 167:1683–1692. [DOI] [PubMed] [Google Scholar]
- 16.Kroegel, C., P. Julius, H. Matthys, J.C. Virchow, Jr., and W. Luttmann. 1996. Endobronchial secretion of interleukin-13 following local allergen challenge in atopic asthma: relationship to interleukin-4 and eosinophil counts. Eur. Respir. J. 9:899–904. [DOI] [PubMed] [Google Scholar]
- 17.Huang, S.K., H.Q. Xiao, J. Kleine-Tebbe, G. Paciotti, D.G. Marsh, L.M. Lichtenstein, and M.C. Liu. 1995. IL-13 expression at the sites of allergen challenge in patients with asthma. J. Immunol. 155:2688–2694. [PubMed] [Google Scholar]
- 18.Ying, S., D.S. Robinson, Q. Meng, J. Rottman, R. Kennedy, D.J. Ringler, C.R. Mackay, B.L. Daugherty, M.S. Springer, S.R. Durham, et al. 1997. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. Association with airway hyperresponsiveness and predominant co-localization of eotaxin mRNA to bronchial epithelial and endothelial cells. Eur. J. Immunol. 27:3507–3516. [DOI] [PubMed] [Google Scholar]
- 19.Matsukura, S., C. Stellato, S.N. Georas, V. Casolaro, J.R. Plitt, K. Miura, S. Kurosawa, U. Schindler, and R.P. Schleimer. 2001. Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am. J. Respir. Cell Mol. Biol. 24:755–761. [DOI] [PubMed] [Google Scholar]
- 20.Yang, M., S.P. Hogan, P.J. Henry, K.I. Matthaei, A.N. McKenzie, I.G. Young, M.E. Rothenberg, and P.S. Foster. 2001. Interleukin-13 mediates airways hyperreactivity through the IL-4 receptor-alpha chain and STAT-6 independently of IL-5 and eotaxin. Am. J. Respir. Cell Mol. Biol. 25:522–530. [DOI] [PubMed] [Google Scholar]
- 21.Zhu, Z., R.J. Homer, Z. Wang, Q. Chen, G.P. Geba, J. Wang, Y. Zhang, and J.A. Elias. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corry, D.B., H.G. Folkesson, M.L. Warnock, D.J. Erle, M.A. Matthay, J.P. Wiener-Kronish, and R.M. Locksley. 1996. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J. Exp. Med. 183:109–117 (erratum published 185:1715). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan, S.P., K.I. Matthaei, J.M. Young, A. Koskinen, I.G. Young, and P.S. Foster. 1998. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J. Immunol. 161:1501–1509. [PubMed] [Google Scholar]
- 24.Mauser, P.J., A. Pitman, A. Witt, X. Fernandez, J. Zurcher, T. Kung, H. Jones, A.S. Watnick, R.W. Egan, W. Kreutner, et al. 1993. Inhibitory effect of the TRFK-5 anti-IL-5 antibody in a guinea pig model of asthma. Am. Rev. Respir. Dis. 148:1623–1627. [DOI] [PubMed] [Google Scholar]
- 25.Nagai, H., S. Yamaguchi, N. Inagaki, N. Tsuruoka, Y. Hitoshi, and K. Takatsu. 1993. Effect of anti-IL-5 monoclonal antibody on allergic bronchial eosinophilia and airway hyperresponsiveness in mice. Life Sci. 53:L243–L247. [DOI] [PubMed] [Google Scholar]
- 26.Leckie, M.J., A. ten Brinke, J. Khan, Z. Diamant, B.J. O'Connor, C.M. Walls, A.K. Mathur, H.C. Cowley, K.F. Chung, R. Djukanovic, et al. 2000. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 356:2144–2148. [DOI] [PubMed] [Google Scholar]
- 27.Djukanovic, R., J.W. Wilson, K.M. Britten, S.J. Wilson, A.F. Walls, W.R. Roche, P.H. Howarth, and S.T. Holgate. 1990. Quantitation of mast cells and eosinophils in the bronchial mucosa of symptomatic atopic asthmatics and healthy control subjects using immunohistochemistry. Am. Rev. Respir. Dis. 142:863–871. [DOI] [PubMed] [Google Scholar]
- 28.McFadden, E.R., Jr. 1994. Asthma: morphologic-physiologic interactions. Am. J. Respir. Crit. Care Med. 150:S23–S26. [DOI] [PubMed] [Google Scholar]
- 29.Mould, A.W., K.I. Matthaei, I.G. Young, and P.S. Foster. 1997. Relationship between interleukin-5 and eotaxin in regulating blood and tissue eosinophilia in mice. J. Clin. Invest. 99:1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothenberg, M.E., J.A. MacLean, E. Pearlman, A.D. Luster, and P. Leder. 1997. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J. Exp. Med. 185:785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamkhioued, B., P.M. Renzi, S. Abi-Younes, E.A. Garcia-Zepada, Z. Allakhverdi, O. Ghaffar, M.D. Rothenberg, A.D. Luster, and Q. Hamid. 1997. Increased expression of eotaxin in bronchoalveolar lavage and airways of asthmatics contributes to the chemotaxis of eosinophils to the site of inflammation. J. Immunol. 159:4593–4601. [PubMed] [Google Scholar]
- 32.Palframan, R.T., P.D. Collins, T.J. Williams, and S.M. Rankin. 1998. Eotaxin induces a rapid release of eosinophils and their progenitors from the bone marrow. Blood. 91:2240–2248. [PubMed] [Google Scholar]
- 33.Jose, P.J., D.A. Griffiths-Johnson, P.D. Collins, D.T. Walsh, R. Moqbel, N.F. Totty, O. Truong, J.J. Hsuan, and T.J. Williams. 1994. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J. Exp. Med. 179:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogan, S.P., A. Mishra, E.B. Brandt, M.P. Royalty, S.M. Pope, N. Zimmermann, P.S. Foster, and M.E. Rothenberg. 2001. A pathological function for eotaxin and eosinophils in eosinophilic gastrointestinal inflammation. Nat. Immunol. 2:353–360. [DOI] [PubMed] [Google Scholar]
- 35.Dent, L.A., M. Strath, A.L. Mellor, and C.J. Sanderson. 1990. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 172:1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webb, D.C., A.N. McKenzie, A.M. Koskinen, M. Yang, J. Mattes, and P.S. Foster. 2000. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J. Immunol. 165:108–113. [DOI] [PubMed] [Google Scholar]
- 37.Wynn, T.A., I. Eltoum, A.W. Cheever, F.A. Lewis, W.C. Gause, and A. Sher. 1993. Analysis of cytokine mRNA expression during primary granuloma formation induced by eggs of Schistosoma mansoni. J. Immunol. 151:1430–1440. [PubMed] [Google Scholar]
- 38.Mahalingam, S., J.M. Farber, and G. Karupiah. 1999. The interferon-inducible chemokines MuMig and Crg-2 exhibit antiviral activity in vivo. J. Virol. 73:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.American Thoracic Society. 1987. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am. Rev. Respir. Dis. 136:225–244. [DOI] [PubMed] [Google Scholar]
- 40.Mattes, J., K. Storm van's Gravesande, U. Reining, K. Alving, G. Ihorst, M. Henschen, and J. Kuehr. 1999. NO in exhaled air is correlated with markers of eosinophilic airway inflammation in corticosteroid-dependent childhood asthma. Eur. Respir. J. 13:1391–1395. [PubMed] [Google Scholar]
- 41.Yoshimoto, T., H. Mizutani, H. Tsutsui, N. Noben-Trauth, K. Yamanaka, M. Tanaka, S. Izumi, H. Okamura, W.E. Paul, and K. Nakanishi. 2000. IL-18 induction of IgE: dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 1:132–137. [DOI] [PubMed] [Google Scholar]
- 42.Woerly, G., N. Roger, S. Loiseau, D. Dombrowicz, A. Capron, and M. Capron. 1999. Expression of CD28 and CD86 by human eosinophils and role in the secretion of type 1 cytokines (interleukin 2 and interferon γ): inhibition by immunoglobulin A complexes. J. Exp. Med. 190:487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takatsu, K., Y. Kikuchi, T. Takahashi, T. Honjo, M. Matsumoto, N. Harada, N. Yamaguchi, and A. Tominaga. 1987. Interleukin 5, a T-cell-derived B-cell differentiation factor also induces cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA. 84:4234–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harada, N., M. Matsumoto, N. Koyama, A. Shimizu, T. Honjo, A. Tominaga, and K. Takatsu. 1987. T cell replacing factor/interleukin 5 induces not only B-cell growth and differentiation, but also increased expression of interleukin 2 receptor on activated B-cells. Immunol. Lett. 15:205–215. [DOI] [PubMed] [Google Scholar]
- 45.Shi, H.Z., A. Humbles, C. Gerard, Z. Jin, and P.F. Weller. 2000. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J. Clin. Invest. 105:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacKenzie, J.R., J. Mattes, L.A. Dent, and P.S. Foster. 2001. Eosinophils promote allergic disease of the lung by regulating CD4(+) TH2 lymphocyte function. J. Immunol. 167:3146–3155. [DOI] [PubMed] [Google Scholar]
- 47.Rothenberg, M.E. 1998. Eosinophilia. N. Engl. J. Med. 338:1592–1600. [DOI] [PubMed] [Google Scholar]
- 48.Nakanishi, K., T. Yoshimoto, H. Tsutsui, and H. Okamura. 2001. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19:423–474. [DOI] [PubMed] [Google Scholar]
- 49.Wild, J.S., A. Sigounas, N. Sur, M.S. Siddiqui, R. Alam, M. Kurimoto, and S. Sur. 2000. IFN-gamma-inducing factor (IL-18) increases allergic sensitization, serum IgE, Th2 cytokines, and airway eosinophilia in a mouse model of allergic asthma. J. Immunol. 164:2701–2710. [DOI] [PubMed] [Google Scholar]
- 50.Campbell, E., S.L. Kunkel, R.M. Strieter, and N.W. Lukacs. 2000. Differential roles of IL-18 in allergic airway disease: induction of eotaxin by resident cell populations exacerbates eosinophil accumulation. J. Immunol. 164:1096–1102. [DOI] [PubMed] [Google Scholar]
- 51.Takamoto, M., K.S. Ovington, C.A. Behn, K. Sugane, I.G. Young, and K.I. Matthaei. 1997. Eosinophilia, parasite burden and lung damage in Toxocara canis infection in C57Bl/6 mice genetically deficient in IL-5. Immunology. 90:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson, P.G., J. Dolovich, A. Girgis-Gabardo, M.M. Morris, M. Anderson, F.E. Hargreave, and J.A. Denburg. 1990. The inflammatory response in asthma exacerbation: changes in circulating eosinophils, basophils and their progenitors. Clin. Exp. Allergy. 20:661–668. [DOI] [PubMed] [Google Scholar]
- 53.Inman, M.D., R. Ellis, J. Wattie, J.A. Denburg, and P.M. O'Byrne. 1999. Allergen-induced increase in airway responsiveness, airway eosinophilia, and bone-marrow eosinophil progenitors in mice. Am. J. Respir. Cell Mol. Biol. 21:473–479. [DOI] [PubMed] [Google Scholar]