Interleukin-5 Expression in the Lung Epithelium of Transgenic Mice Leads to Pulmonary Changes Pathognomonic of Asthma (original) (raw)
Abstract
We have generated transgenic mice that constitutively express murine interleukin (IL)-5 in the lung epithelium. Airway expression of this cytokine resulted in a dramatic accumulation of peribronchial eosinophils and striking pathologic changes including the expansion of bronchusassociated lymphoid tissue (BALT), goblet cell hyperplasia, epithelial hypertrophy, and focal collagen deposition. These changes were also accompanied by eosinophil infiltration of the airway lumen. In addition, transgenic animals displayed airway hyperresponsiveness to methacholine in the absence of aerosolized antigen challenge. These findings demonstrate that lung-specific IL-5 expression can induce pathologic changes characteristic of asthma and may provide useful models to evaluate the efficacy of potential respiratory disease therapies or pharmaceuticals.
The etiology of asthma has remained unclear despite direct attempts to determine factors that lead to pulmonary damage and loss of function. The underlying respiratory inflammation leading to variable airflow limitation and airway hyperresponsiveness to various stimuli is often exacerbated by allergens (1), respiratory tract infections (2), or environmental agents (e.g., air pollutants such as ozone or tobacco smoke) (3). Localized cytokine and chemokine gene expression (4), secretion of low molecular weight inflammatory mediators (5), and the recruitment of specific leukocyte cell types (6) have all been shown to be critical events. Positive correlations with the development of pathology suggest that the process is pluricausal involving several cell types and molecular signaling cascades. In addition, the complex inheritance patterns of genetic predispositions attest to the polygenic nature of the disorder (7).
Many pathophysiological manifestations of asthma are associated with airway infiltration by eosinophils and lymphocytes mediated by cytokines and chemokines. Leukocyte influx, in turn, has been associated with the development of lung dysfunction even in nominal cases of asthma (8). Indeed, the extent of infiltration generally correlates with the severity of disease (8). Antigen-induced mouse models of pulmonary allergic disease have proved particularly informative in the genetic dissection of inflammatory pathways in the lung. Typically, these models involve sensitization with a specific antigen (e.g., ovalbumin) followed by airborne administration of the same antigen (9). Sensitized mice treated with aerosolized allergen develop leukocytic infiltrates of the airway lumen dominated by CD4+ lymphocytes and eosinophils. These mice also develop many of the changes pathognomonic of asthma including airway hyperresponsiveness (AHR)1 and goblet cell hyperplasia with excessive mucus production (10).
The cellular signals leading to airway inflammation, eosinophil infiltration, and AHR remain obscure. Lymphocytes and mast cells have been implicated as requirements for AHR in antigen-challenged mouse models of asthma. SCID mice, which lack both T and B lymphocytes, fail to develop either an airway eosinophilia or AHR after ovalbumin sensitization (11). The depletion of CD4+ lymphocytes either by treatment with anti-CD4 antibodies or MHC class II gene knockout eliminated both eosinophil airway infiltration and AHR in antigen-challenged mice (12). In contrast, depletion of CD8+ T lymphocytes with anti-CD8 antibodies had no effect on lung eosinophil infiltration but eliminated AHR (13, 14). However, recent studies using β2-microglobulin–deficient mice demonstrated no effects on AHR despite the loss of MHC I surface molecules and CD8+ cells (15). Finally, studies with mast cell– deficient mice (W/Wv) indicated that mast cells were not required for either eosinophil airway infiltration or AHR (16).
Collectively, the antigen sensitization and challenge mouse models implicate T lymphocytes as a critical component of the inflammatory response. Subsequent studies have focused on interleukins produced by the TH2 subtype. The necessity of IL-4 and TH2 cells was demonstrated by the failure of IL-4–deficient mice to elicit airway eosinophil infiltration or AHR in response to antigen sensitization and challenge (16). Ovalbumin sensitization and challenge of IL-5–deficient mice showed that these animals also do not develop eosinophil airway infiltration or AHR (17). These cytokine gene knockout studies using aerosolized antigen challenge thus suggest that IL-4 and IL-5 are each important components of proinflammatory cascades that ultimately result in eosinophil airway infiltration and pathophysiological changes characteristic of asthma.
Transgenic mice expressing either IL-4 or IL-5 have provided little additional insights as to the role(s) of these TH2 cytokines in the development of lung inflammatory pathology. IL-4 has been expressed ectopically in several cell and tissue types (18–20) including Clara cells of the airway epithelium (21). Expression of this cytokine in nonlung cell types produces no pulmonary perturbations despite constitutively high serum IL-4 levels. In contrast, lung-specific expression of IL-4 (21) did lead to pathologic pulmonary changes but, surprisingly, these changes were nominal and did not correlate specifically to human disease. Transgenic animal studies ectopically expressing IL-5 have been limited to non-lung cell and tissue types (22–25), and results from these reports also show that none of the animals developed abnormalities in the lung. However, in the most recent of these studies (22), our group has shown that some IL-5 effector functions appear to depend on cell- and tissue-specific expression. This conclusion has potentially important implications in light of clinical observations of asthmatic patients showing the infiltration of the lung by cells expressing high levels of IL-5 (for example see reference 26).
To test the hypothesis that many of the pathologies associated with asthmatic patients result uniquely from lungspecific expression of IL-5, we created transgenic mice that constitutively express IL-5 in the airway lumen using a wellcharacterized lung-specific regulatory element to drive expression in the epithelium of adult animals (27). We report that the resulting transgenic mice develop pathophysiologic changes including eosinophil invasion of peribronchial spaces, epithelial hypertrophy, goblet cell hyperplasia, and increased mucus production. These mice also show evidence of eosinophil recruitment to the airway lumen at levels comparable to asthmatic patients. Moreover, in the absence of antigen-induced inflammation these mice nonetheless display AHR in response to methacholine challenge. Thus, lung-specific expression of IL-5 alone can recreate many of the pathophysiologic conditions associated with allergic respiratory disease.
Materials and Methods
Generation of Transgenic Mice.
A BamHI restriction fragment containing a 5.5-kb IL-5 cDNA/genomic fusion gene (22) was cloned into the BglII site of the plasmid pNNO3 (a kind gift of R. Tizzard, Biogen, Cambridge, MA). A 2.3-kb BamHI fragment containing the promoter region of the rat Clara cell secretory protein CC10 (27) (a kind gift of J. Gitlin, Washington University School of Medicine, St. Louis, MO) was cloned into an upstream BamHI site in the polylinker of this plasmid. This transgenic construct was excised from plasmid sequences by NotI digestion and injected into embryos derived from a cross of F1(CBA/ CaJ × C57BL/6J) females and C57BL/6J males. Transgenic positive founder animals were identified from genomic Southern blots of tail DNA. Subsequent generations of transgenic animals were the result of backcrosses onto the inbred strain C57BL/6J. Animals reported here were maintained in micro-isolator cages housed in a specific pathogen-free (SPF) animal facility. The sentinel cages within the animal colony surveyed negative for viral antibodies and the presence of known mouse pathogens.
RNA Isolation and Northern Blot Analysis.
Total RNA was isolated and formaldehyde-agarose Northern blots were performed as described earlier (28).
Tissue In Situ Hybridization.
Paraffin sections of paraformaldehyde fixed lungs were subjected to in situ hybridization reactions using 35S-labeled IL-5 antisense RNA probes as described earlier (29).
Collection of Bronchial Alveolar Lavage Fluid and Blood Serum for the Determination of IL-5 Levels.
Animals were injected (i.p.) with a lethal dose of ketamine (600 mg/kg of body weight) and xylazine (30 mg/kg of body weight) and placed under a stereodissecting microscope. The trachea was exposed and a cannula was inserted and secured by sutures. The lungs were lavaged four times with 1-ml aliquots of ice-cold PBS with 0.2% fetal calf serum. 3.6–3.8 ml of the instilled lavage fluid were routinely recovered (no difference in recovery was noted between transgenic and wild-type animals). The recovered bronchial alveolar lavage (BAL) fluid was centrifuged (1,000 rpm for 10 min at 4°C) to remove cells, and aliquots of the supernatant were frozen on dry ice and stored at −80°C until use. Peripheral blood (300–500 mm3) was recovered by nicking the tail. Blood was allowed to clot at room temperature for 30 min and serum was recovered by centrifugation (1,000 rpm for 10 min at 4°C), frozen on dry ice, and stored at −80°C until use. IL-5 and IL-4 levels were measured using murine cytokine ELISA kits as described by the manufacturer (Endogen, Inc., Boston, MA).
Hematologic Assays: Hematocrits, Cytospins and Smear Preparations, Cell Counts and Differentials.
Microhematocrits were performed from tail-derived blood. Blood films and marrow brush smears were prepared as described previously (22). Cytospins of cells isolated from collagenase-treated lungs or recovered BAL fluid were prepared using a Shandon Cytospin 3 (Shandon Scientific LTD, Cheshire, England). Cell differentials were performed from slides stained with Leukostat (Fisher Diagnostics, Fisher Scientific, Pittsburgh, PA) or Wright's stain. The specific identification of eosinophils was confirmed by staining representative slides for the cyanide-resistant peroxidase contained in the secondary granule (28) or differential staining with an eosinophil granule protein-specific antisera. Total cell counts were quantified by hemocytometer and, together with the percent cell type by differential, were used to calculate specific cell number.
Tissue Histology.
Before resection, lungs were inflated with 0.5 ml of 10% phosphate-buffered formalin and fixed overnight at room temperature. The fixed tissue samples were embedded into paraffin and sections (4–6 μm) were prepared on TESPA-treated slides for immunofluorescence or acid-washed slides for histochemical staining.
Lung Cell Isolation and Differentials.
Cell suspensions were prepared from collagenase treatment of perfused lungs as previously described (14).
Airway Antigen Sensitization and Challenge.
Mice were sensitized and challenged with aerosolized chicken ovalbumin as previously described (30).
Immunofluorescence of Lung Sections.
Serial sections from formalin-fixed paraffin-embedded tissue were placed onto TESPAtreated slides. The sections were deparaffinized using a xylene/ ethanol/water gradient, treated for 30 min in 0.1% trypsin, 0.1% CaCl (pH 7.5), rinsed well in water, and blocked overnight (>12 h) in PBS, 10% goat serum, 0.1% sodium azide at 4°C. The blocked tissue sections were rinsed in PBS and then rabbit prebleed serum (diluted 1:20) or an anti-murine eosinophil granule major basic protein (mMBP) serum (diluted 1:20) was applied. After the addition of the primary antibody, the slides were incubated for 45 min at 37°C in a humidified chamber. The slides were washed with PBS (3 × 5 min each) and blocked with 0.1% Chromotrope 2R (J.T. Baker, Phillipsburg, NJ) in PBS for 30 min at room temperature. The sections were rinsed in 2–3 changes of PBS and stained with a FITC-conjugated anti-rabbit IgG (diluted 1:40; Sigma Chemical Co., St. Louis, MO) for 30 min at 37°C. The slides were washed in PBS (3 × 5 min each) and coverslips mounted using a mixture of 90% glycerol, 10% PBS, 0.1% _p_-phenylenediamine (Sigma).
Measurement of AHR.
Airway responsiveness was assessed by inducing airflow obstruction with a methacholine aerosol using a noninvasive method (31). This procedure estimates total pulmonary airflow in mice (i.e., the sum of the airflows in the upper and lower respiratory tracts). Minute volume, tidal volume, breathing frequency, and enhanced pause (Penh) were obtained from conscious mice placed in a whole body plethysmograph (model PLY 3115; Buxco Electronics Inc., Troy, NY). Mice were unrestrained and tolerated repetitive measurements with this system. Chamber pressure was measured with a transducer (model TRD 5700) connected to preamplifier modules (model CHA 0150) and analyzed by System XA software (model SFT 1810). The chamber pressure was used as a measure of the difference between thoracic expansion (or contraction) and air volume removed from (or added to) the chamber during inspiration (or expiration). The differential of this function with respect to time produced a pseudo-flow value that is proportional to the difference between the rate of change of the thoracic volume and nasal flow. Mice were placed in each chamber for 2 min and pulmonary airflow obstruction was assessed by measuring Penh using the following formula according to the manufacturer's recommendations: Penh = ([Te/0.3RT] − 1) × (2PEF/3PIF), where Penh = enhanced pause (dimensionless), Te = expiratory time (s), RT = relaxation time (s), PEF = peak expiratory flow (ml/s), and PIF = peak inspiratory flow (ml/s). The breathing pattern was collected for 2 min and an average of each variable was derived from 30 breaths (or 30 s, whichever occurred first). The peak Penh value was recorded. Measurements of methacholine responsiveness were obtained by exposing mice for 1 min to saline, followed by incremental doses (2.5–320 mg/ml) of aerosolized methacholine (model CN-25 Collision MRE type nebulizer; BGI, Inc., Waltham, MA) and monitoring the breathing pattern for 2–3 min after challenge. Dose-response data were plotted and the methacholine dose sufficient to double Penh (ED200) was derived by log-linear interpolation. This procedure was completed within 60 min.
Results
The Generation of Transgenic Mice Constitutively Expressing IL-5 in the Lung Epithelium.
Transgenic mice expressing IL-5 in the lung epithelium were created using the promoter of the rat CC10 gene (27) and a previously described cDNA/genomic fusion of IL-5 (22). Three independent lines (NJ.1659, NJ.1723, NJ.1726) were initially characterized with respect to transgene copy number, peripheral blood cellularity, and lung histology. All three lines of mice had elevated white blood cell counts, including a peripheral eosinophilia, and increases in the peribronchial cellularity of the lung. Transgene copy number varied in each line (NJ.1659 [1], NJ.1723 [10], NJ.1726 [15]) and generally correlated with the extent of the observed pathologies. On this basis, the NJ.1726 transgenic line of mice was chosen for further study of the pathophysiologic effects of expressing IL-5 in the lung epithelium.
NJ.1726 mice were backcrossed to C57BL/6J (+/+) for a minimum of four generations. All data reported are derived from mice 3–8 mo of age. The transgenic animals had live births and numbers of weaned offspring comparable to (+/+) mice. In addition, the transgene was inherited equally among male and female pups, indicating an autosomal insertion. The apparent morbidity and life expectancy of NJ.1726 mice were unchanged relative to transgenenegative littermates. Northern blot analysis showed that expression of IL-5 in transgenic animals was limited to the lung and was not detectable in any of the wild-type tissues examined (Fig. 1). Furthermore, in situ hybridization of adult NJ.1726 lungs shows that principle locations of IL-5 expression included Clara cells of the larger airways (data not shown) and the proximal portion of the pulmonary acinus (i.e., the transitional zone between the conducting airways and the gas exchange areas) as well as cells whose histological characteristics, number, and location suggest that they are alveolar epithelial type II cells (Fig. 2). This expression was found throughout the lung and is consistent with the expression pattern of the rat CC10 gene in adult rat lungs (32).
Figure 1.

Lung-specific IL-5 gene expression. Northern blot of (+/+) and NJ.1726 (Tg) tissue RNA probed with a random-primed 32P-labeled IL-5 cDNA. Each lane contains 15 μg of total RNA. Lane 1, bone marrow; lane 2, liver; lane 3, lung; lane 4, spleen. A photograph of the 18S small ribosomal subunit stained with ethidium bromide is shown to verify the presence of RNA in each lane.
Figure 2.
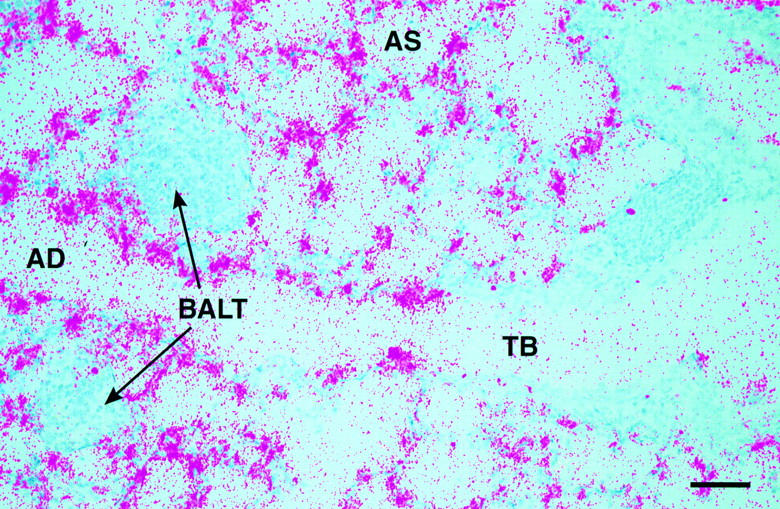
Localization of IL-5 transcripts to the lung epithelium of NJ.1726 mice. An IL-5 antisense RNA probe was synthesized from an IL-5 cDNA (a kind gift of T. Honjo) cloned into the plasmid vector pBluescript KS(+). No signal was detected when a sense probe was hybridized to adjacent sections (data not shown). TB, terminal bronchiole; AD, alveolar duct; AS, alveolar space; BALT, bronchus-associated lymphoid tissue. Bar, 25 μm.
IL-5 levels in the BAL fluid and serum were assessed by ELISA (Table 1). The (+/+) level of IL-5 in both compartments was at or below the level of detection (⩽5pg/ml). In contrast, IL-5 levels in BAL fluid of NJ.1726 mice were dramatically elevated (269 ng/total lavage). An apparent consequence of airway expression at these high levels was displacement of IL-5 into the vasculature such that serum IL-5 was ∼1,700 pg/ml. IL-4 was not detectable (i.e., ⩽5pg/ml) in the BAL fluid of NJ.1726 mice. Moreover, Northern blot comparisons of endogenous pulmonary cytokine and chemokine gene activity in (+/+) and transgenic animals showed that IL-4, RANTES, MCP-3, and eotaxin remained at low and/or nearly undetectable levels in the lungs of NJ.1726 mice (data not shown).
Table 1.
>IL-5 Protein Levels and Its Effects on Peripheral Blood and Femoral Marrow Cellularity
| Mouse* | Hematocrit | IL-5 Present in the BAL | Serum IL-5 | Cell number/mm3 of blood (×10−3) | Cell number/femur (×10−6) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Lym | Mono | Eos | Neu | Total | Ery | Lym | Eos | Neu | ||||
| ng/total lavage | pg/ml | ||||||||||||
| +/+ | 55.3 (4.6)4 | N.D. | N.D. | 9.9 (3.0)4 | 6.1 (3.9) | 1.2 (0.5) | 0.2 (0.1) | 2.5 (1.0) | 27.8 (4.9)4 | 11.0 (2.4) | 5.9 (0.9) | 0.7 (0.8) | 10.1 (5.1) |
| NJ.1726 | 54.3 (4.3)4 | 269 (37)4 | 1696 (449)4 | 41.4 (16.3)4 | 18.8 (10.6) | 2.6 (1.0) | 17.3 (8.7) | 2.7 (1.5) | 30.3 (3.8)4 | 4.1 (1.6) | 3.8 (0.6) | 15.2 (1.8) | 7.3 (3.9) |
IL-5 Induced Changes in Bone Marrow Cell Populations and the Development of a Peripheral Eosinophilia.
The hematopoietic consequences of IL-5 overexpression in the lung and augmentation of blood serum levels are shown in Table 1. NJ.1726 mice have white blood cell (WBC) counts that are moderately elevated relative to (+/+) (approximately fourfold), exceeding 41,000 cells/mm3. This increase results primarily from an expansion of peripheral eosinophils (2 versus 42%) although all other WBC types examined increased in absolute number. NJ.1726 femoral marrow cellularity, however, remained unchanged relative to (+/+) mice. The lack of change associated with the total marrow cell count masks a fundamental shift in cell composition. Eosinophils increased by >20-fold relative to (+/+) and accounted for 50% of the transgenic marrow cells. In contrast, there was a substantive decrease in erythroblasts (60% loss). This decrease, however, did not effect peripheral red blood cell counts as assayed by hematocrit (55.3 versus 54.3) probably reflecting a shift in erythropoiesis from the marrow to an extramedullary site (e.g., spleen, unpublished observations).
Airway Expression of IL-5 Results in Histopathologic Changes in the Lung.
A systematic gross and microscopic survey of adult tissues revealed that, except for mild splenomegaly, the only pathologic manifestations of pulmonary IL-5 overexpression were found in the lungs. The most prominent abnormalities included expansion of bronchus-associated lymphoid tissue (BALT) (33) and the infiltration of peribronchial spaces by leukocytes. The severity of these changes varied greatly and is shown in comparison to (+/+) in the hematoxylin/eosin (H/E) sections of Fig. 3, a–c. BALT aggregates (dominated by mononuclear cells) were surrounded by a lymphoepithelium segregating them from the airway lumen. All NJ.1726 animals examined exhibited these pulmonary changes (n = 11). The majority (54%) showed increases in BALT within the range presented (Fig. 3, b and c). For reasons that remain unclear, ∼27% of 3–8-mo-old animals displayed the extreme form of this pathology (Fig. 3 c). The accumulation of leukocytes was associated with the expansion of the lymphoid tissue around nearly all the larger airways. This expansion was often so great that it resulted in the apparent closure or near occlusion of affected airways (Fig. 3 c, arrows). The appearance of this extreme phenotype was random and did not segregate to specific cages of mice or locations within the mouse colony, nor did it shorten the life-span of transgenic animals. Fig. 3 d shows bronchioles and the surrounding leukocytes at higher magnification. In addition to epithelial hypertrophy, the lymphocytic infiltration was associated with perturbations of the bronchial epithelium and thickening of the epithelial submucosal region of some bronchioles.
Figure 3.
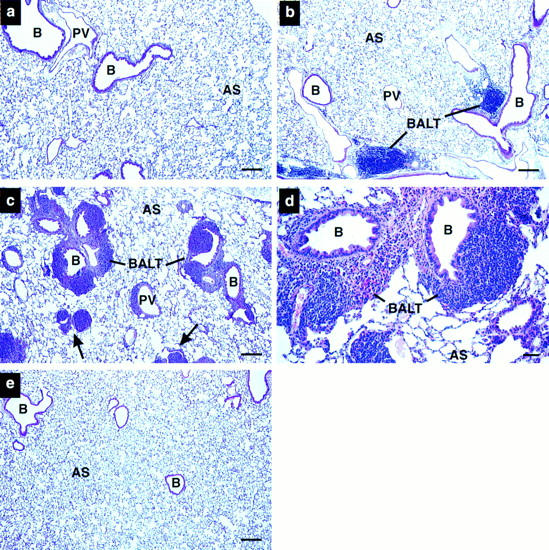
Histopathologic pulmonary changes accompany lung epithelial expression of IL-5. (a) Wild-type, (b–d) NJ.1726, and (e) a transgenic mouse expressing IL-5 from T cells (line NJ.1638 [22]) were stained with hematoxylin and eosin before bright-field photomicroscopy. b and c are representative photographs of the variation in phenotype found in NJ.1726 mice. The arrows in c indicate airways in an NJ.1726 mouse nearly occluded by the expansion of peribronchial lymphoid tissue. The high magnification view of a BALT aggregate (d) shows in greater detail the additional histopathologies associated with these regions. e demonstrates that although T cell–specific expression of IL-5 elevates serum levels to 400–800 pg/ml, no pulmonary changes occurred, and thus the pathologies occurring in NJ.1726 mice are the result of lung-specific IL-5 expression. B, bronchiole; PV, pulmonary blood vessel; AS, alveolar space; BALT, bronchus-associated lymphoid tissue. Bars: (a–c, e) 200 μm; (d) 50 μm.
The leukocyte infiltration associated with NJ.1726 lungs resulted directly from the specific expression of IL-5 in the lung epithelium. Fig. 3 e is a representative H/E section of lung from an age-matched transgenic animal constitutively expressing IL-5 from a T cell–specific promoter (transgenic line NJ.1638 [22]). Serum IL-5 in these mice reaches as high as 800 pg/ml; yet, as shown here, the animals do not exhibit any significant lung pathology (i.e., an increase in peribronchial leukocytes or an expansion of BALT).
Additional histological analyses of NJ.1726 lungs demonstrated that other changes have also occurred as a consequence of airway expression of IL-5. Lung sections stained with alcian blue (pH 2.5) to detect weakly acidic sulphated mucins showed relatively few epithelial goblet cells in (+/+) animals (Fig. 4 a). In contrast, NJ.1726 mice had an increase in the number, distribution, and staining intensity of goblet cells. These increases were evident in the epithelium of large bronchi (Fig. 4 b) as well as smaller more distal bronchioles (Fig. 4 c). Masson's trichrome staining (Fig. 4, d–f) demonstrated that the areas of expanded BALT in NJ.1726 lungs are often accompanied by the deposition of collagen (blue staining extracellular glycoprotein) and hypertrophy of the bronchial epithelium. A higher magnification view of the bronchiole and the surrounding BALT showed that the epithelial hypertrophy involves both ciliated and mucus-secreting cells (Fig. 4 f, arrows). Interestingly, all of these additional histopathologic changes were independent of the severity of the BALT expansion. Goblet cell hyperplasia, epithelial hypertrophy, and focal collagen deposition were observed in nearly all NJ.1726 mice and appear to be tightly linked to IL-5 airway expression.
Figure 4.
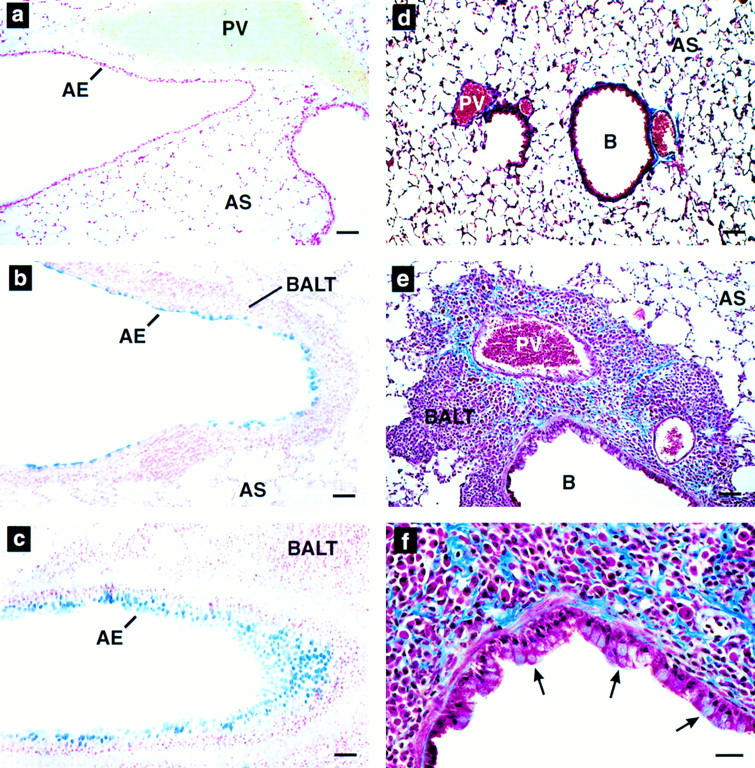
Goblet cell hyperplasia, epithial hypertrophy, and collagen deposition are induced by airway expression of IL-5. (a–c) Alcian blue (pH 2.5) staining of wild-type (a) and NJ.1726 lung sections showing a large bronchus (b) and a smaller more distal bronchiole (c). Intensely blue staining areas are glycoprotein (mucin)-containing goblet cells. (d–e) Masson's trichrome staining of paraffin sections derived from wild-type (d) and NJ.1726 (e and f) lungs. The darkly blue staining extracellular material is collagen. The arrows in f indicate hypertrophy in the bronchial epithelium. AE, airway epithelium; B, bronchiole; PV, pulmonary blood vessel; AS, alveolar space; BALT, bronchus-associated lymphoid tissue. Bars: (a–e) 50 μm; (f) 25 μm..
Lung-specific IL-5 Expression Results in Eosinophil Accumulation in the Peribronchial Spaces and Eosinophil Infiltration of the Airway Lumen.
The total cellularity and composition of the parenchymal infiltrate was determined by analyses of cells recovered from collagenase treatment of perfused lungs. The resulting cell counts and differentials are displayed in Fig. 5 A. The total cellular infiltrate of NJ.1726 lungs increased fourfold relative to wild-type and was dominated by eosinophils (∼50%). The fractional increase of eosinophils occurred with decreases in the percentages of all other WBC types. However, if the increase in total lung parenchymal cells of NJ.1726 is taken into account, then the absolute numbers of all leukocyte populations increased relative to wild-type. Thus, in addition to a 57-fold increase in the numbers of parenchymal eosinophils, the absolute numbers of NJ.1726 lung lymphocytes and monocytes/ macrophages actually increased relative to wild-type 1.8- and 2.1-fold, respectively.
Figure 5.
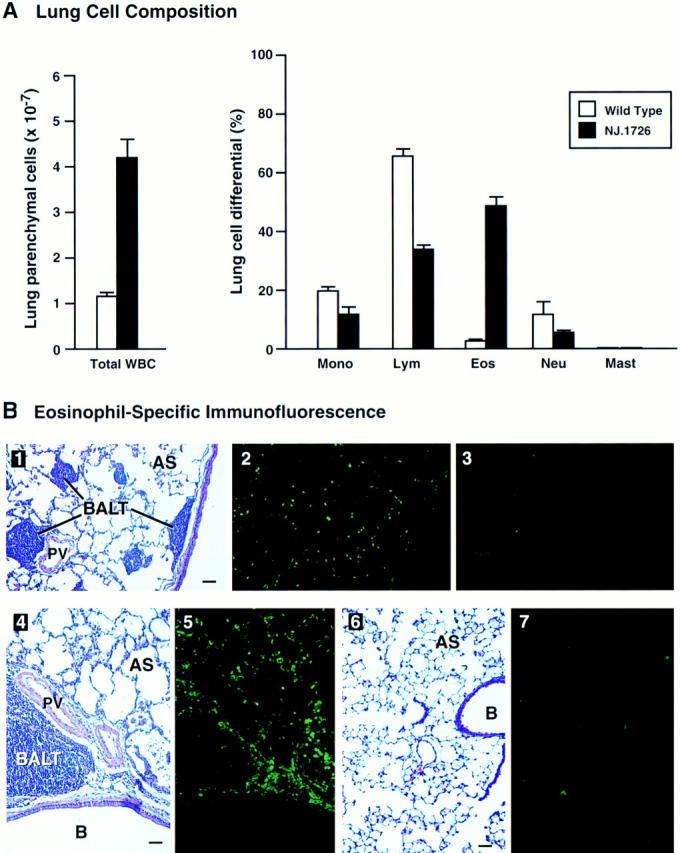
Cell differentials and eosinophil-specific immunofluorescence demonstrate a dramatic peribronchial infiltrate. (A) Collagenase-digested perfused lungs from NJ.1726 mice (n = 4) and transgenic negative littermates (n = 3) were dispersed to single cell suspensions prior to the assessment of lung parenchymal cellularity. The total cell counts of lung parenchymal leukocytes are shown as the histograms on the left. The relative numbers of different types of leukocytes (lung cell differentials) were determined from Wright's stained cytocentrifuge preparations and are shown as the histograms on the right. These data are expressed as the means (±SD) of the percentage of each cell type derived from differentials based on 200 cells. Mono, monocytes and macrophages; Lym, lymphocytes; Eos, eosinophils; Neu, neutrophils; Mast, mast cells. (B) Parenchymal eosinophils were localized within the lung by immunofluorescence using an anti-mMBP polyclonal rabbit antiserum. Serial lung sections (4 μm) were stained with H/E (1, 4, and 6), anti-mMBP serum (2, 5, and 7), and prebleed serum (3) to visualize lung histology and eosinophil infiltration. These photographs represent serial tissue sections from a lung of an NJ.1726 mouse (1–5) or a (+/+) control animal (6 and 7). B, bronchiole; PV, pulmonary blood vessel; AS, alveolar space; BALT, bronchus-associated lymphoid tissue. Bars: 50 μm.
The presence of eosinophils infiltrating the peribronchial spaces was confirmed by immunofluorescence using a polyclonal antisera against the eosinophil granule protein mMBP (Fig. 5 B). This polyclonal antisera was generated from rabbits immunized with purified mMBP isolated from mature peripheral eosinophils (34). The specificity of the antisera was determined by radioimmunoassay and differential binding to eosinophils present in cytospin preparations of WBC (data not shown). Mature eosinophils were common in the peribronchial spaces of NJ.1726 mice and distributed throughout the lung parenchyma (Fig. 5 B, 1–3). Concentrated foci of eosinophils surrounding areas of BALT were also common (Fig. 5 B, 4 and 5). The expanded peribronchial lymph nodes and the respiratory epithelium were virtually devoid of eosinophils. The immunofluorescence data also demonstrated that extensive areas of extracellular major basic protein were not detectable, suggesting that the pathophysiologic changes observed in NJ.1726 mice were not the result of eosinophil degranulation and cationic protein deposition.
Lung-specific IL-5 expression also resulted in the differential recruitment of eosinophils to the airway lumen. Fig. 6 compares the total cellularity and composition of leukocytes found in the BAL fluid of (+/+), NJ.1726, and OVA sensitized and challenged (+/+) mice. The total number of BAL cells in NJ.1726 did not change appreciably relative to (+/+) (1.06 × 105 versus 0.87 × 105 cells, respectively). The absence of an increase in the number of leukocytes in the BAL of transgenic animals is consistent with observations from asthmatic patients (35). Furthermore, this comparison also extends to changes induced in the composition of cells recruited to the BAL. The percentage of BAL eosinophils in NJ.1726 mice increased (<1 versus 7%) to levels similar to those observed in human asthmatic patient populations (35). This result was dramatically different from observations of BAL cells derived from OVA sensitized and challenged (+/+) mice. The respiratory inflammation induced in this mouse model resulted in a significant increase (approximately sixfold) in total BAL cellularity that occurred as a consequence of an increase in the number of eosinophils recruited to the airway lumen (>60% of total BAL cells) as well as smaller increases in neutrophil and lymphocyte numbers.
Figure 6.

Infiltration of the airway lumen by eosinophils did not occur as a consequence of the pulmonary changes in NJ.1726 mice. BAL cells derived from the lungs of 7-mo-old wild-type (+/+) (n = 6), NJ.1726 (n = 3), and, for comparison, control (+/+) animals sensitized (i.p.) and challenged with aerosolized OVA (BAL cells assessed 2 d after aerosol challenge) are displayed as histograms showing total BAL cellularity (left) and the relative numbers of different types of leukocytes (BAL differentials) determined from Wright's stained cytocentrifuge preparations (right). The fractional compositions of each cell type are expressed as percentages derived from differentials of 200 cells. Data represent mean values ±SD. Mφ, macrophages; Lym, lymphocytes; Eos, eosinophils; Neu, neutrophils; Mast, mast cells.
IL-5 Expression in the Lung Results in the Development of AHR.
AHR in response to a cholinergic agonist is a physiologic perturbation characteristic of asthma and other respiratory disorders. In an attempt to correlate the IL-5 induced inflammatory changes in NJ.1726 mice to this response, we measured airway responsiveness to methacholine challenge. NJ.1726 mice, or wild type animals, were exposed to increasing concentrations of aerosolized methacholine and AHR was measured by whole-body plethysmography. Data from transgenic and control animals (n = 11 for each group) were collected as the means of duplicate single-animal measurements and are displayed in graphical form in the left panel of Fig. 7 A. The effective doses of methacholine that achieved 50% maximal responsiveness (ED200) are shown as histograms to the right of the primary data. NJ.1726 mice had a lower threshold dose for methacholine-induced airflow obstruction compared with wildtype controls. In addition, NJ.1726 mice also displayed AHR to methacholine challenge in group mean dose-response data or when the mean of individual NJ.1726 ED200 measurements are compared to the control animals (69 versus 197 mg/ ml, respectively). Our AHR measurements showed surprisingly little variation in response to increasing methacholine concentration among the cohort of animals examined. We also found little difference in the threshold dose for methacholine-induced airflow obstruction among the transgenic animals suggesting that AHR to methacholine challenge is independent of the varying severity of the BALT expansion found in NJ.1726 mice. This increase in AHR was restricted to lung-specific IL-5 expressing animals and was not observed in transgenic mice (line NJ.1638, [22]) constitutively expressing high levels of serum IL-5 (Fig. 7 B). The lack of AHR in these mice suggests that aerosolized methacholine does not induce systemic perturbations of either the upper or lower respiratory tracts in response to serum IL-5 or elevated peripheral eosinophil numbers. The high WBC count (>400,000 cells/mm3; ∼60% eosinophils) and the ubiquitous presence of elevated levels of tissue eosinophils in NJ.1638 mice (e.g., nasal passageways) are particularly significant because they occur in the absence of extensive lung histopathology. Thus, the AHR in mice expressing IL-5 from the airway epithelium appears to correlate with the observed lung histopathologies found in these animals (i.e., lower respiratory tract changes) and not with potential changes that may be induced by IL-5 in the upper respiratory tract.
Figure 7.

AHR in the absence of antigen sensitization and challenge occurs only in lung-specific IL-5–expressing transgenic mice. Airway responsiveness of NJ.1726 mice (A) and transgenic mice expressing IL-5 from a T cell–specific promoter (B) was measured immediately after exposure to aerosolized methacholine. In each case, measurements were made relative to age matched (3–5 mo) transgenic negative littermates. Mice were assessed on two occasions and the average for each animal was obtained. Values reported are group means ±SD (n = 11). These data were used to derive the effective dose200 ED200 levels (i.e., 50% maximal response) shown in the histograms to the left. The ED200 for NJ.1726 transgenic mouse group was significantly different from the control wildtype mice (*P ⩽0.01).
Notably, the airway responsiveness displayed by NJ.1726 mice was achieved in the absence of an inflammatory stimulus (e.g., aerosolized antigen challenge). In relative terms, the increase in AHR observed in naive transgenic mice is comparable to that observed in ovalbumin sensitized and challenged C57BL/6J mouse models of asthma (threefold versus three- to fivefold, respectively [our unpublished observations]).
Discussion
Lung-specific Expression of IL-5: Pulmonary Changes Resembling Allergen-induced Respiratory Inflammation.
Inflammatory responses in the lung are mediated by cytokine and chemokine expression, including IL-5, derived predominately from the airway leukocyte infiltrate (4). We have selectively replicated IL-5 expression from the airway by ectopically expressing this cytokine in the lung epithelium. Although the IL-5 expression in NJ.1726 mice is detectable using an antibody-specific ELISA assay, it is not known whether this ectopically produced IL-5 protein has full biological activities. However, since structure–function studies of ectopically expressed IL-5 in COS cells demonstrated that transfected cells were able to produce functional IL-5 (assayed by proliferation of an IL-5–dependent cell line) (36), it is likely that fully active cytokine can be produced by most cell types.
The prominent changes observed in NJ.1726 mice were an expansion of BALT, goblet cell hyperplasia, epithelial hypertrophy, an increase in peribronchial eosinophils, and the recruitment of eosinophils to the airway lumen. Areas of BALT in the lung are composed mostly of B cells and have been suggested as regions of antigen uptake (33, 37). In NJ.1726 mice, these areas are dominated by mononuclear cells (mostly lymphocytes) and thus the expansion of BALT may be a consequence of IL-5 proliferative effects on existing B lymphocyte populations (22, 38). If IL-5 effector functions rely in part on paracrine mechanisms, then the specific effects associated with the cellularity surrounding larger airways may be an indication of this regulatory loop. The appearance of expanded regions of lymphoid tissue is common to mouse models of respiratory disease (for example see reference 9) and may reflect localized cytokine gene expression. However, this is not a major feature of human disease, perhaps highlighting underlying immunologic differences between rodents and primates that are reflected in physiologic differences in response to pulmonary inflammatory stimuli. In addition to the varying effects on larger airway peribronchial lymph nodes, other histological perturbations also occurred suggesting that chronic expression of IL-5 at an elevated level leads to pathologic changes in the lung. These perturbations (i.e., increased numbers of goblet cells, deposition of collagen among the leukocyte aggregates, and hypertrophy of the airway epithelium) occurred in nearly all transgenic animals and are consistent characteristics of several human inflammatory lung diseases (e.g., asthma [26, 39, 40] and bronchopulmonary aspergillosis [41]) that are associated with increased IL-5 levels in the lung.
The infiltration of the peribronchial interstitium and the airway lumen by eosinophils in the absence of antigen sensitization and challenge was a diagnostic phenotype of the transgenic animals. Since allergic type I hypersensitivity (i.e., aerosolized OVA challenge) in wild-type mice induces a large airway lumen infiltrate dominated by eosinophils, the lack of a large increase in BAL total cellularity in NJ.1726 animals suggests specific differences exist between these models. It is likely that in addition to elevated levels of IL-5 expression, aerosolized OVA challenge also results in the expression of other signals (e.g., chemokines) that are critical for the activation and subsequent migration of eosinophils from the peribronchial spaces.
Different TH2 Cytokines Elicit Distinct Pulmonary Effector Functions: IL-4 versus IL-5.
The ectopic expression in the lung epithelium of the two dominant TH2 cytokines, IL-4 and IL-5, have now been reported. Expression of IL-4 using the rat CC10 promoter produces pathologic changes leading to the recruitment of lymphocytes and both eosin– ophils and neutrophils into the airway lumen (21). In addition, IL-4 expression elicited hypertrophy of the epithelium of the larger airways and increased the baseline airway resistance of transgenic lungs relative to wild-type controls. Surprisingly, the data presented also showed that IL-4 expression in the lung had no demonstrable effect on AHR in response to methacholine challenge.
Since the measured levels of IL-4 in the BAL fluid were only 4% of the IL-5 levels found in NJ.1726 mice, it is difficult to compare, in absolute terms, the effects of IL-4 versus IL-5 airway expression. However, several qualitative conclusions are worth noting: (a) whereas the presence of IL-4 in the BAL of transgenic mice induced a leukocytic airway lumen infiltrate (fivefold relative to wild-type) that included macrophages (44%) and approximately equal numbers (i.e., 15–20%) of lymphocytes, eosinophils and neutrophils, expression of IL-5 at levels 23-fold higher did not alter total BAL cell numbers and induced only an increase in BAL eosinophils. (b) Overexpression of either IL-4 or IL-5 in transgenic mice induced histopathologic pulmonary changes (e.g., epithelial hypertrophy and increases in BALT). The histopathologies reported here for IL-5 transgenic mice are more pronounced than the observed IL-4 pathologies; however, this difference may be a consequence of the relative levels of cytokine. (c) Unlike the IL-4 transgenic mice, the expression of IL-5 in the airway lumen leads to AHR in the absence of antigen sensitization and challenge. Although AHR to methacholine challenge may also be a consequence of the higher level of cytokine expression found in NJ.1726 mice, this pathophysiologic response does correlate with the higher degree of histopathology observed in IL-5 transgenic mice.
IL-5 Is a Link to Signaling Cascades Leading to AHR.
AHR is a complex pathophysiologic response that has been shown to occur as a consequence of several independent pathways. The role of IL-5 gene expression, eosinophil accumulation in the lungs, and the development of AHR has been controversial. In mouse models of respiratory inflammation, many studies have linked IL-5 expression and the recruitment of eosinophils to the lung (41, 42); however, the correlation of IL-5 expression and AHR has been more problematic.
The inability of IL-5–deficient mice to develop AHR and airway eosinophilia after ovalbumin sensitization and challenge suggested that IL-5 was required (17). Moreover, ovalbumin sensitization and challenge of wild-type mice treated with anti–IL-5 antibodies also resulted in the abrogation of the recruitment of eosinophils to the lung and the development of AHR (43). In contrast, other studies with anti–IL-5 antibodies showed that eosinophil airway infiltration can be significantly reduced without any effect on AHR (11, 44). In the most recent study, Corry et. al. (11) have reported that although treatment of mice with antibodies to IL-4 mimics the responses of IL-4–deficient mice, treatment with anti–IL-5 antibodies results in the elimination of eosinophil airway infiltration but had no effect on AHR in response to ovalbumin sensitization. Several explanations have been offered to resolve these differences (e.g., differences in sensitization protocols [43] and inbred mouse strain variation [45]); however, it is most likely that the passive immunization protocols may have incompletely neutralized endogenous IL-5. This conclusion is supported by studies in ovalbumin sensitized and challenged guinea pigs in which low levels of anti–IL-5 antibodies inhibited eosinophil infiltration into the airways without affecting AHR, whereas high levels of anti–IL-5 antibodies eliminated both eosinophil recruitment and AHR (46). This effect, however, is more complex than cells within the lung responding to elevated levels of serum IL-5. Transgenic animals with high serum IL-5 levels derived from several non-lung cell types display no pulmonary pathologies (see Fig. 3 e; 22–24) and measurements of AHR in response to methacholine challenge showed that these mice also do not exhibit bronchial hyperreactivity in the absence of antigen sensitization and challenge (see Fig. 7; 25, 47).
The data reported here show that unlike OVA-induced models of respiratory inflammation, AHR in NJ.1726 mice can be linked to changes in BAL cellularity comparable to those observed in asthmatic patient populations. This phenomenon occurs in the absence of antigen sensitization and challenge and only in transgenic animals where the primary source of IL-5 is the lung. AHR also occurred in all NJ.1726 mice and did not parallel the large variation in BALT expansion found in these animals. This observation suggests that AHR is not linked to changes in BALT and instead may correlate to one of the histopathologies that occur in all transgenic mice (e.g., goblet cell hyperplasia, epithelial hypertrophy, and focal collagen deposition). The observations presented cannot eliminate the possibility of IL-5–induced eosinophil effector functions mediated through the large peribronchial infiltrate or the recruited BAL cells as factors in the development of AHR; however, the induction of this pathophysiologic response in NJ.1726 mice shows that lung-specific IL-5 expression is a sufficient inflammatory signal to induce AHR, airway lumenal infiltration by eosinophils, and histopathologic pulmonary changes. In this paradigm, ectopic lung-specific IL-5 expression reproduces some but not necessarily all of the signals associated with asthmatic inflammation.
Although IL-5 expression in the lung leads to specific pathophysiologic changes such as AHR, the absolute magnitude of these changes are less than those observed in many patients with asthma (48). This suggests either species-specific pathophysiologic differences or the pathologies found in patient populations result only in part because of IL-5 effector functions and that the presence of other inflammatory signals (e.g., chemokine expression, IgE, or masts cells), perhaps working synergistically with IL-5 (49–51), lead to the augmentation of disease symptoms. The combinatorial effect of the pulmonary IL-5 expression found in NJ.1726 mice and exposure to an extrinsic inflammatory stimulus is not yet known. These experiments are of particular interest because of the potential to augment the observed pathologies in IL-5 transgenic mice and reproduce conditions seen in patient populations with severe respiratory disease.
Footnotes
These studies represent a considerable effort on the part of many individuals in addition to the cited authors. We wish to thank Drs. John McDonald and Jeffery Whitsett for their encouragement and assistance in bringing many of us together as part of a productive collaborative effort. We wish to thank the important technical assistance by Trella Mitchell and the help received by the Histology (Anita Jennings) and Transgenic Mouse (Suresh Savarirayan) core facilities. Insightful comments and interpretations of the lung histology were provided by Drs. Thomas Colby and Kevin Leslie. Critical reviews of the manuscript by Drs. John McDonald, Kevin Leslie, Gerald Gleich, and Eric Wieben were invaluable to the clarity of the work presented. We are indebted to the Mayo Clinic Arizona graphic artists Marvin Ruona and Julie Jensen for their dedicated work, Susan Bond and Merrilee Parker for their patience dealing with earlier versions of the manuscript, and our program assistant Beverly K. Pratley without whom we could not function as a productive laboratory.
Funds for these studies were provided by the Mayo Foundation and the Arizona Disease Control Research Commission.
1 Abbreviations used in this paper: AHR, airway hyperresponsiveness; BAL, bronchial alveolar lavage; BALT, bronchus-associated lymphoid tissue; H/E, hematoxylin/eosin; mMBP, anti-murine eosinophil granule major basic protein; Penh, enhanced pause; SPF, specific pathogen-free; WBC, white blood cell.
References
- 1.O'Byrne PM. Allergen-induced airway hyperresponsiveness. J Allergy Clin Immunol. 1988;81:119–127. doi: 10.1016/0091-6749(88)90230-8. [DOI] [PubMed] [Google Scholar]
- 2.Empey DW, Laitinen LA, Jacobs L, Gold WM, Nadel JA. Mechanisms of bronchial hyperreactivity in normal subjects after upper respiratory tract infection. Am Rev Resp Dis. 1976;113:131–139. doi: 10.1164/arrd.1976.113.2.131. [DOI] [PubMed] [Google Scholar]
- 3.McBride DE, Koenig JQ, Luchtel DL, Williams PV, Henderson W., Jr Inflammatory effects of ozone in the upper airways of subjects with asthma. Am J Resp Crit Care Med. 1994;149:1192–1197. doi: 10.1164/ajrccm.149.5.8173759. [DOI] [PubMed] [Google Scholar]
- 4.Krug N, Madden J, Redington AE, Lackie P, Djukanovic R, Schauer U, Holgate ST, Frew AJ, Howarth PH. T-cell cytokine profile evaluated at the single cell level in BAL and blood in allergic asthma. Am J Resp Cell Mol Biol. 1996;14:319–326. doi: 10.1165/ajrcmb.14.4.8600935. [DOI] [PubMed] [Google Scholar]
- 5.Henderson W., Jr Role of leukotrienes in asthma. Ann Allergy. 1994;72:272–278. [PubMed] [Google Scholar]
- 6.Bentley AM, Durham SR, Kay AB. Comparison of the immunopathology of extrinsic, intrinsic and occupational asthma. J Invest Allergol Clin Immunol. 1994;4:222–232. [PubMed] [Google Scholar]
- 7.Daniels SE, Bhattacharrya S, James A, Leaves NI, Young A, Hill MR, Faux JA, Ryan GF, Lesouef PN, Lathrop GM, et al. A genome-wide search for quantitative trait loci underlying asthma. Nature (Lond) 1996;383:247–250. doi: 10.1038/383247a0. [DOI] [PubMed] [Google Scholar]
- 8.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 9.Blyth DI, Pedrick MS, Savage TJ, Hessel EM, Fattah D. Lung inflammation and epithelial changes in a murine model of atopic asthma. Am J Resp Cell Mol Biol. 1996;14:425–438. doi: 10.1165/ajrcmb.14.5.8624247. [DOI] [PubMed] [Google Scholar]
- 10.Fishman, A.P., editor. 1988. Pulmonary Diseases and Disorders. 2nd edition. McGraw-Hill, Inc., New York. 113–118.
- 11.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wienerkronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Resp Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima H, Iwamoto I, Tomoe S, Matsumura R, Tomioka H, Takatsu K, Yoshida S. CD4+ T-lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea. Am Rev Resp Dis. 1992;146:374–377. doi: 10.1164/ajrccm/146.2.374. [DOI] [PubMed] [Google Scholar]
- 14.Hamelmann E, Oshiba A, Paluh J, Bradley K, Loader J, Potter TA, Larsen GL, Gelfand EW. Requirement for CD8(+) T cells in the development of airway hyperresponsiveness in a murine model of airway sensitization. J Exp Med. 1996;183:1719–1729. doi: 10.1084/jem.183.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown DR, Fowell DJ, Corry DB, Wynn TA, Moskowitz MH, Cheever AW, Locksley RM, Reiner SL. β2-microglobulin-dependent NK1.1+ T cells are not essential for T helper cell 2 immune responses. J Exp Med. 1996;184:1295–1304. doi: 10.1084/jem.184.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brusselle G, Kips J, Joos G, Bluethmann H, Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. Am J Resp Cell Mol Biol. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- 17.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tepper RI, Levinson DA, Stanger BZ, CamposTorres J, Abbas AK, Leder P. IL-4 induces allergic-like inflammatory disease and alters T cell development in transgenic mice. Cell. 1990;62:457–467. doi: 10.1016/0092-8674(90)90011-3. [DOI] [PubMed] [Google Scholar]
- 19.Lewis DB, Liggitt HD, Effmann EL, Motley ST, Teitelbaum SL, Jepsen KJ, Goldstein SA, Bonadio J, Carpenter J, Perlmutter RM. Osteoporosis induced in mice by overproduction of interleukin 4. Proc Natl Acad Sci USA. 1993;90:11618–11622. doi: 10.1073/pnas.90.24.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erb KJ, Hanke T, Schimpl A, Hunig T, Stingl G, Elbe A. Impaired survival of T cell receptor V gamma 3+ cells in interleukin-4 transgenic mice. Eur J Immunol. 1995;25:1442–1445. doi: 10.1002/eji.1830250546. [DOI] [PubMed] [Google Scholar]
- 21.Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, Dicosmo B, Tarallo A, Stripp B, Whitsett J, Flavell RA. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung— lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci USA. 1996;93:7821–7825. doi: 10.1073/pnas.93.15.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. Expression of Il-5 in thymocytes T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol. 1997;158:1332–1344. [PubMed] [Google Scholar]
- 23.Dent LA, Strath M, Mellor AL, Sanderson CJ. Eosinophilia in transgenic mice expressing interleukin 5. J Exp Med. 1990;172:1425–1431. doi: 10.1084/jem.172.5.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tominaga A, Takaki S, Koyama N, Katoh S, Matsumoto R, Migita M, Hitoshi Y, Hosoya Y, Yamauchi S, Kanai Y, et al. Transgenic mice expressing a B cell growth and differentiation factor gene (interleukin 5) develop eosinophilia and autoantibody production. J Exp Med. 1991;173:429–437. doi: 10.1084/jem.173.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamoto T, Takatsu K. Evaluation of airway hyperreactivity in interleukin-5 transgenic mice. Int Arch Allergy Immunol. 1995;1:28–30. doi: 10.1159/000237196. [DOI] [PubMed] [Google Scholar]
- 26.Venge J, Lampinen M, Hakansson L, Rak S, Venge P. Identification of IL-5 and RANTES as the major eosinophil chemoattractants in the asthmatic lung. J Allergy Clin Immunol. 1996;97:1110–1115. doi: 10.1016/s0091-6749(96)70265-8. [DOI] [PubMed] [Google Scholar]
- 27.Hackett BP, Gitlin JD. Cell-specific expression of a Clara cell secretory protein-human growth hormone gene in the bronchiolar epithelium of transgenic mice. Proc Natl Acad Sci USA. 1992;89:9079–9083. doi: 10.1073/pnas.89.19.9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horton MA, Larson KA, Lee JJ, Lee NA. Cloning of the murine eosinophil peroxidase gene (mEPO): characterization of a conserved subgroup of mammalian hematopoietic peroxidases. J Leukoc Biol. 1996;60:285–294. doi: 10.1002/jlb.60.2.285. [DOI] [PubMed] [Google Scholar]
- 29.Lee JJ, Radice G, Perkins CP, Costantini FD. Identification and characterization of a novel, evolutionarily conserved gene disrupted by the murine Hβ58 embryonic lethal transgene insertion. Development. 1992;115:277–288. doi: 10.1242/dev.115.1.277. [DOI] [PubMed] [Google Scholar]
- 30.Renz H, Smith HR, Henson JE, Ray BS, Irvin CG, Gelfand EW. Aerosolized antigen exposure without adjuvant causes increased IgE production and increased airway responsiveness in the mouse. J Allergy Clin Immunol. 1992;89:1127–1138. doi: 10.1016/0091-6749(92)90296-e. [DOI] [PubMed] [Google Scholar]
- 31.Hamelmann E, Shwarze J, Takeda K, Oshiba A, Gelfand EW. Measurement of airway hyperresponsiveness (AHR) to inhaled methacholine in free-running allergensensitized mice. Am J Resp Crit Care Med. 1996;153:A625. [Google Scholar]
- 32.Strum JM, Compton RS, Katyal SL, Singh G. The regulated expression of mRNA for Clara cell protein in the developing airways of the rat, as revealed by tissue in situ hybridization. Tissue Cell. 1992;24:461–471. doi: 10.1016/0040-8166(92)90062-c. [DOI] [PubMed] [Google Scholar]
- 33.Bienenstock, J. 1985. Bronchus-associated lymphoid tissue. Int. Arch. Allergy Appl. Immunol. 76(suppl. 1):62–69. [DOI] [PubMed]
- 34.Larson K, Horton M, Madden B, Gleich G, Lee N, Lee J. The identification and cloning of a murine major basic protein gene expressed in eosinophils. J Immunol. 1995;155:3002–3012. [PubMed] [Google Scholar]
- 35.Wardlaw AJ, Dunnette S, Gleich GJ, Collins JV, Kay AB. Eosinophils and mast cells in bronchoalveolar lavage in subjects with mild asthma—relationship to bronchial hyperreactivity. Am Rev Respir Dis. 1988;137:62–69. doi: 10.1164/ajrccm/137.1.62. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Cook R, Dede K, Chaiken I. Single chain human interleukin 5 and its asymmetric mutagenesis for mapping receptor binding sites. J Biol Chem. 1996;271:1817–1820. doi: 10.1074/jbc.271.4.1817. [DOI] [PubMed] [Google Scholar]
- 37.Pabst R, Gehrke I. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? . Am J Resp Cell Mol Biol. 1990;3:131–135. doi: 10.1165/ajrcmb/3.2.131. [DOI] [PubMed] [Google Scholar]
- 38.Harada N, Kikuchi Y, Tominaga A, Takaki S, Takatsu K. BCGFII activity on activated B cells of a purified murine T cell-replacing factor (TRF) from a T cell hybridoma (B151K12) J Immunol. 1985;134:3944–3951. [PubMed] [Google Scholar]
- 39.Busse WW, Sedgwick JB. Eosinophils in asthma. Ann Allergy. 1992;68:286–290. [PubMed] [Google Scholar]
- 40.Sur S, Kita H, Gleich GJ, Chenier TC, Hunt LW. Eosinophil recruitment is associated with IL-5, but not with RANTES, twenty-four hours after allergen challenge. J Allergy Clin Immunol. 1996;97:1272–1278. doi: 10.1016/s0091-6749(96)70195-1. [DOI] [PubMed] [Google Scholar]
- 41.Kurup VP, Choi H, Murali PS, Coffman RL. IgE and eosinophil regulation in a murine model of allergic aspergillosis. J Leukoc Biol. 1994;56:593–598. doi: 10.1002/jlb.56.5.593. [DOI] [PubMed] [Google Scholar]
- 42.Coyle AJ, Erard F, Bertrand C, Walti S, Pircher H, Le Gros G. Virus-specific CD8+ cells can switch to interleukin 5 production and induce airway eosinophilia. J Exp Med. 1995;181:1229–1233. doi: 10.1084/jem.181.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamelmann E, Oshiba A, Loader J, Larsen GL, Larson KA, Gleich GJ, Lee JJ, Gelfand EW. Antiinterleukin-5 antibody prevents airway hyperresponsiveness in a murine model of airway sensitization. Am J Resp Crit Med. 1997;155:819–825. doi: 10.1164/ajrccm.155.3.9117011. [DOI] [PubMed] [Google Scholar]
- 44.Nagai H, Yamaguchi S, Inagaki N, Tsuruoka N, Hitoshi Y, Takatsu K. Effect of anti-IL-5 monoclonal antibody on allergic bronchial eosinophilia and airway hyperresponsiveness in mice. Life Sciences. 1993;53:L243–247. doi: 10.1016/0024-3205(93)90545-e. [DOI] [PubMed] [Google Scholar]
- 45.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–5. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mauser PJ, Pitman A, Witt A, Fernandez X, Zurcher J, Kung T, Jones H, Watnick AS, Egan RW, Kreutner W, et al. Inhibitory effect of the TRFK-5 anti-IL-5 antibody in a guinea pig model of asthma. Am Rev Resp Dis. 1993;148:1623–1627. doi: 10.1164/ajrccm/148.6_Pt_1.1623. [DOI] [PubMed] [Google Scholar]
- 47.Lefort J, Bachelet CM, Leduc D, Vargaftig BB. Effect of antigen provocation of IL-5 transgenic mice on eosinophil mobilization and bronchial hyperresponsiveness. J Allergy Clin Immunol. 1996;97:788–799. doi: 10.1016/s0091-6749(96)80157-6. [DOI] [PubMed] [Google Scholar]
- 48.Smith L, McFadden E., Jr Bronchial hyperreactivity revisited. Ann Allergy Asthma Immunol. 1995;74:454–469. [PubMed] [Google Scholar]
- 49.Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med. 1995;182:1169–1174. doi: 10.1084/jem.182.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eum SY, Haile S, Lefort J, Huerre M, Vargaftig BB. Eosinophil recruitment into the respiratory epithelium following antigenic challenge in hyper-IgE mice is accompanied by interleukin 5-dependent bronchial hyperresponsiveness. Proc Natl Acad Sci USA. 1995;92:12290–12294. doi: 10.1073/pnas.92.26.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabin EA, Kopf MA, Pearce EJ. Schistosoma mansoni egg-induced early IL-4 production is dependent upon IL-5 and eosinophils. J Exp Med. 1996;184:1871–1878. doi: 10.1084/jem.184.5.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]