Resting Memory CD8+ T Cells are Hyperreactive to Antigenic Challenge In Vitro (original) (raw)
Abstract
The characteristics of CD8+ T cells responsible for memory responses are still largely unknown. Particularly, it has not been determined whether different activation thresholds distinguish naive from memory CD8+ T cell populations. In most experimental systems, heterogeneous populations of primed CD8+ T cells can be identified in vivo after immunization. These cells differ in terms of cell cycle status, surface phenotype, and/or effector function. This heterogeneity has made it difficult to assess the activation threshold and the relative role of these subpopulations in memory responses. In this study we have used F5 T cell receptor transgenic mice to generate a homogeneous population of primed CD8+ T cells. In the F5 transgenic mice, peptide injection in vivo leads to activation of most peripheral CD8+ T cells. In vivo BrdU labeling has been used to follow primed T cells over time periods spanning several weeks after peptide immunization. Our results show that the majority of primed CD8+ T cells generated in this system are not cycling and express increased levels of CD44 and Ly6C. These cells remain responsive to secondary peptide challenge in vivo as evidenced by short term upregulation of activation markers such as CD69 and CD44. The activation thresholds of naive and primed CD8+ T cells were compared in vitro. We found that CD8+ T cells from primed mice are activated by peptide concentrations 10–50-fold lower than naive mice. In addition, the kinetics of interleukin 2Rα chain upregulation by primed CD8+ T cells differ from naive CD8+ T cells. These primed hyperresponsive CD8+ T cells might play an important role in the memory response.
Immunological memory is the capacity of the immune system to respond faster and more efficiently against an antigen it has encountered in the past. T cell memory is believed to result from an increase in the frequency of antigen specific T lymphocytes (1, 2). For CD4+ T cells, a change in the activation requirements of memory cells has been reported (3, 4). In these studies, memory CD4+ T cells were identified using different surface markers. However, most of these markers are also expressed by freshly activated cells, making it difficult to distinguish memory T cells from activated T cells. More recent studies have taken advantage of TCR-transgenic mice. These studies have confirmed that primed CD4+ T cells are hyperreactive to antigenic stimulation in vitro (5). However, the contribution of this increased reactivity versus that of clonal expansion to the memory response in vivo is difficult to estimate. Moreover, it may vary depending on the experimental system.
For CD8+ T cells, it is also well established that the frequency of precursor CTL increases after antigenic challenge (1, 6). However, this might not be the only parameter responsible for the memory response because primed CD8+ T cells have also been shown to give a more sustained, albeit slower, in vivo response than naive CD8+ T cells (7).
The cell cycle status of memory CD8+ cells is debatable (8). Few studies have directly measured the proliferation rate of memory CD8+ T cells. When the life span of T cells with a “memory” phenotype was measured using BrdU labeling, a large fraction divided rapidly, whereas another fraction remained in interphase for several weeks (9). A similar dichotomy of cycling versus noncycling cells was found when the cell cycle status of LCMV memory CTL precursors was studied (10). Moreover, these two subsets required different conditions for their activation in vitro. The relationship between the cycling and the noncycling memory CTL precursors as well as their relative longevity has not been studied. They could represent different subsets or differentiation stages of memory T cells requiring different signals for their activation and/or maintenance. The role played by antigen in the maintenance of memory CD8+ T cells is still controversial (11–15), but in some experimental systems maintenance of memory in the absence of antigen persistence is possible (11–13). However, in these conditions it is not known whether memory CTL precursors also show a heterogeneous phenotype and life span.
To identify antigen primed CD8+ T cells, we used F5 TCR transgenic mice. The majority of peripheral T cells in F5 mice are CD8+, and >95% of these express Vβ11. Intraperitoneal injection of influenza virus NP peptide (residues 366–374) in these mice leads to an increase in the number of CD8+ spleen T cells. This is paralleled by CD44 upregulation on all CD8+ T cells (16, 17), which also acquire the ability to lyse peptide-pulsed targets cells ex vivo (16). CD8+ T cells with increased levels of CD44 are still present in the spleen of thymectomized mice up to 6 wk after peptide challenge (17).
In this report, we have characterized these primed CD8+ T cells with respect to surface antigens, life span and reactivity to antigen in vivo and in vitro. We have used BrdU to mark cells proliferating in response to antigen during the primary response. In thymectomized mice, the majority of BrdU labeled CD8+ T cells are still present several weeks after immunization. In normal euthymic mice, BrdU labeled cells are diluted by naive T cells. However, a fraction (20%) of BrdU-labeled CD8+ T cells are still present 11 wk after priming, indicating a long life span. These primed CD8+ T cells express increased levels of CD44 and Ly6C when compared with naive CD8+ T cells. Using thymectomized F5 TCR-transgenic mice we show that primed CD8+ T cells are not anergic as they are responsive to a second in vivo antigenic challenge. The activation threshold of primed and naive CD8+ T cells was measured in vitro. We show that, in the presence of IL-2, primed cells hyperproliferate to all doses of antigen and that their proliferation is triggered at antigen doses 10–50 fold lower than the one triggering naive cells. For the sake of clarity, we call CD8+ T cells surviving >3 wk after immunization primed cells, opposed to CD8+ T cells that are found during the first week after immunization which we call activated.
Materials and Methods
Mice and Immunizations.
C57BL10 and F5 TCR transgenic mice were a gift from D. Kioussis (18). All animals were bred in the institute's animal facility. Thymectomies were performed on 5–7-wk-old mice, which were then allowed to recover for at least 4 wk before antigenic challenge. Immunizations were performed by injecting 50 nmol of the A/NT/60/68 influenza virus nucleoprotein peptide Ala-Ser-Asn-Glu-Asn-Met-Asp-Ala-Met [NP-(366-374)] (Neosystems Laboratoire, Strasbourg, France) in PBS into the peritoneal cavity. Control mice were either not treated or injected with PBS alone.
In Vivo BrdU Labeling.
Mice were given BrdU (_Sigma Chemical_s Co., St. Louis, MO) at 1 mg/ml in drinking water. The BrdU-containing water was protected from light and changed daily. Mice were given BrdU for 5 or 7 d, starting 1 d before peptide injection.
Fluorescence Staining and Flow Cytometry Analysis.
Spleen cell suspensions were applied to Ficoll–Hypaque (Lympholyte M; Cedarlane Laboratories Ltd., Hornby, Canada) gradient centrifugation. Cells were then washed twice with medium, resuspended in PBS containing 2% horse serum and 0.1% NaN3, and then 106 cells were incubated with antibody for 45 min at 4°C. After two more washes with PBS/horse serum/NaN3, cells were incubated in the presence of second layer reagents (avidin-PE or avidin-tricolor; (CALTAG Laboratories, South San Francisco, CA) for 30 min at 4°C. Cells were washed twice and analyzed on a FACScan® (Becton Dickinson & Co., Mountain View, CA). The following antibodies were used: YTS169.4-PE (anti CD8), IM7.8.1-FITC (anti CD44), CG16-biotin (anti-CD5) from Caltag Laboratories; HI.2F3-biotin (anti CD69), Jo2-biotin (anti Fas), 5H4-FITC (anti–IL2Rβ), 4G3-PE (anti–IL2Rγ) from PharMingen (San Diego, CA); PC61.5.3-biotin (anti–IL2Rα) from Cedarlane Laboratories Ltd.; IM7.8.1-biotin (anti CD44), Mel-14-biotin (anti–l-selectin), RR3-15-biotin (anti Vβ11), RA3-2C2-biotin (anti-CD45RA), 23G2-biotin (anti-CD45RB), FD441.8-biotin (anti–LFA-1), YN1/ 1.7-biotin (anti–ICAM-1), 145-2C11-FITC (anti CD3), and 1434-2-biotin (anti-Ly6.2C) were prepared in house.
Staining for BrdU incorporation was conducted as previously described (9) with minor modifications. In brief, cells were stained for surface markers as above, resuspended in cold 0.15 M NaCl solution, and fixed by injection into cold 95% ethanol. After a 30 min incubation on ice, cells were washed once with PBS and resuspended in PBS, 0.01% Tween 20, and 1% paraformaldehyde. After a 1-h incubation at room temperature, cells were washed with 0.15 M NaCl, and the DNA was partially digested with DNase I (Appligene, Illkirch, France) in 25 mM CaCl2, 5mM MgCl2, and 10 mM Hepes, pH 7.4. Cells were then washed with 0.15 M NaCl before adding the anti-BrdU antibody (B44-FITC; Becton Dickinson & Co.). After overnight incubation with anti-BrdU, cells were washed with PBS and analyzed on the FACScan®.
Cell Culture.
Spleen cells were cultured in DMEM (GIBCO BRL, Gaithersburg, MD) supplemented with 2 mM l-glutamine (GIBCO BRL), 10 μg/ml gentamycin (GIBCO BRL), 6% FCS (Boehringer Mannheim, Mannheim, Germany), 10 mM Hepes, and 50 μM 2-ME. F5 transgenic spleen cells (5 × 104 nucleated cells/well) were activated in 96-well plates with various concentrations of nucleoprotein peptide in the presence of 2 × 105 irradiated (3,000 rad) C57BL10 spleen cells in the presence or absence of 2.5% supernatant containing IL-2 (19). On day 4, cells were pulsed for 16 h with 0.5 μCi [3H]thymidine/well (2.0 Ci/mmol; Amersham) and harvested on day 5 unless otherwise indicated. For inhibition by anti-CD8 mAb (YTS 169), 1 μg/ml of purified antibody was added to the cultures at the day of activation.
Results
The Majority of F5 CD8+ T Cells Incorporate BrdU in Response to Peptide Challenge.
To determine the proportion of CD8+ T lymphocytes proliferating in a primary response to peptide, we used in vivo BrdU labeling. Results presented in Fig. 1 show that the majority of F5 spleen CD8+ T cells incorporated BrdU in their DNA 3 d after peptide injection. The same CD8+BrdU+ T cell population also expressed high levels of CD44. In control mice, only a small percentage of spleen CD8+ T cells incorporated BrdU and expressed CD44, consistent with the view that naive cells are CD44− noncycling cells (20). Similar results were obtained in thymectomized and nonthymectomized animals, indicating that the BrdU is incorporated in peripheral T cells and not in immature thymocytes (Fig. 1). Together these data indicate that the majority of the peripheral T cell pool can be activated and enters the cell cycle in response to peptide stimulation in vivo.
Figure 1.

In vivo BrdU incorporation by transgenic CD8+ T cells after peptide stimulation. Thymectomized and euthymic mice were either immunized with 50 nmol peptide on day 0 (Activated) or received only PBS (Naive). BrdU was given in the drinking water for 4 d starting 1 d before peptide injection. 3 d after peptide challenge, spleen cells were double stained for BrdU and CD8 or triple stained for BrdU, CD8, and CD44. Results for one representative mouse are shown. The percentage of BrdU+ and BrdU− CD8+ T cells is given in the first column. The total number (×106) of BrdU+CD8+ cells is given in brackets. The expression of CD44 by BrdU labeled cells is shown for gated CD8+ T cells. Results for one representative mouse out of two naive or four primed mice are shown.
Primed CD8+ T Cells Are Maintained In Vivo.
To investigate the maintenance of primed F5 CD8+ T cells in vivo, we have followed BrdU+ labeled peripheral CD8+ T cells over time. Pulse–chase experiments were performed. Mice were given BrdU continuously for 5 d starting 1 d before priming. The percentage of CD8+ BrdU+ T cells was measured at different days after immunization.
Results presented in Fig. 2 a show that in thymectomized mice, ∼85% of CD8+ T cells were still labeled with BrdU 21 d after peptide priming. In euthymic mice, the percentage of BrdU+ cells was lower (55 instead of 85%). This difference is likely to be due to dilution by BrdU− thymic emigrants, because this increase in BrdU− cells was not found in thymectomized mice. In addition, these BrdU− cells expressed low levels of CD44, which correlates with a naive phenotype (data not shown).
Figure 2.


Primed CD8+ T cells are long lived. The maintenance of BrdU labeled CD8 T cells was measured at different times after peptide stimulation and BrdU labeling. Thymectomized or euthymic mice were either immunized with 50 nmol peptide on day 0 (primed) or received only PBS (naive). BrdU was given in the drinking water for 5 d starting 1 d before peptide injection. Spleen cells were double stained for BrdU and CD8. Results for one representative mouse are shown. (a) The percentage of BrdU+ and BrdU− CD8+ T cells in thymectomized and euthymic mice 21 d after peptide stimulation. (b) The percentage of BrdU+ and BrdU− CD8+ T cells in euthymic mice 35 and 77 d after peptide priming. The total number (×106) of BrdU+CD8+ cells is given in brackets. Results for one representative mouse out of two naive or four primed mice are shown.
To confirm the maintenance of primed cells in an environment where they are in competition with naive cells, we studied their survival in euthymic mice 5 and 11 wk after priming (Fig. 2 b). The percentage of CD8+BrdU+ cells declined slowly with time. However, primed CD8+BrdU+ cells still represented 20% of the total CD8+ T cell population after 80 d. Together these data show that a substantial number of primed CD8+ T cells are maintained in the absence of division for up to 11 wk in animals in which the thymus provides a constant supply of naive CD8+ T cells to the periphery. To avoid dilution by naive CD8+ T cells, thymectomized mice were generally used for the characterization of primed CD8+ T cells. For activated CD8+ T cells, we used either thymectomized or euthymic mice because in the 1st wk after priming CD8+ T cells obtained in these mice are indistinguishable in terms of phenotype or function (data not shown).
Primed CD8+ T Cells Express CD44 and Ly6C.
To further characterize the surface phenotype of the primed CD8+ T cell population, we studied the expression of a number of markers that are usually associated with an activated or memory phenotype. We saw no differences in expression of CD8, Vβ11, CD3, CD69, IL-2Rα, IL-2Rβ, IL-2Rγ, ICAM-1, LFA-1, CD45RA, CD45RB, l-selectin, Fas, or CD5 when comparing CD8+ T cells from naive and primed mice 6 wk after peptide injection (Fig. 3 a). However, all of these markers were up- or downregulated on recently activated CD8+ T cells 1 or 3 d after activation (Fig. 3 b). In these experiments, CD44 was the only marker that was upregulated by a subset of primed CD8+ T cells (Fig. 3 a).
Figure 3.


Surface phenotype of primed, naive and recently activated CD8+ T cells. The expression of CD8, Vβ11, CD3, CD69, IL-2Rα, IL-2Rβ, IL-2Rγ, ICAM-1, LFA-1, CD45RA, CD45RB, l-selectin, Fas, and CD5 by naive, activated, or primed CD8 spleen cells was measured by double staining. Cells were gated for CD8+ at the acquisition level. Results for one representative mouse out of a minimum of four are shown. (a) CD8+ T cells (- - -) from naive thymectomized mice were compared with CD8+ T cells (—) from thymectomized mice primed 6 wk earlier. (b) CD8+ T cells from naive mice (- - -) were compared with recently activated CD8+ T cells (—). Expression of CD8, CD3, and Vβ11 was measured 3 d after peptide stimulation. All other markers were analyzed on day 1.
We also measured the expression of Ly6C, which has been described by others as a good marker for memory CD8+ T cells (21). Using triple staining for CD8, CD44, and Ly6C, we found that a large proportion of primed CD8+ T cells expressed intermediate levels of CD44 and high levels of Ly6C in peptide primed thymectomized mice (Fig. 4 a). This CD44intLy6Chigh CD8+ T cell population was not seen in naive thymectomized animals. Ly6C was also not expressed by recently activated CD8+ T cells, which have already upregulated CD44 (Fig. 4 a). In addition to these cells, a CD8+CD44highLy6Chigh T cell population was detected in primed and naive thymectomized mice. These cells also expressed a large number of the other activation markers (data not shown). Their origin is unknown, but peptide priming is not necessary for their generation because they are also present in naive mice. These results indicate that CD44intLy6Chigh CD8+ T cells are the major subset of primed cells generated in this system.
Figure 4.

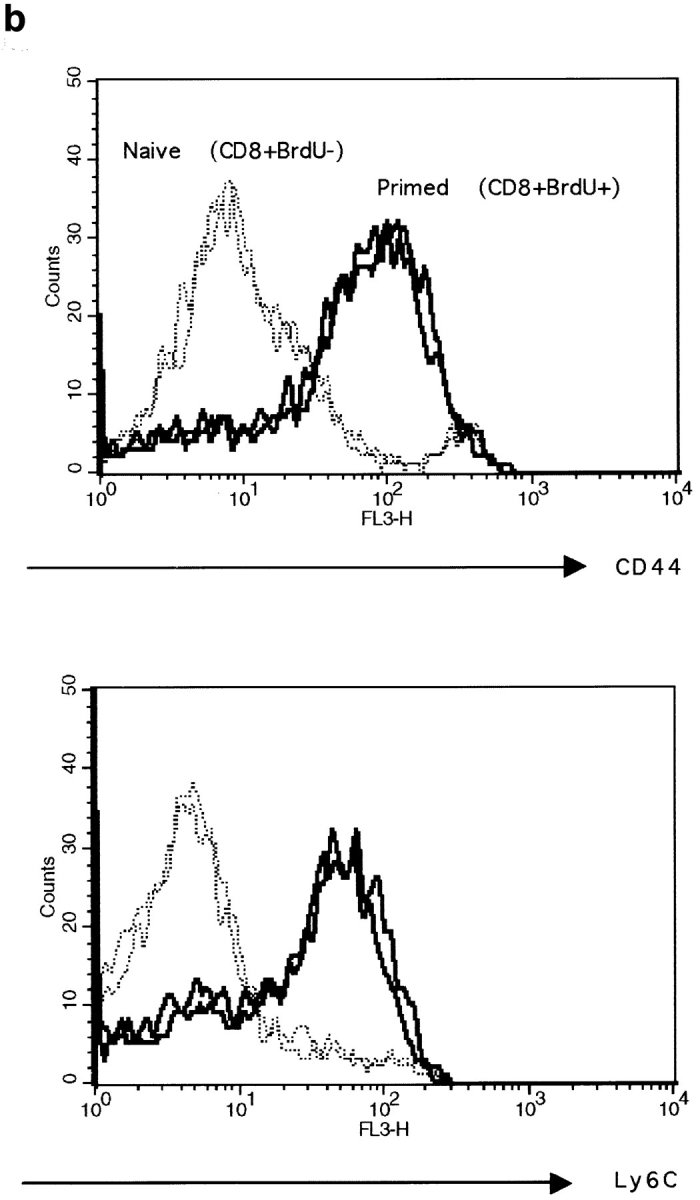
Expression of Ly6C and CD44 by naive, activated, or primed CD8+ T cells. (a) Spleen cells from thymectomized mice primed 5 wk earlier or from naive thymectomized mice and from euthymic mice primed 1 d earlier (activated) or from naive euthymic mice (naive) were triple stained for CD44, Ly6C, and CD8. Cells were gated for CD8+ at the acquisition level. The expression of CD44 and Ly6C is shown. The percentage of CD8+ cells expressing intermediate levels of CD44 and intermediate to high levels of Ly6C is given. The total number (×106) of CD8+CD44intLy6Chigh cells is given in brackets. Results for one mouse out of a minimum of three are shown. (b) The expression of CD44 and Ly6C by BrdU+CD8+ T cells from primed euthymic mice (—) was measured 11 wk after peptide stimulation. The CD8+ BrdU− T cells (- - -) from the same mice are shown as a control. CD8 cells were labeled with BrdU for 5 d starting 1 d before peptide stimulation. Cells were gated for CD8+BrdU− or CD8+BrdU+ at the acquisition level. Results are shown for two mice per group.
The phenotype of primed CD8+ T cells found in thymectomized mice was confirmed in euthymic mice. CD8+ T cells from euthymic mice primed and pulsed with BrdU 11 wk earlier were triple stained for CD8, BrdU, and CD44 or Ly6C. Data presented in Fig. 4 b confirm that primed CD8+ BrdU+ T cells expressed increased levels of CD44 and Ly6C.
These results confirm that in this system, primed CD8+ T cells are noncycling and can be phenotypically distinguished from naive or freshly activated CD8+ T cells by differential expression of the surface markers CD44 and Ly6C.
Primed CD8+ T Cells Can Be Reactivated In Vivo.
Because in some systems peptide challenge can also lead to anergy (22), we studied the in vivo response to peptide of primed CD8+ T lymphocytes. Primed and naive thymectomized mice were challenged with peptide as described in Fig. 5 a, and the changes of surface phenotype as well as the proliferation of CD8+ T cells were measured.
Figure 5.



In vivo primed CD8+ T cells are responsive to secondary challenge with peptide. (a) Experimental plan of the in vivo CD8+ T cells restimulation. (b) 1 d after peptide stimulation, spleen cells were double stained for CD8 and CD69 or CD8 and CD44. The cell size of CD8+ T cells as well as the expression of CD69 or CD44 by these cells are shown. Results for one naive and two primed mice are shown. (c) The percentage of CD8+ T cells proliferating in response to peptide challenge was measured by BrdU incorporation. Spleen cells were double stained for BrdU and CD8. The percentage of BrdU+ and BrdU− CD8+ T cells is given. The total number (×106) of BrdU+CD8+ cells is given in brackets. Results for one representative mouse out of two are shown.
Resting naive and primed CD8+ T cells expressed similar levels of CD69 and had a low forward scatter profile. In contrast, as previously shown, primed CD8+ T cells expressed intermediate levels of CD44. 1 d after peptide challenge, all CD8+ T cells from primed or naive mice became blast size and upregulated the early activation marker CD69 as well as CD44 (Fig. 5 b), indicating that the majority of CD8+ T cells are stimulated by the peptide.
We next determined whether cells from primed mice proliferated in vivo. Primed and naive thymectomized mice were injected with peptide, and dividing cells were labeled with BrdU. The proportion of CD8+BrdU+ T cells was determined 6 d later. Results presented in Fig. 5 c show that the same proportion of naive and primed CD8+ T cells incorporated BrdU in response to peptide challenge (73 and 75%, respectively). In contrast, in the absence of peptide challenge, only a small fraction of naive and primed splenic T cells incorporated BrdU (15 and 8%, respectively) and these cells were essentially all CD44high cells (data not shown). This observation further supports results discussed above that suggest that similarly to naive CD8+ T cells, primed CD44intCD8+ T cells are slowly or noncycling cells.
Together, these results show that the majority of primed CD8+ T cells are not anergic because they respond to peptide restimulation in vivo.
Primed CD8+ T Cells Are Hyperreactive to Peptide In Vitro.
We have compared the activation threshold of naive and primed CD8+ T cells in vitro. Spleen cells from naive and primed mice were activated with different peptide concentrations, and proliferation was measured by thymidine uptake. In the absence of IL-2, naive and primed cells proliferated equally well in response to a range of peptide doses (Fig. 6 a). In the presence of IL-2, both cell populations were activated by lower peptide concentrations. However, in these conditions, primed cells responded to lower doses of peptide and showed a stronger proliferative capacity than their naive counterparts.
Figure 6.

In vitro proliferative responses of CD8 T cells from primed and naive thymectomized mice. (a) 5 × 104 spleen cells were activated with different concentrations of peptide in the presence or absence of IL-2 as described in Materials and Methods. Cell proliferation was measured by [3H]thymidine uptake after 4 d in culture. Results for two mice per group are shown. The percentages of CD8+ T cells in the spleen of primed or naive mice were comparable, 23 and 17% versus 21 and 18%, respectively. This experiment was performed at least four times with similar results. (b) The proliferation rate of primed and naive CD8+ T cells was measured at different times after activation with 0.1 nM peptide in the presence of IL-2. (c and d) The activation of CD8+ T cells by different concentrations of peptide was inhibited by anti-CD8 mAb (1 μg/ml final concentration in the wells). Proliferation was measured after 4 d of culture. The proliferative response of one naive and one primed mouse in the presence or absence of anti-CD8 mAb is shown in c. The percentage inhibition in the presence of anti-CD8 antibodies was calculated for each dose of peptide from the following formula: ([proliferation in the absence of anti-CD8 − proliferation in the presence of anti-CD8]/proliferation in the absence of anti-CD8) ×100. The percentage inhibition for two naive and two primed mice is shown in d.
To verify that the hyperreactivity in the presence of IL-2 was not due to different kinetics of proliferation, we monitored thymidine incorporation at different days after in vitro peptide stimulation. As can be seen in Fig. 6 b, primed cells were hyperreactive to peptide during the entire duration of the culture.
In other CD8 systems, in vitro proliferation or cytolytic activity of naive CTL precursors was inhibited by an antiCD8 mAb, whereas primed CTL precursors were resistant to anti-CD8 inhibition (23, 24). We therefore tested the inhibitory effect of anti-CD8 antibodies on the CD8+ T cell proliferative response to peptide in the presence of IL-2. Results presented in Fig. 6, c and d, show that primed CD8+ T lymphocytes are more resistant to anti CD8 mAb inhibition than naive cells at high antigen concentrations. At lower concentrations of peptide, both naive and primed cells were completely inhibited. These results show that in the presence of IL-2, primed CD8+ T cells have a lower stimulation threshold than naive CD8+ T cells. Finally, because the hyperreactivity of primed CD8+ T cells was dependent on the presence of IL-2, it was possible that the expression of IL-2R was differentially regulated in naive and primed CD8+ T cells. The responsiveness of T cells to IL-2 is mainly regulated through the expression of IL-2Rα (25). Therefore, we measured the expression of the IL-2Rα chain by primed and naive CD8+ T cells after in vitro activation by peptide. Fig. 7 shows that 21 h after activation, the majority of naive or primed CD8+ T cells have upregulated their IL-2Rα chain. This is followed by a further increase in IL-2Rα chain expression, with primed cells showing a more rapid upregulation than naive CD8+ T cells. Indeed, after 2 d of culture 25% of primed cells compared with 8% of naive cells have already further upregulated their IL-2Rα chain. A similar but only transient upregulation of the IL-2Rα chain was observed when CD8+ T cells were activated in the absence of IL-2, suggesting that the maintenance of high levels of IL-2Rα chain expression is dependent on IL-2. These results indicate that the hyperreactivity of primed CD8+ T cells in the presence of IL-2 correlates with a more rapid upregulation of the IL-2Rα chain by these cells.
Figure 7.

Expression of IL-2Rα on naive and primed CD8+ T cells after activation with peptide in vitro. 106 spleen cells were activated with 1 nM peptide in the presence or absence of 2.5% IL-2. Expression of IL-2Rα by CD8+ T cells was measured by double staining after 21, 45, or 69 h of stimulation. Cells were gated for CD8+ at the acquisition level. The results for two representative naive (. . . .) or primed mice (—) out of six are shown. Fresh resting CD8+ T cells from a naive mouse were used as a negative control (· –· –·).
Discussion
We have described an experimental system in which a population of resting primed CD8+ T cells can be generated in vivo. These CD8+ T cells express a defined phenotype different from naive or recently activated CD8+ T cells, are capable of responding to peptide in vivo, and are hyperreactive to antigen in vitro.
TCR transgenic mice or peptide immunization have been used with mixed success to study the differentiation of memory T cells in vivo. Although peptides have been used successfully to induce CD8-mediated immunity in several systems (26–29), they have also been shown to induce unresponsiveness rather than immunity (22). A number of parameters could account for these differences. First, different affinities of peptide for MHC and TCR for peptide– MHC complexes could influence the level of T cell triggering, which in turn might play a role in the fate of activated T cells (30). This has been clearly illustrated for CD4+ T cells by Liblau and colleagues (31), who have shown in two different strains of TCR transgenic mice that opposite results are obtained with different peptides but similar immunization protocols.
Second, the route of administration as well as the use of adjuvant has also been shown to influence the outcome of the response (27, 32). In the F5 system, we have tested different routes of peptide delivery (i.e., intraperitoneal, in NaCl, intraperitoneal in IFA, or subcutanously in CFA) and have not found any differences in the changes of phenotype of spleen or LN CD8+ T cells during the primary response (data not shown). This indicates that in F5 TCR transgenic mice, the TCR used leads to CD8+ T cell activation by peptide independently of the immunization route.
For CD4+ T cells, it has been reported that primed cells generated by in vivo immunization of TCR transgenic mice differ from CD4+ cells primed in a polyclonal environment (5). We have successfully used the F5 TCR transgenic mice to generate a peptide primed CD8+ T cell population that shows in vitro hyperreactivity to peptide. These cells express levels of CD44 and Ly6C that differentiate them not only from naive transgenic CD8+ T cells but also from freshly activated transgenic CD8+ T cells. This is in agreement with the phenotype of memory CD8+ T cells obtained in other systems (21, 23). In contrast, l-selectin is expressed by our primed CD8+ T cells. In other systems, this marker is downregulated by primed CD8+ T cells (7, 33), although in some systems a population of l-selectin+ primed CD8+ T cells is also found (10, 33). This indicates that phenotypically different subsets of primed CD8+ T cells can be generated in vivo. Such a heterogeneity is also found when the cell cycle status or the ex vivo CTL activity of primed CD8+ T cells is studied (9). Up to now it has not been possible to match up these different properties of primed CD8+ T cells. This is mainly due to the fact that in most systems cells that were primed in the course of the primary response cannot be traced and identified later on. Moreover, antigen priming quite often leads to a mixed population of primed cells in terms of phenotype, cell cycle status, or function. The advantage of the system we describe is that it allows the characterization of one subset of primed CD8+ T cells that may contribute to the memory response. These resting primed CD8+ T cells are not a transgenic artefact, because CD8+ T cells with a similar phenotype (i.e., CD44int) and cell cycle status have been described in nontransgenic animals (9). However, these nontransgenic cells have not been characterized functionally.
A population of CD44highLy6ChighCD8+ cells was also observed in thymectomized TCR transgenic mice (see Fig. 4 a). These cells are also found in nonthymectomized mice, where they represent a minority of CD8+ T cells (<5% or 0.5–1.0 × 106 cells per spleen). After thymectomy there is a three to five-fold increase in the number of these cells whether the mice have been primed or not, indicating that the expansion of this population is independent of peptide priming. These cells show a phenotype similar to recently activated cells (i.e., they are CD8low, Vβ11low, CD3low, IL-2Rβhigh, ICAM-1high, LFA-1high; data not shown), and in vivo they proliferate more than naive T cells, adding to the hypothesis that they are activated CD8+ T cells (see Fig. 1, CD44 versus BrdU in naive animals). The origin of these cells as well as the ligand that drives their proliferation are not known. Some of these cells could express a second receptor. However, because cells with a similar phenotype also exist in F5 mice back-crossed to a Rag−/− background, it is likely that at least a subset of them only express the F5 TCR.
Primed cells generated in vivo after immunization with peptide were not anergic, because their response to peptide rechallenge was indistinguishable from the response of naive cells in terms of phenotype or proliferation. In these in vivo experiments, primed cells did not show increased proliferation compared with naive cells. This could be due to the experimental conditions because the amount of peptide used to prime mice is 10–50-fold higher than the minimum needed to activate all CD8+ T cells in vivo (based on CD44 upregulation). Therefore it is possible that primed CD8+ T cells could respond differently from naive CD8+ T cells if mice were immunized with suboptimal doses of peptide.
The activation threshold of naive and primed cells was compared in vitro. In the absence of IL-2, naive and primed cells were activated by the same peptide concentrations and proliferated to the same extent. In contrast, in the presence of IL-2, the activation of naive cells was significantly increased, indicating that F5 CD8+ T cells are helper dependent at low peptide concentrations. More notable, under the same culture conditions, primed cells showed a strong hyperreactivity to peptide because they responded to peptide concentrations 10–50-fold lower than those required to activate naive T cells. The increased proliferation of primed cells was not due to the sole action of IL-2 because they did not express detectable levels of IL-2 receptors or proliferate to IL-2 in the absence of peptide. T cell responsiveness to IL-2 is mainly regulated via the expression of the IL-2Rα chain (25). This expression is initiated by TCR engagement and can be upregulated by IL-2 and a number of other interleukins or costimulatory molecules (25, 34–36). The expression of IL-2Rα chain by primed and naive T cells was measured at different times after activation. A similar increase in IL-2Rα chain expression was observed in both populations after 24 h of culture. However, further upregulation was more efficient on primed cells. These results are difficult to interpret because the experiments we have performed do not allow us to determine if the increased efficiency in Il-2Rα chain expression by primed cells is the cause or the consequence of the primed CD8+ T cells' hyperreactivity. However, they indicate that the initial IL-2Rα induction by the TCR is similar in naive and primed cells. The in vitro hyperreactivity of primed CD8+ T cells could result either from a different coupling of the TCR and/or from an overall increased avidity after the upregulation of CD44, Ly6C, or other molecules. Difference in the coupling of TCR has been demonstrated for anergic T cells, where the activation threshold is significantly increased after uncoupling of the TCR from the rasraf-MapK pathway (37, 38). The CD44 and Ly6C upregulation could also be part of the hyperreactive phenotype because both molecules are able to costimulate T cells (39, 40). These different hypotheses are currently being tested.
Acknowledgments
We thank Dimitris Kioussis for the F5 transgenic mice, Oberdan Leo for the Ly6C antibody, and Patrick Bertolino for the LFA-1 antibody. We also thank Chantal Rabourdin Combe and Janet Maryanski for helpful comments on the manuscript.
Footnotes
M. Pihlgren and P. Dubois are supported by fellowships from the Ligue Nationale Contre le Cancer and the Association Française contre les Myopathies, respectively. This work was supported by institutional grants from the Centre National de la Recherche Scientifique and Ministere de l'éducation et de la Recherche and by additional support from the Association pour la Recherche sur le Cancer, Association Française Contre les Myopathies, and the Comité Départemental de la Ligue Nationale Française contre le Cancer.
References
- 1.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science (Wash DC) 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 2.McHeyzer-Williams MG, Davis MM. Antigenspecific development of primary and memory T cells in vivo. Science (Wash DC) 1995;268:106–111. doi: 10.1126/science.7535476. [DOI] [PubMed] [Google Scholar]
- 3.Horgan KJ, Van Seventer GA, Shimizu Y, Shaw S. Hyporesponsiveness of naive (CD45RA+) human T cells to multiple receptor-mediated stimuli but augmentation of responses by co-stimuli. Eur J Immunol. 1990;20:1111–1118. doi: 10.1002/eji.1830200525. [DOI] [PubMed] [Google Scholar]
- 4.Sanders ME, Makgoba MW, June CH, Young HA, Shaw S. Enhanced responsiveness of human memory T cells to CD2 and CD3 receptor-mediated activation. Eur J Immunol. 1989;19:803–808. doi: 10.1002/eji.1830190504. [DOI] [PubMed] [Google Scholar]
- 5.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 6.Zinkernagel R. Immunology taught by viruses. Science (Wash DC) 1996;271:173–178. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]
- 7.Bruno L, Kirberg J, von Boehmer H. On the cellular basis of immunological T cell memory. Immunity. 1995;2:37–43. doi: 10.1016/1074-7613(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 8.Zimmermann C, Brduscha-Riem K, Blaser C, Zinkernagel R, Pircher H. Visualization, characterization, and turnover of CD8+memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razvi ES, Welsh RM, McFarland HI. In vivo state of antiviral CTL precursors. Characterization of a cycling cell population containing CTL precursors in immune mice. J Immunol. 1995;154:620–632. [PubMed] [Google Scholar]
- 11.Mullbacher A. The long-term maintenance of cytotoxic T cell memory does not require persistence of antigen. J Exp Med. 1994;179:317–321. doi: 10.1084/jem.179.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature (Lond) 1994;369:648–652. doi: 10.1038/369648a0. [DOI] [PubMed] [Google Scholar]
- 13.Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus-specific CD8+T-cell memory determined by clonal burst size. Nature (Lond) 1994;369:652–654. doi: 10.1038/369652a0. [DOI] [PubMed] [Google Scholar]
- 14.Oehen S, Waldner H, Kundig TM, Hengartner H, Zinkernagel RM. Antivirally protective cytotoxic T cell memory to lymphocytic choriomeningitis virus is governed by persisting antigen. J Exp Med. 1992;176:1273–1281. doi: 10.1084/jem.176.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray D, Matzinger P. T cell memory is shortlived in the absence of antigen. J Exp Med. 1991;174:969–974. doi: 10.1084/jem.174.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mamalaki C, Norton T, Tanaka Y, Townsend AR, Chandler P, Simpson E, Kioussis D. Thymic depletion and peripheral activation of class I major histocompatibility complex-restricted T cells by soluble peptide in T-cell receptor transgenic mice. Proc Natl Acad Sci USA. 1992;89:11342–11346. doi: 10.1073/pnas.89.23.11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pihlgren M, Lightstone L, Mamalaki C, Rimon G, Kioussis D, Marvel J. Expression in vivo of CD45RA, CD45RB and CD44 on T cell receptor-transgenic CD8+T cells following immunization. Eur J Immunol. 1995;25:1755–1759. doi: 10.1002/eji.1830250640. [DOI] [PubMed] [Google Scholar]
- 18.Mamalaki C, Elliot J, Norton T, Yannoutsos N, Townsend AR, Chandler P, Simpson E, Kioussis D. Positive and negative selection in transgenic mice expressing a T-cell receptor specific for influenza nucleoprotein and endogenous superantigen. Dev Immunol. 1993;3:159–174. doi: 10.1155/1993/98015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4, or 5, using modified cDNA expression vectors. Eur J Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 20.von Boehmer H, Hafen K. The life span of naive α/β T cells in secondary lymphoid organs. J Exp Med. 1993;177:891–896. doi: 10.1084/jem.177.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walunas TL, Bruce DS, Dustin L, Loh DY, Bluestone JA. Ly-6C is a marker of memory CD8+T cells. J Immunol. 1995;155:1873–1883. [PubMed] [Google Scholar]
- 22.Kyburz D, Aichele P, Speiser DE, Hengartner H, Zinkernagel RM, Pircher H. T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur J Immunol. 1993;23:1956–1962. doi: 10.1002/eji.1830230834. [DOI] [PubMed] [Google Scholar]
- 23.Budd RC, Cerottini J-C, MacDonald HR. Phenotypic identification of memory cytolytic T lymphocytes in a subset of Lyt-2+cells. J Immunol. 1987;138:1009–1013. [PubMed] [Google Scholar]
- 24.Cai Z, Sprent J. Resting and activated T cells display different requirements for CD8 molecules. J Exp Med. 1994;179:2005–2015. doi: 10.1084/jem.179.6.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waldmann T. The multi-subunit interleukin-2 receptor. Annu Rev Biochem. 1989;58:875–911. doi: 10.1146/annurev.bi.58.070189.004303. [DOI] [PubMed] [Google Scholar]
- 26.Aichele P, Hengartner H, Zinkernagel RM, Schulz M. Antiviral cytotoxic T cell response induced by in vivo priming with a free synthetic peptide. J Exp Med. 1990;171:1815–1820. doi: 10.1084/jem.171.5.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schild H, Deres K, Wiesmüller K-H, Jung G, Rammensee H-G. Efficiency of peptides and lipopeptides for in vivo priming of virus-specific cytotoxic T cells. Eur J Immunol. 1991;21:2649–2654. doi: 10.1002/eji.1830211102. [DOI] [PubMed] [Google Scholar]
- 28.Schulz M, Zinkernagel RM, Hengartner H. Peptide-induced antiviral protection by cytotoxic T cells. Proc Natl Acad Sci USA. 1991;88:991–993. doi: 10.1073/pnas.88.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oldstone MB, Tishon A, Eddleston M, de la Torre JC, McKee T, Whitton JL. Vaccination to prevent persistent viral infection. J Virol. 1993;67:4372–4378. doi: 10.1128/jvi.67.7.4372-4378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valitutti S, Müller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liblau RS, Pearson CI, Shokat K, Tisch R, Yang XD, McDevitt HO. High-dose soluble antigen: peripheral T-cell proliferation or apoptosis. Immunol Rev. 1994;142:193–208. doi: 10.1111/j.1600-065x.1994.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 32.Dyall R, Vasovic LV, Molano A, Nikolic-Zugic J. CD4-independent in vivo priming of murine CTL by optimal MHC class I-restricted peptides derived from intracellular pathogens. Int Immunol. 1995;7:1205–1212. doi: 10.1093/intimm/7.8.1205. [DOI] [PubMed] [Google Scholar]
- 33.Walker PR, Ohteki T, Lopez JA, MacDonald HR, Maryanski JL. Distinct phenotypes of antigen-selected CD8 T cells emerge at different stages of an in vivo immune response. J Immunol. 1995;155:3443–3452. [PubMed] [Google Scholar]
- 34.Pimentel-Muinos F, Munoz-Fernandez A, Fresno M. Control of T lymphocyte activation and IL-2 receptor expression by endogenously secreted lymphokines. J Immunol. 1994;152:5714–5722. [PubMed] [Google Scholar]
- 35.Ledbetter J, Imboden J, Schieven G, Grosmaire L, Rabinovitch P, Lindsten T, Thompson C, June C. CD28 ligation in T-cell activation: evidence for two signal transduction pathways. Blood. 1990;75:1531–1539. [PubMed] [Google Scholar]
- 36.Kumaki K, Armitage R, Ahdieh M, Park L, Cosman D. Interleukin-15 up-regulates interleukine-2 receptor α chain but down-regulates its own high-affinity binding sites on human T and B cells. Eur J Immunol. 1996;26:1235–1239. doi: 10.1002/eji.1830260608. [DOI] [PubMed] [Google Scholar]
- 37.Li W, Whaley C, Mondino A, Mueller D. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+T cells. Science (Wash DC) 1996;271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 38.Fields P, Gajewski T, Fitch F. Blocked Ras activation in anergic CD4+T cells. Science (Wash DC) 1996;271:1276–1278. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 39.Huet S, Groux H, Caillou B, Valentin H, Prieur AM, Bernard A. CD44 contributes to T cell activation. J Immunol. 1989;143:798–801. [PubMed] [Google Scholar]
- 40.Wegener AM, Letourneur F, Hoeveler A, Brocker T, Luton F, Malissen B. The T cell receptor/CD3 complex is composed of at least two autonomous transduction modules. Cell. 1992;68:83–95. doi: 10.1016/0092-8674(92)90208-t. [DOI] [PubMed] [Google Scholar]