Specific T Helper Cell Requirement for Optimal Induction of Cytotoxic T Lymphocytes against Major Histocompatibility Complex Class II Negative Tumors (original) (raw)
Abstract
This study shows that induction of tumor-specific CD4+ T cells by vaccination with a specific viral T helper epitope, contained within a synthetic peptide, results in protective immunity against major histocompatibility complex (MHC) class II negative, virus-induced tumor cells. Protection was also induced against sarcoma induction by acutely transforming retrovirus. In contrast, no protective immunity was induced by vaccination with an unrelated T helper epitope. By cytokine pattern analysis, the induced CD4+ T cells were of the T helper cell 1 type. The peptide-specific CD4+ T cells did not directly recognize the tumor cells, indicating involvement of cross-priming by tumor-associated antigen-presenting cells. The main effector cells responsible for tumor eradication were identified as CD8+ cytotoxic T cells that were found to recognize a recently described immunodominant viral gag-encoded cytotoxic T lymphocyte (CTL) epitope, which is unrelated to the viral env-encoded T helper peptide sequence. Simultaneous vaccination with the tumor-specific T helper and CTL epitopes resulted in strong synergistic protection. These results indicate the crucial role of T helper cells for optimal induction of protective immunity against MHC class II negative tumor cells. Protection is dependent on tumor-specific CTLs in this model system and requires cross-priming of tumor antigens by specialized antigen-presenting cells. Thus, tumor-specific T helper epitopes have to be included in the design of epitope-based vaccines.
Adequate T helper cell activation is essential in the initiation of an immune reaction. The inability to control tumor outgrowth can be due to inadequate T helper responses underlying poor tumor-specific immunity. In the cellular immune response, specialized APCs process protein and present antigenic peptide fragments in MHC class II molecules to CD4+ T helper lymphocytes. These provide “help” to effector cells via the production of cytokines. Although tumor cells can directly present endogenously processed antigenic peptide in surface MHC class I molecules to CD8+ CTL precursors, initiation of tumor-specific CTL responses is likely to involve indirect presentation of tumor antigens by specialized APCs.
Evidence for a role of T helper cell–mediated immunity comes from studies with genetically modified tumor cells. CD4+ cells can be directly activated by transfection of MHC class II α and β chain genes in mouse tumor cells (1–4). These cells become immunogenic, lose their tumorigenicity, and even induce protection against wild-type MHC class II negative tumors, indicating that direct MHC class II presentation of tumor expressed antigens can induce efficient anti–tumor responses.
A central role of CD4+ T cells emerged from studies of immunity against FMR (Friend, Moloney, Rauscher)1 murine leukemia virus (MuLV) type tumors by Greenberg (5). Transfer of purified polyclonal T cells from FBL (Friend MuLV-induced erythroleukemia cell line) vaccinated mice in naive animals can protect these mice against subsequent tumor challenge. Both purified CD4+ and CD8+ T cells transfer protection to FBL tumors (6). FBL cells do not express MHC class II molecules, but CD4+ T cells can protect mice even in the absence of CD8+ T cells. In this case, macrophages seem to play an important effector role. CD8+ T cells can only be effective if CD4+ T cells are present or if exogenous IL-2 is administered. Neither B cells nor NK cells seem to exert a significant role in the FBL sytem. These data suggest involvement of APCs, presenting tumor antigens, and a crucial regulatory role of Ths, which was strongly supported by experiments performed in Friend MuLV env-transgenic mice (7). These mice were rendered tolerant for env-specific Th responses and it was not possible to protect these mice against FBL tumors by vaccination.
Immune responsiveness to MuLV is classically regulated by the genes of the H-2 (MHC) complex (8). In particular, the H-2b haplotype confers resistance, and studies using H-2 recombinant and H-2 mutant mouse strains have mapped the protective effects to the class II I-Ab locus (9, 10). This MHC class II association indicates an important role of T helper cells influencing both CTL activity as well as class switching of antiviral antibodies from IgM to IgG. The H-2 I-Ab phenotype protects against early lymphomagenesis. The identification of two Friend MuLV env-derived epitopes, presented by MHC class II, I-Ab and I-Eb/d, respectively, indicated that tumor-directed T helper immunity is virus specific (11). The few lymphomas that arise in H-2b mice have abrogated viral antigen or (more rarely) MHC class I expression (12), indicating that CTLs also play a crucial role. CTLs have been proven to recognize viral antigens, both gag and env proteins encode CTL epitopes (13). We have identified a Kb-presented, env-derived Moloney and Rauscher CTL epitope that is subdominant in C57BL/6 mice making use of the Db mutant BM13 mouse strain (14). The Db-presented gag-leader (gag-L) derived immunodominant CTL epitope for the FMR type of MuLV has been identified only recently (15).
Vaccination with a synthetic peptide comprising a relevant T cell epitope is a powerful method to induce highly specific T cells. Protective vaccination using CTL peptide epitopes has been achieved in pathogenic viral models (16, 17) and tumor models (18–20). Peptide vaccination in IFA led to measurable specific CTL induction and protective immunity against virulent virus or tumor cells. Importantly, peptide vaccination can also be applied succesfully for therapy of established tumors by presenting the peptide in IFA, on RMA-S cells, or on activated dendritic cells (21, 22).
We now report the induction of tumor-protective immunity by a single vaccination with a tumor-specific MuLV env-encoded T helper peptide. Strong protection can be achieved against highly aggressive tumor cells that lack MHC class II expression. This indicates the requirement of cross-priming of tumor antigens by local APCs. We show that CD8+ T cells, recognizing the gag-L–encoded CTL epitope, are crucial effector cells that are efficiently activated with help from peptide-primed tumor-specific CD4+ T cells. Vaccination with a mixture of the T helper peptide and the immunodominant CTL epitope resulted in synergistic, long-term tumor protection.
Materials and Methods
Mice.
C57BL/6 (B6 Kh, H-2b) mice were bred under specific pathogen-free conditions at TNO-PG (Leiden, The Netherlands).
Cell Lines, Viruses, and Antibodies.
771 is an MCF1233-induced B cell lymphoma cell line from a C57BL/10 mouse neonatally inoculated with MCF1233 MuLV as described (23). RMA is a mutagenized derivative of RBL-5, a Rauscher MuLV-induced T lymphoma cell line of C57BL/6 origin (24). EL-4 is a chemically induced (non-MuLV–induced) T cell lymphoma of C57BL/6 origin. HeLaDb is a stable transfectant of the human cervix carcinoma cell line HeLa, expressing the murine MHC class I molecule Db.
Moloney murine sarcoma and leukemia virus complex (MoMSV) was prepared and injected in the left thigh intramuscularly for sarcoma induction as described (25). Moloney Abelson virus was used for infection of B6 LPS blasts as described (25).
Monoclonal antibodies used for immunofluorescence staining and FACScan® (Becton Dickinson, Mountain View, CA) analysis were: anti-Kb (B8.24.3), anti-Db (28.14.8S), anti–I-Ab (17.227 and Y3P), anti-CD4 (H129.19), anti-CD8 (53.6.7), and anti-CD3 (500A2).
Monoclonal antibodies used for in vivo depletion of the CD4+ and CD8+ T cell subsets were GK1.5 and 2.43, respectively. Purified antibodies were administered by intraperitoneal injection of 100 μg of antibody in 0.2 ml PBS, in schedules that are specified in the figure legends.
Peptides.
Peptides were synthesized on multiple peptide synthesizer (442; ABIMED, Langenfeld, Germany) as previously described (26). Peptides were analyzed for purity by reverse phase HPLC and lyophilized. The T helper epitope peptide env-H19 EPLTSLTPRCNTAWNRLKL (11) (and its length variants) and the env-derived CTL epitope peptide env-Kb8 SSWDFITV (14) were dissolved in PBS. The dominant gag-L–derived CTL epitope peptide was used as a 10-mer (gag-L–Db10) or a 9-mer (gag-L–Db9) (L)CCLCLTVFL (15) was dissolved in DMSO (20 μl/mg peptide) and diluted in PBS. The Sendai virus nucleoprotein CTL epitope SV9 (FAPGNYPAL) was used as a control Kb-binding peptide (16). The ovalbumin T helper peptide OvaH (ISQAVHAAHAEINEAGR) (27) was used as a control I-Ab–binding peptide. This peptide induces strong CD4 responses in C57BL/6 mice. Stock solutions of 5 mg/ml were stored at −80°C.
Peptide Vaccination and Tumor Challenge.
C57BL/6 mice, aged 7–10 wk, were vaccinated with a single dose of synthetic peptide (0.1–100 μg/mouse) in 50% (vol/vol) emulsion with IFA administered in a 0.2-ml depot subcutaneously. Control groups were similarly injected with a 50% (vol/vol) emulsion of PBS and IFA. After 14 d, the mice were challenged with 103 RMA tumor cells in 0.2 ml PBS, 0.1% (wt/vol) BSA, intraperitoneally. Weights of the mice were monitored every other day. Mice were killed if their weight increased for >25%. Statistical analysis on the survival data was performed using the log rank test. Significance was defined as probability values below 0.05.
T Cell Culture.
CD4+ T cells were obtained from T helper peptide immunized mice by culturing spleen cells (3 × 106 cells/ well of 24-well plates) in the presence of 10 μg/ml T helper peptide in complete medium in the absence of exogenous IL-2. Bulk cultures after 1 wk were mainly (70–80%) CD4+ as determined by FACScan® analysis. The bulk culture was restimulated using irradiated B6 spleen cells in the presence of 10 μg/ml T helper peptide without IL-2. Clones were isolated by limiting dilution and tested for peptide specificity.
T helper clone 3A12 (CD4+) and CTL clone 2H9 (CD8+) was isolated from a bulk spleen cell culture of tumor-protected mice immunized with 11-mer MuLV T helper peptide (H11.1) that were challenged with 103 live RMA tumor cells. Spleen cells were restimulated with mitomycin-C (50 μg/ml for 1 h at 37°C) -treated and irradiated (25 Gy) RMA tumor cells. After 1 wk, bulk cultures were seeded under limiting dilution conditions and clones were randomly isolated and tested for specific proliferative capacity and cytotoxicity against RMA tumor cells.
All cells were cultured in Iscove's modified Dulbecco's medium (GIBCO Biocult, Glasgow, UK) supplemented with 5% FCS (tumor cells) or 10% FCS (CTL), penicillin, (100 IU/ml) and 20 μM 2-mercaptoethanol. T cell clones were cultured in the presence of 20 IU/ml human recombinant IL-2.
Proliferation Assays.
Proliferation assays were carried out in 96-well U-bottomed plates using CD4+ T cells as responder cells at 4 × 104, 2 × 104, 104, and 5 × 103 cells/well. Several stimulator cells were tested: irradiated (25 Gy) spleen cells (105 cells/well), with or without T helper peptide (5–10 μg/ml), with or without tumor cells (mitomycin-C–treated and irradiated RMA cells; 2 × 104 cells/well). All assays were performed in the absence of exogenous IL-2. After 3 d, the cultures were pulsed with [3H]thymidine (New England Nuclear, Boston, MA; 0.5 μCi/well) for 5 h. The results are expressed as mean cpm of triplicate cultures. Standard deviations were <10% of the mean.
Cytotoxicity Assays.
CTL assays were performed as described (23). In short, 2 × 103 Na2 51CrO4-labeled target cells were incubated with effector cells for 5–6 h at 37°C. The culture supernatant was harvested and counted for released radioactivity. The percentage of specific 51Cr-release was calculated as a ratio of 100 × (cpm experimental release − cpm spontaneous release)/(cpm maximal release [1% Triton X-100] − cpm spontaneous release). All assays were carried out in triplicate. For target cell sensitization, 51Cr-labeled target cells were incubated with peptide 1 h before addition of the CTLs. Peptides remained present during the assay.
Cytokine Assays.
Bioactive IL-2 production was determined using the IL-2–dependent cell line CTLL2. 50 μl supernatant of specifically stimulated CD4+ T cell clones (3-d stimulation with irradiated [25 Gy] 771 [I-Ab+] cells with and without env-H19 peptide) was incubated with 5 × 103 CTLL2 cells in a total volume of 100 μl. As a standard, human recombinant IL-2 was used. After 24 h, the wells were pulsed with [3H]thymidine (New England Nuclear; 0.5 μCi/well) for 5 h. All determinations were performed in triplicate and standard deviations were <10% of the mean.
The presence of cytokine transcripts of IFN-γ, IL-2, IL-3, IL-4, IL-5, and IL-6 were tested by reverse transcriptase PCR using cytokine-specific primer sets as described (28). CD4+ T cell clones were specifically stimulated with env-H19 peptide-loaded 771 cells for 2 and 16 h and total RNA and cDNA was prepared as described (29). Controls were CD4+ T cells incubated with 771 cells without peptide, CD4+ T cell alone, and 771 cells with peptide without CD4+ T cells. Control reactions without reverse transcriptase were always negative for any primer set.
RMA-S MHC Class I Peptide-binding Assay.
The RMA-S assay was performed as described previously (18). In short, RMA-S cells were cultured for 36 h at 26°C and were added to serial dilutions of peptide in serum-free medium. After a 4-h incubation at 37°C, cells were washed and incubated with the Db-specific mAb 28.14.8S for 30 min on ice. After washing, the cells were incubated with FITC-conjugated goat anti–mouse IgG F(ab′)2 fragments for 30 min on ice and immunofluorescence was analyzed using a FACScan® flow cytometer.
Results
Peptide Vaccination with T Helper Epitope Peptide Leads to Protective Immunity.
MuLV-encoded epitope peptides were tested for their capacity to induce tumor-protective immune responses in vivo. Synthetic peptide diluted in saline and emulsified in IFA was injected subcutaneously. The subdominant 8-mer CTL epitope env-Kb (SSWDFITV; reference 14) and the T helper 19-mer peptide env-H19 (EPLTSLTPRCNTAWNRLKL), originally described as a Friend MuLV T helper epitope (11) sharing identical sequences with Moloney and Rauscher MuLV but not with AKV/MCF type MuLV, were used to vaccinate immunocompetent C57BL/6 mice. After 14 d, the mice were challenged intraperitoneally with the Rauscher MuLV-induced tumor cell line RMA. RMA is an aggressively growing T cell lymphoma, lethal for mice within 4 wk when as few as 1000 cells are administered. RMA expresses MHC class I Kb and Db but is negative for MHC class II expression even after IFN-γ treatment, as monitored by staining with specific antibodies and Northern blotting analysis (data not shown). RMA cells isolated from the peritoneal cavity 3 wk after inoculation in C57BL/6 mice were tested for MHC expression and found to have retained their I-Ab negative phenotype in vivo (data not shown). Fig. 1 shows the weight increase of individual mice in time, reflecting ascites tumor growth. The subdominant CTL epitope env-Kb8, despite being expressed by RMA cells (14), did not induce a protective response (Fig. 1 B). In contrast, vaccination with the env-H19 T helper peptide (Fig. 1 C) led to inhibition of RMA tumor growth in a significant number of mice. Combined vaccination with the T helper and the subdominant env-Kb8 CTL epitopes (Fig. 1 D) did not significantly increase survival beyond that seen with the T helper peptide alone, within the first 40 d after tumor challenge. Fig. 2 shows 100-d survival of mice vaccinated with MuLV CTL and T helper peptides compared with an irrelevant ovalbumin T helper peptide (OvaH; reference 27). Vaccination with the OvaH peptide alone or in combination with the env-Kb8 CTL epitope did not induce detectable protection, whereas the MuLV T helper peptide induced long-term protection in 40% of the mice (Fig. 2 A). The combination vaccine (env-Kb8 and env-H19 mixture) induced long-term protection of 70% of the animals. This difference was not found to be statistically significant (P = 0.28). The nonrelated CTL and T helper epitopes Sendai virus SV-Kb9 and OvaH, respectively, did not induce measurable protective immunity. The recently described gag-L–derived, Db-presented dominant CTL epitope (L)CCLCLTVFL (15) was tested in a similar way (Fig. 2 B). The gag-L–Db peptide is able to induce protection in 20–40% of the mice either as a 9 or 10 mer. The mixture of the env T helper peptide and the gag-L CTL epitope peptide resulted in very efficient protection leading to long-term survival of 90% of the mice. The requirement for induction of tumor-specific T helper cells is shown in Fig. 2 C. Vaccination with a mixture of OvaH with the gag-L–Db9 peptide resulted in significantly less efficient protection than the combination vaccine of MuLV env-derived T helper peptide with the gag-L peptide. These results show that tumor-specific T helper peptide vaccination as such can induce efficient protective immunity against MHC class II negative tumor cells. This protective effect can be further enhanced by vaccination of T helper peptide in combination with CTL epitope-bearing peptides.
Figure 1.

T helper peptide vaccination protects mice against intraperitoneal tumor induction. C57BL/6 mice were vaccinated with synthetic peptide (100 μg/mouse) in a 50% PBS/IFA (vol/vol) emulsion subcutaneously at day −14 and challenged with 1,000 RMA cells intraperitoneally at day zero. The weights of the individual mice were monitored every other day and the percent weight increase is depicted. Each line is an individual mouse. (A) Six mice were injected with an emulsion of IFA with PBS without peptide as a negative control. (B) Six mice were vaccinated with the subdominant Kb-presented MuLV env–gp70 CTL epitope peptide env-Kb8 (SSWDFITV). (C) Seven mice were vaccinated with the I-Ab–presented 19-mer MuLV env–gp70 T helper epitope peptide env-H19 (EPLTSLTPRCNTAWNRLKL). (D) 10 mice were vaccinated with a mixture of env-Kb8 and env-H19 (both 100 μg) in a single vaccine depot.
Figure 2.
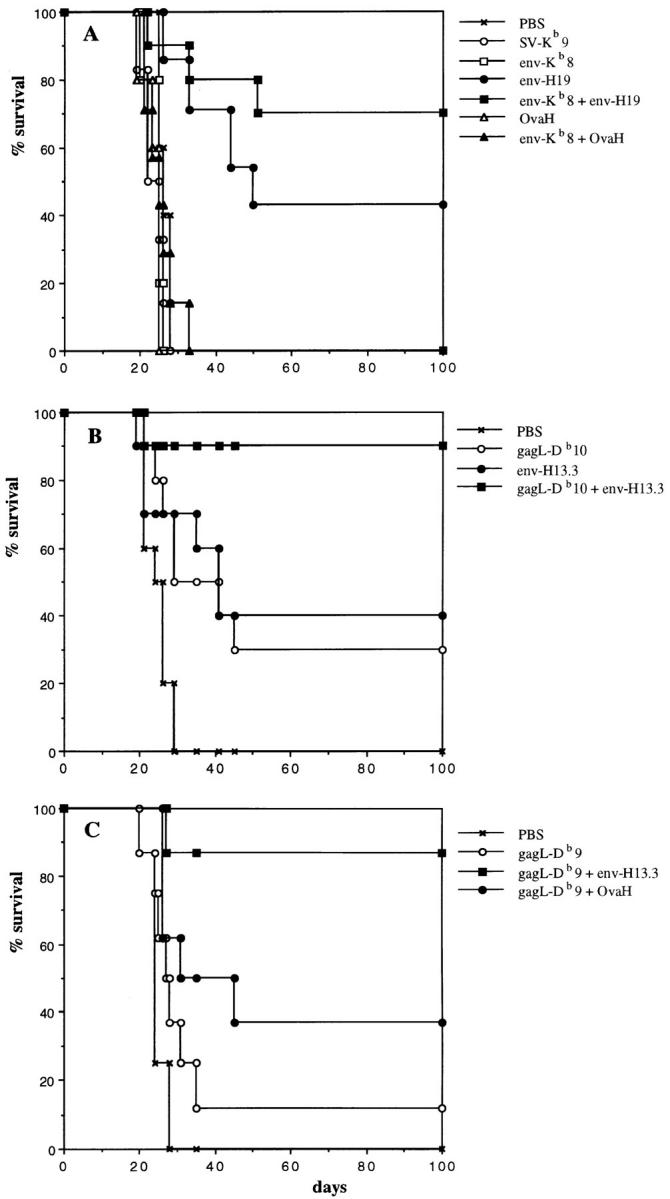
(A) Tumor-specific T helper peptide vaccination increases long-term survival of mice challenged with tumor cells. C57BL/6 mice were vaccinated with synthetic peptides as described in the legend of Fig. 1. All groups received IFA (50% vol/vol) with or without 100 μg peptide subcutaneously. The percentage of surviving mice is depicted. Mice were killed when the weight increase was >25%. PBS, nonimmunized control (n = 6); SV-Kb9, nonrelated Sendai virus nucleoprotein Kb-presented CTL epitope (FAPGNYPAL; n = 6); env-Kb8, MuLV env–gp70 subdominant CTL epitope (n = 7); env-H19, 19-mer MuLV env–gp70 T helper epitope (n = 7); env-Kb8 + env-H19, mixture vaccine of MuLV CTLs and T helper epitopes (n = 10); OvaH, nonrelated ovalbumin I-Ab–presented T helper epitope (ISQAVHAAHAEINEAGR; n = 6); env-Kb8 + OvaH, mixture of MuLV subdominant CTL epitope and ovalbumin T helper epitope. Significance of differences (log rank test): PBS versus env-H19: P = 0.04; PBS versus env-Kb8 + env-H19: P = 0.0015; env-H19 versus env-Kb8 + env-H19: P = 0.28 (not significant; n.s.). (B) Combination of T helper peptide and the immunodominant gag-L CTL epitope induces highly efficient tumor protection. C57BL/6 mice were immunized with synthetic peptides in IFA subcutaneously (50 μg/mouse) at day −14 and challenged with 103 RMA tumor cells intraperitoneally. Long-term survival was monitored. gag-L-Db10, gag-L CTL epitope (LCCLCLTVFL); env-H13.3, env-gp70 T helper peptide (SLTPRCNTAWNRL). All groups: n = 10. Significance of differences (log rank test): PBS versus gag-L-Db10: P = 0.0078; PBS versus env-H13.3: P = 0.0078; PBS versus gag-L-Db10 + env-H13.3: P = 0.0001; gag-L-Db10 versus gag-L-Db10 + env-H13.3: P = 0.0108; env-H13.3 versus gag-L-Db10 + env-H13.3: P = 0.025. (C) Efficient protection by dominant CTL epitope requires specific help. Similar experiment using the 9-mer dominant CTL epitope gag-L–Db9 in combination with either the MuLV T helper peptide env-H13.3 or the nonrelated OvaH T helper peptide. All groups: n = 8. Significance of differences (log rank test): gag-L-Db9 versus gag-L-Db9 + OvaH: P = 0.169 (n.s.); gag-L-Db9 versus gag-L-Db9 + env-H13.3: P = 0.003; gag-L-Db9 + OvaH versus gag-L-Db9 + env-H13.3: P = 0.04; PBS versus gag-L-Db9 + env-H13.3: P = 0.0004.
In addition, we tested the ability of the helper peptide to protect mice against virus-induced tumorigenesis next to the in vitro–established tumor cell line RMA. The effect of peptide vaccination on sarcoma induction by MoMSV was determined by measuring thigh thickness. This retrovirus complex characteristically induces acute transformation of muscle cells and rapid tumor growth (within 2 wk), and an efficient T cell response can control tumor growth (nude mice die because of progressive tumor growth, data not shown) as evidenced by tumor regression within a month (Fig. 3 A). Vaccination with the T helper peptide significantly reduced tumor induction (five out of seven mice remained tumor-free) as well as tumor size (Fig. 3 B). Here we have used a 13-mer peptide and only 10 μg/mouse. This indicates that preimmunized MuLV-specific T helper cells can also control tumor growth by acutely transforming retroviruses.
Figure 3.
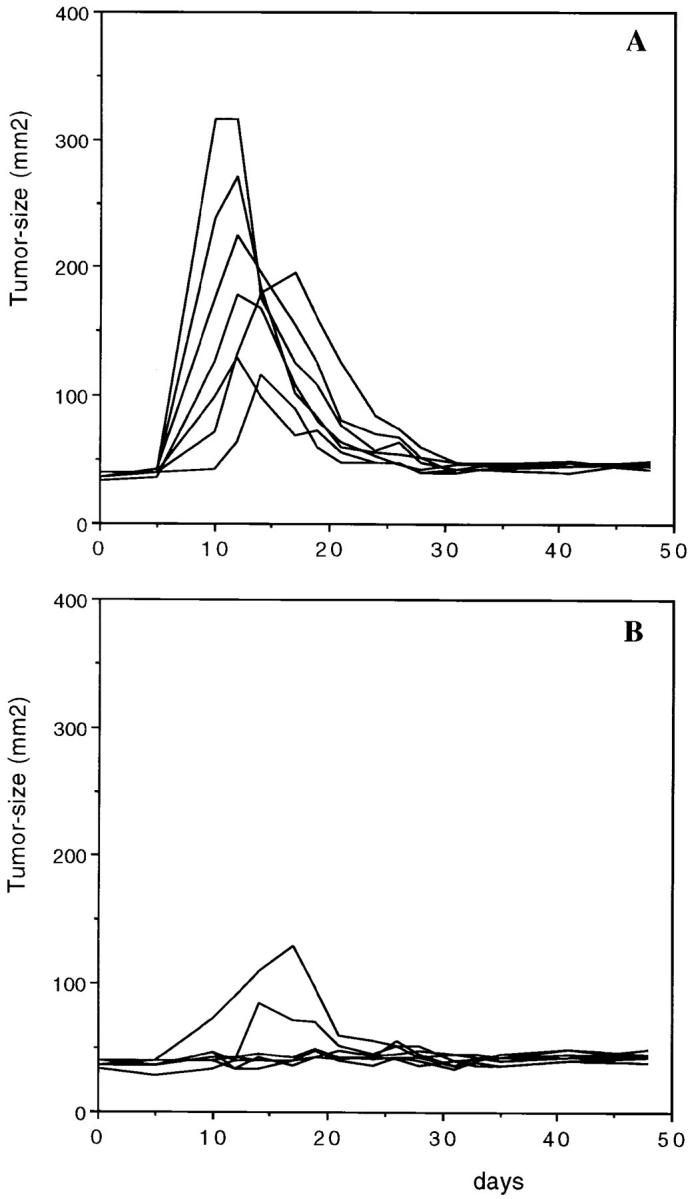
T helper peptide vaccination protects mice against retrovirus-induced sarcoma. C57BL/6 mice were vaccinated at day −14 with (A) PBS-IFA subcutaneously (n = 7) and (B) 13-mer MuLV T helper peptide H13.3 (SLTPRCNTAWNRL), 10 μg/mouse in IFA subcutaneously (n = 7). At day zero mice were injected in the left thigh muscle with 103 focus-forming units of MoMSV. Mice were monitored for tumor size using a caliper measuring two perpendicular diameters. Each line represents one individual mouse.
Optimization of Peptide Length and Dose.
The MuLV T helper peptide was originally identified as a 20 mer using Friend MuLV-specific CD4+ T cells, and the optimally recognized sequence was found to be a 13-mer LTSLTPRCNTAWN (30). We investigated the minimal peptide length that was still able to protect mice against the Rauscher MuLV-induced lymphoma RMA. Table 1 shows the comparison of several length variants of the T helper peptide on their tumor-protective capacity in C57BL/6 mice. These data show that the minimal peptide that still protects is the 10-mer LTPRCNTAWN and longer peptides with additional flanking residues can also protect. Titration of four of these peptides (Table 2) shows that longer peptides can still protect at 1 μg/mouse, whereas the 10-mer core peptide can only protect with doses as low as 10 μg/ mouse, indicating that longer peptides are more efficient in triggering the proper T helper cell population. The 13-mer peptide H13.3 (SLTPRCNTAWNRL) was selected for further studies.
Table 1.
In Vivo Tumor-protective Capacity of Length Variants of MuLV env–gp70 T Helper Peptide
| Peptide | Sequence | Surviving mice at day 40 |
|---|---|---|
| H19 | EPLTSLTPRCNTAWNRLKL | 5/7 |
| H17 | LTSLTPRCNTAWNRLKL | 5/7 |
| H15.2 | SLTPRCNTAWNRLKL | 3/7 |
| H13.2 | TPRCNTAWNRLKL | 2/7 |
| H10.2 | CNTAWNRLKL | 0/7 |
| H9.2 | NTAWNRLKL | 0/7 |
| H15.1 | LTSLTPRCNTAWNRL | 5/7 |
| H13.3 | SLTPRCNTAWNRL | 6/7 |
| H11.2 | TPRCNTAWNRL | 1/7 |
| H13.1 | LTSLTPRCNTAWN | 4/7 |
| H11.1 | SLTPRCNTAWN | 5/7 |
| H10.1 | LTPRCNTAWN | 4/7 |
| H9.1 | TPRCNTAWN | 1/7 |
Table 2.
Optimization of T Helper Peptide Dose for In Vivo Tumor Protection
| Peptide | Peptide dose | |||
|---|---|---|---|---|
| 100 μg | 10 μg | 1 μg | 0.1 μg | |
| H19 | 3/6 | 4/6 | 5/6 | 0/6 |
| H13.3 | 6/6 | 5/6 | 4/6 | 1/6 |
| H10.1 | 5/6 | 4/6 | 1/6 | 0/6 |
| H9.1 | n.t. | 1/6 | 0/6 | 0/4 |
Mechanism of T Helper Peptide-induced Tumor Protection.
These results show that a single vaccination with a synthetic peptide, which was reported as a T helper epitope, leads to substantial tumor protection. We investigated the contribution of CD4+ and CD8+ T cells in this model both in vivo and in vitro. C57BL/6 mice were treated with antibodies against CD4 and CD8. Antibody treatment resulted in depletion of >95% of the respective T cell subsets as monitored by FACScan® analysis of spleen cells (data not shown). Fig. 4 A shows the dramatic effect of CD4 depletion during the peptide immunization phase before tumor challenge. Anti-CD4 treatment almost completely abrogates the protective effect of peptide vaccination, indicating the necessity of the presence of CD4+ T cells during the induction period. In addition, mice were treated with anti-CD4 and anti-CD8 during the effector period, i.e., just after RMA tumor challenge. Fig. 4 B shows that depletion of either CD4 or CD8 positive cells completely abrogates the survival of mice that were preimmunized with the T helper peptide. This indicates that both CD4+ and CD8+ T cells are essential in the effector phase, suggesting that upon tumor challenge, tumor-specific T helper cells are required for prompt activation of CD8 positive effector cells.
Figure 4.

Both CD4+ and CD8+ T cells are involved in the protective mechanism. C57BL/6 mice were treated with anti-CD4 (GK1.5) and anti-CD8 (2.43) monoclonal antibodies leading to selective depletion (>95%) of these T cell subsets. (A) Mice were vaccinated with 100 μg of H19 in IFA subcutaneously (day −14) and treated five times with 100 μg GK1.5 monoclonal antibody in 200 μl PBS intraperitoneally during the vaccination period (days −14, −13, −10, −7, and −4) or as a control with PBS. At day 0, 103 RMA cells were injected intraperitoneally. Significance of differences (log rank test): PBS versus env-H19: P = 0.0002; env-H19 + anti-CD4 versus env-H19: P = 0.0009; PBS versus env-H19 + anti-CD4: P = 0.577 (n.s.). (B) Mice were vaccinated with 10 μg T helper peptide H13.3 in IFA subcutaneously at day −14 and 103 RMA cells were injected intraperitoneally at day 0. Antibody treatment (three times with 100 μg of GK1.5 or 2.43 intraperitoneally) was done during the effector phase (days 0, 3, and 7). Significance of differences (log rank test): PBS versus env-H13.3: P = 0.0027; env-H13.3/anti-CD4 versus env-H13.3: P = 0.0013; env-H13.3/anti-CD8 versus env-H13.3: P = 0.0068.
The T cell response to the synthetic peptide was investigated by in vitro analysis of T cells obtained from peptide-vaccinated mice. Spleen cells from mice that were vaccinated with env-H19 synthetic peptide were restimulated after 14 d with env-H19 peptide-loaded spleen cells in vitro. Massive outgrowth of CD4 positive cells was observed and several clones obtained after limiting dilution were tested for specific proliferation and cytokine production. We were not able to isolate peptide-specific CD8+ T cells from H19 peptide-immunized mice. The CD4+ T cell clones specifically proliferated on H19 peptide and shorter peptide length variants in an I-Ab–restricted fashion (as evidenced by a stimulator cell panel study, data not shown). Most of the clones (7/10) were also able to recognize 15-mer peptides but not shorter variants, and 3/10 clones were also able to recognize shorter peptides, 13- and 11-mer peptides (not shown). No specific proliferation nor cytotoxicity of these clones directly to RMA cells, with or without IFN-γ treatment, could be observed that was expected as the cells lack MHC class II (data not shown). Five of these clones were tested for peptide-specific cytokine production profile. All clones produced IL-2 IFN-γ, but not IL-4, IL-3, IL-5, and IL-6 (data not shown), indicating a Th1 type of T helper response.
In addition, we isolated tumor-specific T cell cultures from mice that were protected from tumor challenge. Spleen cells from env-H11.1 peptide-vaccinated mice that survived RMA tumor challenge after 6 wk were restimulated with RMA cells in vitro. From these T cell cultures we were able to isolate both CD4+ and CD8+ T cell clones. Fig. 5 A shows the peptide and tumor specificity of CD4+ clone 3A12. Although it is not able to recognize the MHC class II negative RMA cells directly, it specifically proliferates to the combination of RMA cells with APCs present in autologous spleen cells, indicating specific recognition of processed RMA-derived antigen. The peptide fine specificity of clone 3A12 is shown in Fig. 5 B. The recognition of shorter length variants of the peptide is in agreement with the observed protective ability of the shorter peptides (Table 1). Fig. 5 C shows the specific proliferative response of CD4+ clone 3A12 to endogenously processed viral antigen using Moloney MuLV-infected LPS blasts.
Figure 5.
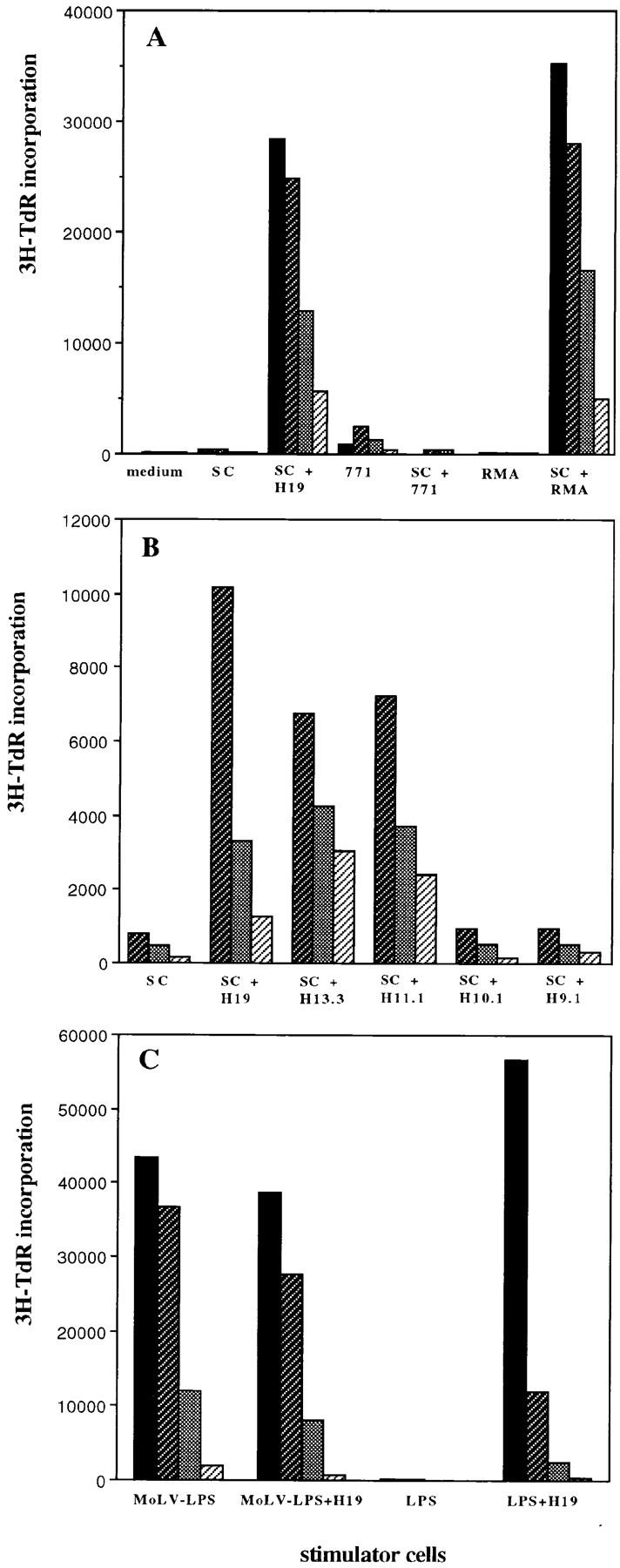
MuLV-specific CD4+ T cells can be isolated from T helper peptide-immunized mice. CD4+ T cell clones were isolated from T helper peptide-immunized and RMA-challenged mice and tested in a 3-d proliferation assay. Values represent means of triplicate measurements. (A) CD4+ clone 3A12 was isolated from a spleen of a protected mouse, 2 mo after tumor challenge, that was previously immunized with the 11-mer T helper H11.1 peptide. Spleen cells were in vitro restimulated with mitomycin-treated and irradiated RMA tumor cells and CD4+ T cell clones were isolated by limiting dilution. Shaded bars from black to gray: 4 × 104, 2 × 104, 104, and 5 × 103 cells/well, respectively. env-H19 peptide was used at 10 μg/ml. SC, irradiated spleen cells (105/well). RMA cells were mitomycin treated, irradiated, and used at 2 × 104 cells/well with or without irradiated spleen cells. 771 is a B cell lymphoma cell line isolated from a tumor induced by the AKV type MCF1233 MuLV that lacks the T helper epitope sequence in env (2 × 104 irradiated cells/well). (B) Peptide fine specificity of CD4+ clone 3A12. Peptides were used at 5 μg/ml. Shaded bars from dark to light gray: 2 × 104, 104, and 5 × 103 cells/well, respectively. (C) Moloney MuLV specificity of clone 3A12. LPS B cell blasts were induced using C57BL/6 spleen cells in the presence or absence of Moloney MuLV (MoLV-LPS). After 3 d, the LPS blasts were isolated, irradiated, and used as stimulator cells (LPS, 105 cells/well) for clone 3A12 in the presence or absence of H19 peptide (10 μg/ml).
From the same T cell bulks we were able to isolate RMA-specific CD8+ T cell clones. These clones were cytotoxic for RMA cells and Db-restricted. Representative clone 2H9 is shown in Figure 6 A and was found to be specific for the recently identified common FMR immunodominant CTL epitope that is present in the leader sequence of gag (15), but not for another upstream gag-L epitope (31; Fig. 6 B). No reactivity was found with the T helper peptide to which the mice were vaccinated, not even with 9- and 10-mer peptides with potential Db-binding characteristics (Fig. 6 B). These peptides have undetectable MHC class I–binding affinity as tested with the RMA-S peptide-binding assay (data not shown). This indicates that no direct CTL priming occurs after vaccination with the T helper peptide vaccine. Tumor-specific T helper cells are specifically activated by the peptide that will regulate, depending on cross-priming of tumor antigens, efficient generation of tumor-specific CTLs. Vaccination of mice with a combination of the T helper peptide and the dominant CTL epitope resulted in very efficient protection (Fig. 2 B). These data indicate that specific CTLs are the main effector cells that require tumor-specific T cell help for efficient antitumor immunity in this system.
Figure 6.


Cytotoxic T cells isolated from protected mice recognize the immunodominant MuLV gag-L peptide. CD8+ T cell bulk cultures were isolated from spleen cells of a C57BL/6 mouse that was immunized with the H11.1 T helper peptide and survived RMA tumor challenge. 2H9 was a representative clone isolated from this RMA-specific bulk culture by limiting dilution. (A) Cytotoxicity assay using RMA cells and HeLa cells selectively transfected with the MHC class I Db molecule as target cells. HeLaDb cells were incubated with the gag-L (CCLCLTVFL) peptide (1 μg/ml) during the assay. (B) No reactivity of 2H9 CTL clone with the T helper peptide sequence. EL-4 cells (H-2b) were incubated with different synthetic peptides (1 μg/ml). gag-L–Db9 is the dominant gag-L epitope CCLCLTVFL; gag-L–SIV9, another upstream gag-leader sequence reported to comprise a CTL epitope SIVLCCLCL (31).
Discussion
In this study, we show that a single vaccination with a synthetic peptide comprising a retrovirus-encoded MHC class II–presented T helper epitope can induce tumor-protective immunity against MHC class II negative tumor cells. This finding underlines a crucial regulatory function of CD4+ T cells in recruiting tumor-directed effector cells and the necessity of MHC class II–mediated antigen presentation by, most likely, specialized APCs. In this model, CTLs appear to be the effector cells that eradicate the tumor cells in vivo since CD8 depletion during the effector phase resulted in abolition of protection, and peptide vaccination with the relevant CTL epitope led to measurable protective responses. In a related study we have investigated the protective capacity of three CTL epitopes in relation to their respective CTL precursor frequencies in C57BL/6 mice (Van Hall, T., N. van de Rhee, E. Mengedé, R. Offringa, C.J.M. Melief, and F. Ossendorp, manuscript submitted for publication). This study indicated that the immunodominance of the CTL epitope is related to its protective capacity and is in line with the current findings.
MuLV env-gp70 T helper peptide vaccination resulted in long-term protection of 40–50% of the mice and a delay in tumor growth in 80–90% of the animals in contrast to irrelevant ovalbumin T helper peptide vaccination. Although protection is not complete, this indicates a significant specific immune response since RMA is an extremely aggressive tumor cell line in immunocompetent animals. Strikingly, a combination vaccine containing the immunodominant MuLV gag-L CTL epitope and the T helper peptide induces highly protective responses leading to long-term survival of 90% of the mice. Vaccination with the immunodominant CTL epitope by itself also induces protection, but is generally far less effective (∼30% of the mice survive). Some bystander help was observed upon administration of the immunodominant CTL epitope in combination with the ovalbumin T helper peptide (Fig. 2 C), although the difference was not found to be statistically significant from vaccination with the CTL epitope alone. Bystander help can be beneficial for CTL induction as shown by others (32). This might be due to cytokines produced by Ova-specific T helper cells during the priming phase leading to increased numbers of CTL precursors. In the effector phase after tumor challenge the Ova-specific T helper cells will not be activated.
CTL induction in vivo has classically been reported to be T helper dependent (33), but synthetic peptide vaccination studies using the minimal CTL epitope sequence were either shown to be T helper independent (16) or dependent (34, 35) in different virus systems. Here, we report induction of protective CTL-mediated immunity by vaccination with a T helper epitope by itself or in combination with a CTL epitope indicating T helper dependence for optimal protection. The requirement for physical linkage between CTL and the T helper peptide as shown for HIV (36) is not essential since a single vaccination with a mixture of T helper and CTL epitopes in IFA induces efficient protective immunity.
Recent experiments by Bennett et al. (37) indicate that induction of CTL responses by cross-priming requires cognate T cell help, involving presentation of both CTL and helper epitopes on the same APC. We consider these findings in line with our study. Local specific help appears to be crucial for efficient CTL-mediated tumor control. This can only be expected when APCs present tumor-derived antigenic fragments to tumor-specific T helper cells. This explains why nonrelated bystander (OVA-specific) help is far less efficient. In conclusion, optimal virus or tumor vaccines should preferably contain both antigen-specific CTLs and T helper epitopes.
Vaccination with the MuLV-env–gp70 T helper peptide in IFA leads to activation of CD4+ T cells that exhibit a Th1 type cytokine production pattern as established in vitro at the clonal level. The Th1 type appears to be crucially related to the cellular (cytotoxic) response leading to tumor cell destruction. The essential rules for peptide vaccination inducing the required T helper type are presently not understood. In future experiments, we will purposely direct the T helper type response using specific antibodies and cytokines and monitor the effects on tumor protection.
Greenberg (5), in adoptive transfer experiments of FBL-specific CD4+ T cells, found macrophages to be a main effector population next to CTLs. We have performed adoptive transfer experiments with our specific CTL and Th clones in RMA tumor-bearing nude mice (our unpublished observations). We were able to control RMA tumor growth by intravenous infusion of the gag-L–specific CTL clone in combination with subcutaneous IL-2 or in combination with the T helper clone. In contrast, the T helper clone by itself could only partially control tumor outgrowth in these T cell–deficient mice, possibly by activating macrophages into tumoricidal effector cells (5). Therefore, we conclude that CTLs are important effector cells in this system, but we do not exclude other effector mechanisms operating in vivo. Macrophages and/or other professional APCs of the dendritic cell lineage are likely to play an important role in the cross-presentation of tumor antigens. Vaccination studies using vaccinia virus containing a chimeric Friend MuLV env gene resulted in CD4- and CD8-dependent protection against Friend virus–induced erythroid proliferation (38). Miyazawa et al. (39) showed protective effects of env-gp70 T helper peptide vaccination against Friend virus, especially by a I-Eb/k–binding peptide. Here, the mechanism was reported to be control of infection by induction of virus-neutralizing antibodies or direct effects of the CD4+ T cells. In our study, using an established tumor cell line, peptide vaccination and CD8 depletion studies strongly indicate a dominant role for Rauscher MuLV-specific CTL.
We have explored the possibility of using the helper and CTL epitope-containing synthetic peptides for therapeutic use in mice bearing RMA tumors or when tumor cells were administered at the same day of vaccination. Thus far, using the IFA protocol, we were not able to control this extremely aggressive tumor cell line in a therapeutic set up. Apparently, the naive immune system cannot control this rapidly dividing, established tumor cell line. Other ways of efficient activation of the immune system, i.e., using peptide-loaded dendritic cells (22), will be investigated in this model.
Peptide vaccines are generally extremely potent in vivo, but peptide administration can lead to either induction of protective responses or (unwanted) induction of specific T cell tolerance. This has been found for CTL epitopes in both virus (40) and tumor models (41, 42). Peptide vaccination with the LCMV CTL epitope can lead to induction of immunity, but is dose and route dependent. For immunization rather than tolerance induction, only a single dose of peptide in a quite narrow window is required (40). In the adenovirus tumor model, peptide-induced tolerization is dose independent (41, 42). In the MuLV system in which we use an identical peptide administration protocol, both T helper and CTL peptide vaccination appear to lead to activation rather than functional deletion of specific T cells. Our study with an intraperitoneally growing tumor shows that T helper peptide administration in IFA is protective across a wide dose of peptides of varying lengths. However, T helper peptides can induce tolerance as shown by the ovalbumin peptide (43), dependent on the mode of peptide delivery. Our findings indicate that T helper peptide vaccination with a single subcutaneous dose in IFA leads to immunization rather than tolerization.
The use of T helper epitopes in vaccines can have potential advantages. First, T helper cells might play an important role not only for optimal induction of CTL responses as reported in this paper, but also for maintainance of CTL memory as reported for LCMV (44, 45) and γ-herpesvirus (46). Secondly, the regulatory role of T helper cells allows recruitment of several effector systems next to CTLs that one can make use of to specifically attack tumor cells. Utilization of specific cytokines, antibodies, or adjuvants allows one to selectively tilt the response towards cells of the Th1 or Th2 type. And thirdly, T helper peptides are usually more promiscuous in binding to MHC class II molecules than CTL epitopes are for MHC class I. This opens the possibility that T helper–based vaccines are less allele dependent and thereby widely applicable. Inclusion of CTL epitopes next to T helper epitopes in vaccines nevertheless seems unavoidable for optimal action. Finally, these findings illustrate the importance of identification of tumor-specific T helper epitopes, even if tumor cells lack MHC class II expression.
Footnotes
We wish to thank Drs. R. Offringa, F. Koning, and T. Ottenhoff for critically reading the manuscript, Dr. J.W. Drijfhout for synthesis of peptides, Dr. R. Schipper for statistical analysis, and G. Schijff (TNO-PG) for excellent biotechnical assistance.
This study was financed by the Netherlands Cancer Foundation grants 93-560 and 97-1451.
Address correspondence to F. Ossendorp, Department of Immunohematology and Bloodbank, University Hospital Leiden, Bldg. 1, E3-Q, PO Box 9600, 2300 RC Leiden, The Netherlands. Phone: 31-71-5263800; Fax: 31-71-5216751; E-mail: ossendorp@rullf2.medfac.leidenuniv.nl
1
Abbreviations used in this paper: FMR, Friend, Moloney, Rauscher; gag-L, gag-leader; gp, glycoprotein; MoMSV, Moloney murine sarcoma and leukemia virus complex; MuLV, murine leukemia virus; n.s., not significant.
References
- 1.Ostrand-Rosenberg S, Thakur A, Clements V. Rejection of mouse sarcoma cells after transfection of MHC class II genes. J Immunol. 1990;144:4068–4071. [PubMed] [Google Scholar]
- 2.James R, Edwards S, Hui K, Bassett P, Grosveld F. The effect of class II gene transfection on the tumorigenicity of the H-2knegative mouse leukemia cell line K36.16. Immunology. 1991;72:213–218. [PMC free article] [PubMed] [Google Scholar]
- 3.Chen P, Aanathaswamy H. Rejection of K1735 murine melanoma in syngeneic hosts requires expression of MHC class I antigens and either class II antigens or IL-2. J Immunol. 1993;151:244–255. [PubMed] [Google Scholar]
- 4.Yoshimura A, Shiku H, Nakayama E. Rejection of an IA+ variant line of FBL-3 leukemia by cytotoxic T lymphocytes with CD4+ and CD4−CD8−T cell receptor-αβ phenotypes generated in CD8-depleted C57BL/6 mice. J Immunol. 1993;150:4900–4910. [PubMed] [Google Scholar]
- 5.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281–355. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 6.Klarnet JP, Kern DE, Okunu K, Holt C, Lilly F, Greenberg PD. FBL-reactive CD8 cytotoxic and CD4 helper T cells recognize distinct Friend murine leukemia virus–encoded antigens. J Exp Med. 1989;169:457–467. doi: 10.1084/jem.169.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu J, Kindsvogel W, Busby S, Bailey MC, Shi Y, Greenberg PD. An evaluation of the potential to use tumor-associated antigens as targets for antitumor T cell therapy using transgenic mice expressing a retroviral tumor antigen in normal lymphoid tissues. J Exp Med. 1993;177:1681–1690. doi: 10.1084/jem.177.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lilly F, Boyse EA, Old LJ. Genetic basis of susceptibility to viral leukemogenesis. Lancet. 1964;ii:1207–1209. doi: 10.1016/s0140-6736(64)91043-8. [DOI] [PubMed] [Google Scholar]
- 9.Vasmel WLJ, Zijlstra M, Radaszkiewicz T, Leupers CJM, De Goede REY, Melief CJM. Major histocompatibility class II regulated immunity to murine leukemia virus protects against early T- but not late B-cell lymphomas. J Virol. 1988;62:3156–3166. doi: 10.1128/jvi.62.9.3156-3166.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesebro B, Miyazawa B, Britt WJ. Host genetic control of spontaneous and induced immunity to Friend murine retrovirus infection. Annu Rev Immunol. 1990;8:477–499. doi: 10.1146/annurev.iy.08.040190.002401. [DOI] [PubMed] [Google Scholar]
- 11.Iwashiro M, Kondo T, Shimizu T, Yamagishi H, Takahashi K, Matsubayashi Y, Masuda T, Otaka A, Fujii N, Ishimoto A, et al. Multiplicity of virus-encoded helper T-cell epitopes expressed on FBL-3 tumor cells. J Virol. 1993;67:4533–4542. doi: 10.1128/jvi.67.8.4533-4542.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasmel WLE, Sijts EJAM, Leupers CJM, Matthews EA, Melief CJM. Primary virus–induced lymphomas evade T cell immunity by failure to express viral antigens. J Exp Med. 1989;169:1233–1254. doi: 10.1084/jem.169.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plata F, Langlade-Demoyen P, Abastado JP, Berbar T, Kourilsky P. Retrovirus antigens recognized by cytotoxic T lymphocytes activate tumor rejection in vivo. Cell. 1987;48:231–240. doi: 10.1016/0092-8674(87)90426-0. [DOI] [PubMed] [Google Scholar]
- 14.Sijts AJAM, De Bruijn ML, Ressing ME, Nieland JD, Mengedé EAM, Boog CJ, Ossendorp F, Kast WM, Melief CJM. Identification of an H-2 Kb–presented Moloney murine leukemia virus cytotoxic T lymphocyte epitope that displays enhanced recognition in H-2 Dbmutant bm13 mice. J Virol. 1994;68:6038–6046. doi: 10.1128/jvi.68.9.6038-6046.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Qin H, Chesebro B, Cheever MA. Identification of a gag-encoded cytotoxic T lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus–induced tumors. J Virol. 1996;70:7773–7782. doi: 10.1128/jvi.70.11.7773-7782.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kast WM, Roux L, Curren J, Blom HJJ, Voordouw AC, Meloen RH, Kolakofski D, Melief CJM. Protection against lethal Sendai virus infection by in vivo priming of virus-specific cytotoxic T lymphocytes with an unbound peptide. Proc Natl Acad Sci USA. 1991;88:2283–2287. doi: 10.1073/pnas.88.6.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulz M, Zinkernagel RM, Hengartner H. Peptide-induced antiviral protection by cytotoxic T cells. Proc Natl Acad Sci USA. 1991;88:991–993. doi: 10.1073/pnas.88.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feltkamp MCW, Smits HL, Vierboom MPM, Minnaar RP, De Jongh BM, Drijfhout JW, Ter J, Schegget, Melief CJM, Kast WM. Vaccination with a cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human Papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23:2242–2249. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 19.Schild H, Norda M, Deres K, Falk K, Rötschke O, Wiesmüller K-H, Jung G, Rammensee H-G. Fine specificity of cytotoxic T lymphocytes primed in vivo either with virus or synthetic lipopeptide vaccine or primed in vitro with peptide. J Exp Med. 1991;174:1665–1668. doi: 10.1084/jem.174.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minev BR, McFarland BJ, Spiess PJ, Rosenberg SA, Restifo NP. Insertion signal sequence fused to minimal peptides elicits specific CD8+T-cell responses and prolongs survival of thymoma-bearing mice. Cancer Res. 1994;54:4155–4161. [PMC free article] [PubMed] [Google Scholar]
- 21.Mandelboim O, Vadai E, Fridkin M, Katz-Hillel A, Feldman M, Berke G, Eisenbach L. Regression of established murine carcinoma metastases following vaccination with tumour-associated antigen peptides. Nat Med. 1995;1:1179–1183. doi: 10.1038/nm1195-1179. [DOI] [PubMed] [Google Scholar]
- 22.Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CJM, Ildstad ST, Kast WM, Deleo AB, Lotze MT. Bone marrow–derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1:1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 23.Sijts AJAM, Ossendorp F, Mengedé EAM, van den Elsen PJ, Melief CJM. Immunodominant mink cell focus-inducing murine leukemia virus (MuLV)-encoded CTL epitope, identified by its MHC class I binding motif, explains MuLV-type specificity of MCF-directed cytotoxic T lymphocytes. J Immunol. 1994;152:106–116. [PubMed] [Google Scholar]
- 24.Ljunggren H-G, Kärre K. Host resistance directed selectively against H-2 deficient lymphoma variants. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stukart MJ, Vos A, Melief CJM. Cytotoxic T cell response against lymphoblasts infected with Moloney (Abelson) murine leukemia virus. Methodological aspects and H-2 requirements. Eur J Immunol. 1981;11:251–257. doi: 10.1002/eji.1830110316. [DOI] [PubMed] [Google Scholar]
- 26.Gausepohl, H., M. Kraft, C. Boulin, and R.W. Frank. 1990. Automated multiple peptide synthesis with BOP activation. In Proc. of the 11th American Peptide Symposium. J.E. Rivier and G.R. Marshall, editors. ESCOM, Leiden. 1003–1004.
- 27.Shimonkevitz R, Colon S, Kappler JW, Marrack P, Grey HM. Antigen recognition by H-2–restricted T cells. II. A tryptic ovalbumin peptide that substitutes for processed antigen. J Immunol. 1984;133:2067–2074. [PubMed] [Google Scholar]
- 28.Bouaboula M, Legoux P, Pességué B, Delpech B, Dumont X, Piechaczyk M, Casellas P, Shire D. Quantitation of cytokine gene expression using a polymerase chain reaction method involving co-amplification with an internal multi-specific control. J Biol Chem. 1992;267:21830–21838. [PubMed] [Google Scholar]
- 29.Ossendorp F, Eggers M, Neisig A, Ruppert T, Groettrup M, Sijts A, Mengedé E, Kloetzel P, Neefjes J, Koszinowski U, Melief C. A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity. 1996;5:115–124. doi: 10.1016/s1074-7613(00)80488-4. [DOI] [PubMed] [Google Scholar]
- 30.Shimizu T, Uenishi H, Teramura Y, Iwashiro M, Kuribayashi K, Tamamura H, Fujii N, Yamagishi H. Fine specificity of a virus-encoded helper T-cell epitope expressed on FBL-3 tumor cells. J Virol. 1994;68:7704–7708. doi: 10.1128/jvi.68.12.7704-7708.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondo T, Uenishi H, Shimizu T, Hirama T, Iwashiro M, Kuribayashi K, Tamamura H, Fujii N, Fujisawa R, Yamagishi H. A single retroviral gag precursor signal peptide recognized by FBL-3 tumor-specific cytotoxic T lymphocytes. J Virol. 1995;69:6735–6741. doi: 10.1128/jvi.69.11.6735-6741.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, Grey H. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 33.Keene JA, Forman J. Helper activity is required for the induction of cytotoxic T lymphocytes. J Exp Med. 1982;155:768–782. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X-M, Zheng B, Liew FY, Brett S, Tite J. Priming of influenza virus–specific cytotoxic T lymphocytes in vivo by short synthetic peptides. J Immunol. 1991;147:3168–3173. [PubMed] [Google Scholar]
- 35.Fayolle C, Deriaud E, Leclerc C. In vivo induction of cytotoxic T cell response by a free synthetic peptide requires CD4+T cell help. J Immunol. 1991;147:4069–4073. [PubMed] [Google Scholar]
- 36.Shirai M, Pendleton CD, Ahlers J, Takeshita T, Newman M, Berzofsky JA. Helper-cytotoxic T lymphocyte (CTL) determinant linkage required for priming of HIV CD8+CTL in vivo with peptide vaccine constructs. J Immunol. 1994;152:529–556. [PubMed] [Google Scholar]
- 37.Bennett SRM, Carbone FR, Karamalis F, Miller JFAP, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+T cell help. J Exp Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasenkrug KJ, Brooks DM, Nishio J, Chesebro B. Differing T-cell requirements for recombinant retrovirus vaccines. J Virol. 1996;70:368–372. doi: 10.1128/jvi.70.1.368-372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazawa M, Fujisawa R, Ishihara C, Takei YA, Shimizu T, Uenishi H, Yamagishi H, Kuribayashi K. Immunization with a single T helper cell epitope abrogates Friend virus–induced early erythroid proliferation and prevents late leukemia development. J Immunol. 1995;155:748–758. [PubMed] [Google Scholar]
- 40.Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. J Exp Med. 1995;182:261–266. doi: 10.1084/jem.182.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toes REM, Blom RJJ, Offringa R, Kast WM, Melief CJM. Functional deletion of tumor-specific cytotoxic T lymphocytes induced by peptide immunization can lead to the inability to reject tumors. J Immunol. 1996;156:3911–3918. [PubMed] [Google Scholar]
- 42.Toes REM, Offringa R, Blom RJJ, Melief CJM, Kast WM. Peptide vaccination can lead to enhanced tumor outgrowth through specific T-cell tolerance induction. Proc Natl Acad Sci USA. 1996;93:7855–7860. doi: 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide induced T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 44.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Von Herrath MG, Yokoyama M, Dockter J, Oldstone MBA, Whitton JL. CD4-deficient mice have reduced levels of memory cytotoxic T lymphocytes after immunization and show diminished resistance to subsequent virus challenge. J Virol. 1996;70:1072–1079. doi: 10.1128/jvi.70.2.1072-1079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cardin RD, Brooks JW, Sarawar SR, Doherty PC. Progressive loss of CD8+ T cell-mediated control of a γ-herpesvirus in the absence of CD4+T cells. J Exp Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]