GenBank (original) (raw)
Abstract
GenBank (R) is a comprehensive database that contains publicly available nucleotide sequences for more than 260 000 named organisms, obtained primarily through submissions from individual laboratories and batch submissions from large-scale sequencing projects. Most submissions are made using the web-based BankIt or standalone Sequin programs and accession numbers are assigned by GenBank staff upon receipt. Daily data exchange with the European Molecular Biology Laboratory Nucleotide Sequence Database in Europe and the DNA Data Bank of Japan ensures worldwide coverage. GenBank is accessible through NCBI's retrieval system, Entrez, which integrates data from the major DNA and protein sequence databases along with taxonomy, genome, mapping, protein structure and domain information, and the biomedical journal literature via PubMed. BLAST provides sequence similarity searches of GenBank and other sequence databases. Complete bimonthly releases and daily updates of the GenBank database are available by FTP. To access GenBank and its related retrieval and analysis services, begin at the NCBI Homepage: www.ncbi.nlm.nih.gov
INTRODUCTION
GenBank (1) is a comprehensive public database of nucleotide sequences and supporting bibliographic and biological annotation, built and distributed by the National Center for Biotechnology Information (NCBI), a division of the National Library of Medicine (NLM), located on the campus of the US National Institutes of Health (NIH) in Bethesda, MD, USA.
NCBI builds GenBank primarily from the submission of sequence data from authors and from the bulk submission of expressed sequence tag (EST), genome survey sequence (GSS), and other high-throughput data from sequencing centers. The US Office of Patents and Trademarks also contributes sequences from issued patents. GenBank, the European Molecular Biology Laboratory Nucleotide Sequence Database (EMBL) (2) in Europe, and the DNA Databank of Japan (DDBJ) (3) comprise the International Nucleotide Sequence Database Collaboration (INSDC), and are members of a long-standing collaboration in which data is exchanged daily to ensure a uniform and comprehensive collection of sequence information. NCBI makes the GenBank data available at no cost over the Internet, via FTP and via a wide range of Web-based retrieval and analysis services which operate on the GenBank data (4).
ORGANIZATION OF THE DATABASE
From its inception, GenBank has doubled in size about every 18 months. The traditional GenBank divisions contain over 80 billion nucleotide bases from more than 76 million individual sequences, with 15 million new sequences added in the past year. Contributions from Whole Genome Shotgun (WGS) projects supplement the data in the traditional divisions to bring the total beyond 190 billion bases. Complete genomes (www.ncbi.nlm.nih.gov/Genomes/index.html) continue to represent a rapidly growing segment of the database, with some 200 of more than 570 complete microbial genomes in GenBank deposited over the past year. The number of eukaryote genomes for which coverage and assembly are significant continues to increase as well, with over 190 assemblies now available, including that of the reference human genome.
Sequence-based taxonomy
Database sequences are classified and can be queried using a comprehensive sequence-based taxonomy (www.ncbi.nlm.nih.gov/sites/entrez?db=taxonomy) developed by NCBI in collaboration with EMBL and DDBJ and with the valuable assistance of external advisers and curators. More than 260 000 named species are represented in GenBank and new species are being added at the rate of over 1700 per month. About 12% of the sequences in GenBank are of human origin and 8% of all sequences are human expressed sequence tags (ESTs). The top species in GenBank in terms of number of bases are Homo sapiens (12.7 billion bases), Mus musculus (8.3 billion), Rattus norvegicus (5.8 billion), Bos taurus (3.8 billion), Zea mays (3.6 billion), Danio rerio (2.8 billion), Sus scrofa (1.9 billion), Oryza sativa (1.5 billion), Strongylocentrotus purpuratus (1.4 billion), Xenopus tropicalis (1.1 billion) and Pan troglodytes (940 million).
GenBank records and divisions
Each GenBank entry includes a concise description of the sequence, the scientific name and taxonomy of the source organism, bibliographic references and a table of features (www.ncbi.nlm.nih.gov/collab/FT/index.html) listing areas of biological significance, such as coding regions and their protein translations, transcription units, repeat regions and sites of mutations or modifications.
The files in the GenBank distribution have traditionally been partitioned into ‘divisions’ that roughly correspond to taxonomic groups such as bacteria (BCT), viruses (VRL), primates (PRI) and rodents (ROD). In recent years, divisions have been added to support specific sequencing strategies. These include divisions for expressed sequence tag (EST), genome survey (GSS), high-throughput genomic (HTG), high-throughput cDNA (HTC) and environmental sample (ENV) sequences, making a total of 18 divisions. For convenience in file transfer, the GenBank data is partitioned into multiple files, currently more than 1300, for the bimonthly GenBank releases on NCBI's FTP site.
Expressed sequence tags (ESTs)
ESTs continue to be a major source of new sequence records and gene sequences, comprising over 25 billion nucleotide bases in GenBank release 161. Over the past year, the number of ESTs has increased by over 19% to a total of 45.5 million sequences representing more than 1370 different organisms. The top organisms represented in the EST division are Homo sapiens (8.1 million records), Mus musculus (4.9 million), Bos taurus (1.5 million), Sus scrofa (1.5 million), Danio rerio (1.4 million) and Arabidopsis thaliana (1.3 million). As part of its daily processing of GenBank EST data, NCBI identifies through BLAST searches all homologies for new EST sequences and incorporates that information into the companion database, dbEST (www.ncbi.nlm.nih.gov/dbEST/index.html) (5). The data in dbEST is processed further to produce the UniGene database (www.ncbi.nlm.nih.gov/sites/entrez?db=unigene) of more than 1.5 million gene-oriented sequence clusters representing over 85 organisms and described more fully in Ref. (4).
Sequence-tagged sites (STSs), genome survey sequences (GSSs) and environmental sample sequences (ENV)
The STS division of GenBank (www.ncbi.nlm.nih.gov/dbSTS/index.html) contains over 930 000 sequences, including anonymous STSs based on genomic sequence as well as gene-based STSs derived from the 3′ ends of genes and ESTs. These STS records usually include mapping information.
The GSS division of GenBank (www.ncbi.nlm.nih.gov/dbGSS/index.html) has grown over the past year by 29% to a total of 21 million records for over 670 organisms and contributes over 13.5 billion nucleotide bases. GSS sequences are the products of as many as 80 different experimental techniques, including ‘metagenomic’ surveys of sequences arising from biological communities. However, about half of all GSS records are single reads from Bacterial Artificial Chromosomes (‘BAC-ends’) used in a variety of genome sequencing projects. The most highly represented species in the GSS division, including metagenomic surveys, are marine metagenome (2.6 million records), Zea mays (2.1 million), Mus musculus (1.8 million) and Homo sapiens (1.1 million). The human data has been used (www.ncbi.nlm.nih.gov/projects/genome/clone/) along with the STS records in tiling the BACs for the Human Genome Project (6).
The ENV division of GenBank accommodates non-WGS sequences obtained via environmental sampling methods in which the source organism is unknown. Records in the ENV division contain ‘ENV’ in the keyword field and use an‘/environmental_sample’ qualifier in the source feature. As of GenBank release 161, the ENV division of GenBank contained over 600 000 sequences, comprising 403 million base pairs.
High-throughput genomic (HTG) and high-throughput cDNA (HTC) sequences
The HTG division of GenBank (www.ncbi.nlm.nih.gov/HTGS/) contains unfinished large-scale genomic records, which are in transition to a finished state (7). These records are designated as Phase 0–3 depending on the quality of the data. Upon reaching Phase 3, the finished state, HTG records are moved into the appropriate organism division of GenBank. As of release 161 of GenBank, the HTG division comprised 18 billion base pairs of sequence, an increase of more than 2 billion bases over the past year.
The HTC division of GenBank accommodates high-throughput cDNA sequences. HTCs are of draft quality but may contain 5′UTRs and 3′UTRs, partial coding regions and introns. HTC sequences which are finished and of high quality are moved to the appropriate organism GenBank division. GenBank release 161 contained more than 429 000 HTC sequences totaling 570 million bases. A project generating HTC data is described in Ref. (8).
Whole Genome Shotgun (WGS) sequence
More than 101 billion bases of WGS sequence appear in GenBank as sets of WGS contigs, many of them bearing annotations originating from a single sequencing project. These sequences are issued accession numbers consisting of a 4-letter project ID, followed by a two-digit version number and a 6-digit contig ID. Hence, the WGS accession number ‘AAAA01072744’ is assigned to contig number ‘072744’ of the first version of project ‘AAAA’. Whole Genome Shotgun (WGS) sequencing projects have contributed some 25 million contigs to GenBank, a 39% increase over last year's total. These primary sequences have been used to construct 4.1 million large-scale assemblies of scaffolds and chromosomes. WGS project contigs for Homo sapiens, Pan trodlodytes, Macacca mulatta, Equus caballus, Canis familiaris, Drosophila, Saccharomyces and 800 other organisms and environmental samples are available. For a complete list of WGS projects with links to the data, see (www.ncbi.nlm.nih.gov/projects/WGS/WGSprojectlist.cgi).
Although WGS project sequences may be annotated, many low-coverage genome projects do not contain annotation. Because these sequence projects are ongoing and incomplete, these annotations may not be tracked from one assembly version to the next and should be considered preliminary.
Submitters of WGS sequences, and genomic sequences in general, are urged to use a new set of evidence tags of the form‘/experimental=_text_’ and‘/inference=_TYPE:text_’, where‘_TYPE_’ is one of a number of standard inference types and ‘_text_’ is made up of structured text. These new qualifiers replace ‘evidence=experimental’ and ‘evidence=non-experimental’, respectively, which are no longer supported.
Special Record types
Third Party Annotation (TPA)
Third Party Annotation (TPA) records support the reporting of published sequence annotation by a scientist other than the original submitter of the primary sequence record in DDBJ/EMBL/GenBank. TPA records fall into one of two categories, ‘experimental’, in which case there is direct experimental evidence for the existence of the annotated molecule, and ‘inferential’, in which case the experimental evidence is indirect. TPA sequences may be created by assembling a number of primary sequences. The format of a TPA record (e.g. BK000016) is similar to that of a conventional GenBank record but includes the label ‘TPA:’ at the beginning of each Definition Line and the keywords ‘Third Party Annotation; TPA’ in the Keywords field. The Comment field of TPA records lists the primary sequences used to assemble the TPA sequence; the Primary field provides the base ranges of the primary sequences that contribute to the TPA sequence.
Over 5500 TPA records are contained in GenBank release 161, including 2170 for Drosophila melanogaster, 960 for Homo sapiens, 330 for Oryza sativa and 290 for Mus musculus. TPA sequences are not released to the public until their accession numbers or sequence data and annotation appear in a peer-reviewed biological journal. TPA submissions to GenBank may be made using either BankIt or Sequin. For more information on TPA, see (www.ncbi.nlm.nih.gov/Genbank/TPA.html).
GenBank CON records for assemblies of smaller records
Although many genomes, such as bacterial genomes, are represented in GenBank as single sequences, it is desirable from the standpoints of data transfer and analysis to break some very long sequences, such as portions of eukaryotic genomes, into smaller segments. In these cases, CON division records for the entire sequence are produced that contain assembly instructions to allow the seamless display and download of the full sequence. Many CON records also include annotations.
BUILDING THE DATABASE
The data in GenBank, and the collaborating databases EMBL and DDBJ, is submitted primarily by individual authors to one of the three databases, or by sequencing centers as batches of EST, STS, GSS, HTC, WGS or HTG sequences. Data is exchanged daily with DDBJ and EMBL so that the daily updates from NCBI servers incorporate the most recently available sequence data from all sources.
Direct electronic submission
Virtually all records enter GenBank as direct electronic submissions (www.ncbi.nlm.nih.gov/Genbank/index.html), with the majority of authors using the BankIt or Sequin programs. Many journals require authors with sequence data to submit the data to a public database as a condition of publication.
GenBank staff can usually assign an accession number to a sequence submission within two working days of receipt, and do so at a rate of almost 1600 per day. The accession number serves as confirmation that the sequence has been submitted and allows readers of articles, in which the sequence is cited, to retrieve the data. Direct submissions receive a quality assurance review that includes checks for vector contamination, proper translation of coding regions, correct taxonomy and correct bibliographic citations. A draft of the GenBank record is passed back to the author for review before it enters the database. Authors may ask that their sequences be kept confidential until the time of publication. Since GenBank policy requires that the deposited sequence data be made public when the sequence or accession number is published, authors are instructed to inform GenBank staff of the publication date of the article in which the sequence is cited in order to ensure a timely release of the data. Although only the submitting scientist is permitted to modify sequence data or annotations, all users are encouraged to report lags in releasing data or possible errors or omissions to GenBank at (update@ncbi.nlm.nih.gov).
NCBI works closely with sequencing centers to ensure timely incorporation of bulk data into GenBank for public release. GenBank offers special batch procedures for large-scale sequencing groups to facilitate data submission, including the program ‘tbl2asn’, described at (www.ncbi.nlm.nih.gov/Sequin/table.html).
Submission using BankIt
About a third of author submissions are received through NCBI's Web-based data submission tool, BankIt (www.ncbi.nlm.nih.gov/BankIt). Using BankIt, authors enter sequence information directly into a form and add biological annotation such as coding regions or mRNA features. Free-form text boxes, list boxes and pull-down menus allow the submitter to further describe the sequence without having to learn formatting rules or restricted vocabularies. Before creating a draft record in GenBank flat file format for the submitter to review, BankIt validates submissions, flagging many common errors and checks for vector contamination using a variant of BLAST called Vecscreen. BankIt is the tool of choice for simple submissions, especially when only one or a small number of records is to be submitted (7). BankIt can also be used by submitters to update their existing GenBank records.
Submission using Sequin and tbl2asn
NCBI also offers a standalone multi-platform submission program called Sequin (www.ncbi.nlm.nih.gov/Sequin/index.html) that can be used interactively with other NCBI sequence retrieval and analysis tools. Sequin handles simple sequences such as a cDNA, as well as segmented entries, phylogenetic studies, population studies, mutation studies, environmental samples and alignments for which BankIt and other Web-based submission tools are not well-suited. Sequin has convenient editing and complex annotation capabilities and contains a number of built-in validation functions for quality assurance. In addition, Sequin is able to accommodate large sequences, such as that of the 5.6 Mb Escherichia coli genome, and read in a full complement of annotations via simple tables. Versions for Macintosh, PC and Unix computers are available via anonymous FTP at (ftp.ncbi.nih.gov) in the ‘sequin’ directory. Once a submission is completed, submitters can e-mail the Sequin file to the address (gb-sub@ncbi.nlm.nih.gov).
Submitters of large, heavily annotated genomes may find it convenient to use ‘tbl2asn’, referenced above under ‘Direct submission’, to convert a table of annotations generated via an annotation pipeline into an ASN.1 (Abstract Syntax Notation One) record suitable for submission to GenBank.
Submission of barcode sequences
The Consortium for the Barcode of Life (CBOL) is an international initiative to develop DNA barcoding as a tool for characterizing species of organisms using a short, usually a 648 bp DNA sequence derived from a portion of the cytochrome oxidase subunit I gene. NCBI, in collaboration with CBOL, (www.barcoding.si.edu/index.htm) has created an online tool for the bulk submission of barcode sequences to GenBank (www.ncbi.nlm.nih.gov/BankIt/websub/?tool=barcode) that allows users to upload files containing a batch of sequences with associated source information. It is anticipated that this tool will be used for other types of bulk submissions in the near future.
Sequence identifiers and accession numbers
Accession.Version
Each GenBank record, consisting of both a sequence and its annotations, is assigned a unique identifier, the accession number that is shared across the three collaborating databases (GenBank, DDBJ, EMBL) and remains constant over the lifetime of the record even when there is a change to the sequence or annotation. Each version of the DNA sequence within a GenBank record is also assigned a unique NCBI identifier, called a ‘gi’, that appears on the VERSION line of GenBank flat file records following the accession number. A third identifier of the form ‘Accession.version’, also displayed on the VERSION line of flat file records, contains the information present in both the gi and accession numbers. An entry appearing in the database for the first time has an ‘Accession.version’ identifier equivalent to the ACCESSION number of the GenBank record followed by ‘.1’ to indicate the first version of the sequence for the record, e.g.:
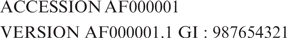
When a change is made to a sequence in a GenBank record, a new gi number is issued to the sequence and the version extension of the ‘Accession.version’ identifier is incremented. The accession number for the record as a whole remains unchanged and the older sequence remains available under the old ‘Accession.version’ identifier and gi.
A similar system tracks changes in the corresponding protein translations. These identifiers appear as qualifiers for CDS features in the FEATURES portion of a GenBank entry, e.g./protein_id=’AAA00001.1’. Protein sequence translations also receive their own unique gi number, which appears as a second qualifier on the CDS feature, e.g.:
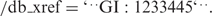
Ensuring stable access to sequence data
A convenient way to share the data among a set of collaborators is to post the data to a locally maintained Web site. However, if original data and updates are not simultaneously submitted to a central repository, significant problems can arise.
The access lifetime of the data may be reduced
The ephemeral nature of much of the content on the Web is part of the common experience. In one attempt to quantify content lifetime, 360 randomly selected web pages were tracked for a period of four years, and a half-life of only two years was measured for the set (9). While a well-maintained web page can certainly persist for longer than two years, the relatively short half-life reported for this set of pages is worth noting.
The full biological context of the data may not be realized
Even during the accessible lifetime of locally posted sequence data, the full biological context of a sequence may not be realized, if the sequence cannot be conveniently compared to others—perhaps derived from distantly related organisms that are beyond the scope of the host web page.
Existing data in heavily used, centralized databases will become outdated
If updates to sequences contained within centralized databases are made to a local page, but not also made to corresponding records in a central database, the newer data will not reach the wider research community and much of its impact will be lost.
Submission of sequence data to a centralized repository solves these problems
Centralized databases, such as GenBank and the other members of the INSDC, ensure stable access to sequence data by providing versioned releases available by FTP, Web interfaces to a uniform data set and archival redundancy. Combining new data with that of other researchers worldwide within a central database provides a broad biological context that stimulates discovery—keeping each sequence up to date magnifies the utility of all the sequences in the database.
RETRIEVING GENBANK DATA
The Entrez system
The sequence records in GenBank are accessible via Entrez (www.ncbi.nlm.nih.gov/sites/gquery), a flexible database retrieval system that covers 35 biological databases. Entrez databases contain DNA and protein sequences derived from GenBank and other sources, genome maps, population, phylogenetic and environmental sequence sets, gene expression data, the NCBI taxonomy, protein domain information and protein structures from the Molecular Modeling Database, MMDB (10). Each database is linked to the scientific literature via PubMed and PubMed Central.
Associating sequence records with sequencing projects
The ability to identify all GenBank records submitted by a specific group or those with a particular focus, such as metagenomic surveys, is essential for the analysis of large volumes of sequence data. The use of organism or submitter names as a means to define such a set of sequences is unreliable. The Genome Project Database, developed at NCBI and subsequently adopted across the INSDC, allows sequencing centers to register projects under a unique project identifier, enabling reliable linkage between sequencing projects and the data they produce.
A new ‘PROJECT’ line appearing in GenBank flat files identifies the sequencing projects with which a GenBank sequence record is associated. The PROJECT line may contain multiple identifiers of the form ‘type’ and ‘value’, respectively, separated by a semicolon. As an example, the PROJECT line below associates a GenBank sequence record with Genome Project (www.ncbi.nlm.nih.gov/sites/entrez?db=genomeprj) record ‘18787’.
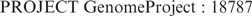
Genome Project record ‘18787’ provides details of the progress made in the effort to sequence Anolis carolinensis (the green anole) (www.broad.mit.edu/models/anole/). Within the Entrez system, such a sequence record is linked directly to the appropriate Genome Project record; conversely, Genome Project records link back to associated sequence records.
BLAST sequence-similarity searching
Sequence-similarity searches are the most fundamental and frequent type of analysis performed on the GenBank data. NCBI offers the BLAST (www.ncbi.nlm.nih.gov/BLAST/) family of programs to detect similarities between a query sequence and database sequences (11,12). BLAST searches may be performed on NCBI's Web site (13), or via a set of standalone programs distributed by FTP. BLAST is discussed in a separate article in this issue (4).
Obtaining GenBank by FTP
NCBI distributes GenBank releases in the traditional flat file format as well as in the ASN.1 format used for internal maintenance. The full bimonthly GenBank release and the daily updates, which also incorporate sequence data from EMBL and DDBJ, are available by anonymous FTP from NCBI at (ftp.ncbi.nih.gov) or (www.ncbi.nlm.nih.gov/Ftp/) as well as from a mirror site at the University of Indiana (ftp://bio-mirror.net/biomirror/genbank/). The full release in flat file format is available as compressed files in the directory, ‘genbank’ with a non-cumulative set of updates contained in ‘daily-nc’. A script is provided in the ‘tools’ directory of the GenBank FTP site to convert a set of daily updates into a cumulative update.
MAILING ADDRESS
GenBank, National Center for Biotechnology Information, Building 38A, Room 3N-301-B, 8600 Rockville Pike, Bethesda, MD 20894, USA.+1 301 496 2475 +1 301 480 9241.
ELECTRONIC ADDRESSES
info@ncbi.nlm.nih.gov NCBI Home Page.
gb-sub@ncbi.nlm.nih.gov Submission of sequence data to GenBank.
update@ncbi.nlm.nih.gov Revisions to, or notification of release of ‘confidential’ GenBank entries.
info@ncbi.nlm.nih.gov General information about NCBI and services.
CITING GENBANK
If you use the GenBank database in your published research, we ask that this article be cited.
ACKNOWLEDGEMENTS
Funding to pay the Open Access publication charges for this article was provided by the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2007;35(Database issue):21–25. doi: 10.1093/nar/gkl986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kulikova T, Akhtar R, Aldebert P, Althorpe N, Andersson M, Baldwin A, Bates K, Bhattacharyya S, Bower L, et al. EMBL Nucleotide Sequence Database in 2006. Nucleic Acids Res. 2007;35(Database issue):16–20. doi: 10.1093/nar/gkl913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugawara H, Abe T, Gojobori T, Tateno Y. DDBJ working on evaluation and classification of bacterial genes in INSDC. Nucleic Acids Res. 2007;35(Database issue):13–15. doi: 10.1093/nar/gkl908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkm1000. This issue (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boguski MS, Lowe TM, Tolstoshev CM. dbEST – database for ‘expressed sequence tags’. Nat. Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 6.Smith MW, Holmsen AL, Wei YH, Peterson M, Evans GA. Genomic sequence sampling: a strategy for high resolution sequence-based physical mapping of complex genomes. Nat. Genet. 1994;7:40–47. doi: 10.1038/ng0594-40. [DOI] [PubMed] [Google Scholar]
- 7.Kans J, Ouellette B. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins chapter Submitting DNA Sequences to the Databases. New York, NY: John Wiley and Sons, Inc.; 2001. pp. 65–81. [Google Scholar]
- 8.Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409:685–690. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- 9.Koehler W. Web page change and persistence – a four-year longitudinal study. J. Am. Soc. Inf. Sci. Technol. 2002;53:162–171. [Google Scholar]
- 10.Wang Y, Addess KJ, Chen J, Geer LY, He J, He S, Lu S, Madej T, Marchler-Bauer A, et al. MMDB: annotating protein sequences with Entrez's 3D-structure database. Nucleic Acids Res. 2007;35(Database issue):298–300. doi: 10.1093/nar/gkl952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Schäffer AA, Miller W, Madden TL, Lipman DJ, Koonin EV, Altschul SF. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res. 1998;26:3986–3990. doi: 10.1093/nar/26.17.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34(Web Server issue):6–9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]