Conditional Lethality, Division Defects, Membrane Involution, and Endocytosis in mre and mrd Shape Mutants of Escherichia coli (original) (raw)
Abstract
Maintenance of rod shape in Escherichia coli requires the shape proteins MreB, MreC, MreD, MrdA (PBP2), and MrdB (RodA). How loss of the Mre proteins affects E. coli viability has been unclear. We generated Mre and Mrd depletion strains under conditions that minimize selective pressure for undefined suppressors and found their phenotypes to be very similar. Cells lacking one or more of the five proteins were fully viable and propagated as small spheres under conditions of slow mass increase but formed large nondividing spheroids with noncanonical FtsZ assembly patterns at higher mass doubling rates. Extra FtsZ was sufficient to suppress lethality in each case, allowing cells to propagate as small spheres under any condition. The failure of each unsuppressed mutant to divide under nonpermissive conditions correlated with the presence of elaborate intracytoplasmic membrane-bound compartments, including vesicles/vacuoles and more-complex systems. Many, if not all, of these compartments formed by FtsZ-independent involution of the cytoplasmic membrane (CM) rather than de novo. Remarkably, while some of the compartments were still continuous with the CM and the periplasm, many were topologically separate, indicating they had been released into the cytoplasm by an endocytic-like membrane fission event. Notably, cells failed to adjust the rate of phospholipid synthesis to their new surface requirements upon depletion of MreBCD, providing a rationale for the “excess” membrane in the resulting spheroids. Both FtsZ and MinD readily assembled on intracytoplasmic membrane surfaces, and we propose that this contributes significantly to the lethal division block seen in all shape mutants under nonpermissive conditions.
As in many other bacterial species, maintenance of cell shape in Escherichia coli requires an intact peptidoglycan (murein) layer of the envelope and, at least, five well-conserved proteins (PBP2, RodA, MreB and -C, and D) that are each required to prevent a cell shape conversion from rod to prolate spheroid or sphere (for reviews, see references 9, 13, 14, 40, 55, and 90).
PBP2 and RodA are encoded by the mrdA (pbpA) and mrdB (rodA) genes residing in the mrd (murein D) operon (75, 76). Penicillin binding protein 2 (PBP2) is a bitopic integral cytoplasmic membrane (CM) species with a large periplasmic domain that possesses murein dd-transpeptidase activity, binds β-lactams, and has a particularly high affinity for the amidino-penicillin amdinocillin (mecillinam) (FL1060) (42, 74). PBP2 is unique among the PBPs in E. coli in that it is specifically required for cylindrical murein synthesis during cell elongation but dispensable for septal murein synthesis during cell constriction. Its counterpart, PBP3 (FtsI), is similarly unique in that it is specifically required for septal murein synthesis but dispensable for cylindrical murein synthesis (28, 40, 74). RodA belongs to a family of polytopic membrane proteins which also includes the division protein FtsW (39, 56), and evidence suggests that RodA and FtsW are required for proper functioning of PBP2 and PBP3, respectively (31, 40-42, 58).
MreB, -C, and -D are encoded by the mre operon. MreB is the sole known bacterial actin (79) in E. coli. As in Bacillus subtilis (26, 46, 73) and Caulobacter crescentus (32, 35), MreB localizes just underneath the CM in a spiral/banded-like pattern along the length of the cell (50, 71) and is implicated in both shape maintenance and chromosome segregation (48, 85). MreC is a bitopic CM protein and MreD a polytopic one (49, 53, 84). A crystal structure of the large periplasmic domain of Listeria monocytogenes MreC revealed a dimer with some structural similarities to chymotrypsins, though it is unlikely to be a protease (80). Affinity purification and bacterial two-hybrid analyses indicate that MreC interacts with MreD as well as with several of the high-molecular-weight murein synthases (PBPs), including the PBP2 homologues in C. crescentus and B. subtilis (29, 49, 80). An interaction between MreC and PBP2 is further supported by colocalization experiments with C. crescentus and Rhodobacter sphaeroides cells (30, 72). Like MreB, PBP2, MreC, and MreD appear to accumulate in a spotty or helical fashion along the cell envelope in E. coli, B. subtilis, and/or C. crescentus (27, 29, 30, 32, 52), and these localization patterns are reminiscent of those of new murein insertion in the cylindrical portion of B. subtilis cells (22, 78). These and other observations support models in which the helical organization of bacterial actins in the cytoplasm topologically constrain murein synthase and/or hydrolase activities in the periplasm, resulting in growth of the sacculus as a cylinder in between periods of cell constriction (15, 22, 32). How the location of MreB polymers in the cytoplasm is coupled to that of (mostly) periplasmic MreC/PBP complexes is unclear. Coupling could be quite direct in E. coli, as suggested by an MreC-MreB interaction in a bacterial two-hybrid assay (49), but this is probably not the case in C. crescentus and R. sphaeroides (30, 72).
How loss of rod shape affects the ability of E. coli to propagate has been a confusing issue. Loss of the Mre proteins has variously been reported to yield stably propagating spheres (50, 61, 70, 84) or to be lethal (49, 86), unless cells are supplied with extra copies of ftsQAZ (49). Inactivation of PBP2 and/or RodA, by treatment of wild-type (wt) cells with amdinocillin or by mutation of mrdA or mrdB, typically results in the formation of giant nondividing spheroids/spheres, which eventually lyse and die (5, 62, 76, 82). However, PBP2− cells were found to stably propagate as smaller dividing spheres under several conditions, including (i) simultaneous increases in the essential division proteins FtsQ, -A, and -Z (59, 82); (ii) a low growth rate (4, 47); and/or (iii) an increase in the level of the alarmone ppGpp above a certain threshold (12, 47, 81). RodA− spheres were similarly reported to survive on poor medium or upon overexpression of ftsQAZ. (5, 28, 82).
Taking care not to select for secondary suppressing alterations, we created sets of mre and mrd mutants in two genetic backgrounds and compared their properties. Our results indicate that unsuppressed cells lacking either of the Mre proteins behave very similarly to those lacking PBP2 and/or RodA. Thus, like mrd cells (28, 47, 82), mre cells were conditionally viable in that they propagated stably as small dividing spheres at low growth rates on poor media but formed giant nondividing spheroids at higher growth rates. The lethality of mre cells at higher growth rates could be partially suppressed by a supply of an overactive form of (p)ppGpp synthase (RelA′). In addition, we found that increased expression of just FtsZ was sufficient to suppress the lethality of both mre and mrd mutants on rich medium.
One striking feature, common to all unsuppressed shape mutants under nonpermissive conditions, was the extensive invasion and elaboration of the CM into the spheroid's cytoplasm. Some of these elaborations were continuous with the exterior CM, while others were topologically separate. Several lines of evidence indicate that the latter derived from the exterior CM by endocytic-like membrane fission events that release periplasm-filled vesicles in the cytoplasm. Interestingly, MreBCD-depleted spheres synthesized phospholipid at about the same rate per unit of cell mass as wt rods, providing a rationale for the “excess” membrane in their interiors. This failure to properly adjust membrane lipid synthesis to actual surface requirements under nonpermissive conditions is likely to contribute to the accompanying lethal division defect in the shape mutants. Both FtsZ and MinD assembled aberrantly on internal membrane systems, suggesting that the latter directly interferes with proper assembly of a division apparatus on external segments of the CM by diverting significant fractions of division proteins into nonproductive assemblies.
MATERIALS AND METHODS
E. coli plasmids and phages.
The most relevant plasmids, phages, and strains used in this study are listed in Table 1 and depicted in Fig. 1.
TABLE 1.
E. coli strains, plasmids, and phages used in this study
| Strain or plasmid | Relevant genotypeb | ori | Source or reference |
|---|---|---|---|
| Strainsa | |||
| DY329 | _rph_1 IN(_rrnD_-rrnE) Δ(argF_-lac)U169 nadA::Tn_10 gal490 λ_cI_857 Δ(_cro_-bioA) | 91 | |
| PA340 | _argH1 thr_-_1 leuB_6 _ghd_-_1 gltB_3_1 thi_-_1 lacY1 gal_-6 _xyl_-7 _ara_-_14 mtl_-2 malA1 rpsL9 tonA2 | 85 | |
| PA340-129 | PA340, _hisG1 mreB_-129 | 85 | |
| PA340-678 | PA340, _hisG1 mre_-_678_[Δ(_mreB_-rng)] | 85 | |
| PB103 | dadR trpE trpA tna | 25 | |
| FB2* | PB103, mreB<>aph | This work | |
| FB2supc | PB103, mreB<>aph sup? | This work | |
| FB9* | PB103, mreB<>frt | This work | |
| FB10* | PB103, mreC<>aph | This work | |
| FB11* | PB103, mreD<>aph | This work | |
| FB12* | PB103, mreBCD<>aph | This work | |
| FB13* | PB103, mreBC<>aph | This work | |
| FB14* | PB103, mreCD<>aph | This work | |
| FB15* | PB103, mreC<>frt | This work | |
| FB16* | PB103, mreD<>frt | This work | |
| FB17* | PB103, mreBCD<>frt | This work | |
| FB18* | PB103, mreBC<>frt | This work | |
| FB19* | PB103, mreCD<>frt | This work | |
| FB37* | PB103, mrdAB<>aph | This work | |
| FB39* | PB103, mrdAB<>frt | This work | |
| MG1655 | ilvG rfb50 rph1 | 36 | |
| TB12 | MG1655, lacIZYA<>aph | 8 | |
| TB28 | MG1655, lacIZYA<>frt | 8 | |
| FB21* | TB28, mreB<>aph | This work | |
| FB22* | TB28, mrdB<>aph | This work | |
| FB30* | TB28, mreBCD<>aph | This work | |
| FB38* | TB28, mrdAB<>aph | This work | |
| FB40* | TB28, mrdAB<>frt | This work | |
| Plasmids | |||
| pCH235 | bla lacI_q P_lac::_mreD_-LE | ColE1 | This work |
| pCH244 | bla lacI_q P_lac::mreB mreC mreD yhdE | ColE1 | This work |
| pCX16 | aadA sdiA | pSC101 | 87 |
| pDR3 | bla lacI_q P_lac::ftsZ | ColE1 | 7 |
| pDR144 | bla lacI_q P_lac::sfiA | ColE1 | 7 |
| pFB112 | tet sdiA | ColE1 | This work |
| pFB118 | bla lacI_q P_lac::mreB | ColE1 | This work |
| pFB120 | bla lacI_q P_lac::_mreC_-LE | ColE1 | This work |
| pFB121 | bla lacI_q P_lac::_mreC mreD_-LE | ColE1 | This work |
| pFB124 | _aadA c_I857(Ts) PλR::_mreC mreD_-LE | pSC101 | This work |
| pFB128 | _aadA c_I857(Ts) PλR::_mreD_-LE | pSC101 | This work |
| pFB142 | bla lacI_q P_lac::_mreB mreC_-LE | ColE1 | This work |
| pFB149 | bla lacI_q P_lac::_mreB mreC mreD_-LE | ColE1 | This work |
| pFB174 | cat araC PBAD::_mreB mreC mreD_-LE | pACYC | This work |
| pFB194 | _aadA c_I857(Ts) PλR::mrdB(rodA) | pSC101 | This work |
| pGB2 | aadA | pSC101 | 18 |
| pMLB1113 | bla lacI_q P_lac::lacZ | ColE1 | 24 |
| pTB6 | bla lacI_q P_lac::ss_torA_-_gfp_-T7 | ColE1 | 6 |
| pTB63 | tet ftsQ ftsA ftsZ | pSC101 | 6 |
| pTB182 | aadA ftsQ ftsA ftsZ | pSC101 | This work |
| pTB188 | aadA PλR::ftsZ | pSC101 | This work |
| pYT11 | bla lacI_q P_tac::relA(1-454)-LE | ColE1 | This work |
| Phages | |||
| λCH178 | imm_21_bla lacI_q P_lac::zipA(1-183)-gfp | λ | 45 |
| λCH221 | imm_21_bla lacI_q P_lac::_gfp_-T7-mrdB | λ | This work |
| λCH235 | imm_21_bla lacI_q P_lac::_mreD_-LE | λ | This work |
| λCH268 | imm_21_bla lacI_q P_lac::_gfp_-T7-zapA | λ | This work |
| λDR122 | imm_21_bla lacI_q P_lac::_gfp_-T7-minD minE | λ | 67 |
| λFB120 | imm_21_bla lacI_q P_lac::_mreC_-LE | λ | This work |
| λFB185 | imm_21_bla lacI_q P_lac::mrdB(rodA) | λ | This work |
| λFB190 | imm_21_bla lacI_q P_lac::mrdA(pbpA) | λ | This work |
| λTB59 | imm_21_bla lacI_q P_lac::mrdA mrdB | λ | This work |
FIG. 1.

Genetic constructs. Shown are the E. coli mre (A) and mrd (B) loci, chromosomal deletion-replacements, and inserts present on plasmids and phages. Portions of the chromosome that were replaced with an aph cassette, or an frt scar sequence remaining after eviction of the cassette, are indicated by brackets. Numbers next to brackets refer to the base pairs replaced, counting from the start of mreB (A) or mrdA (B). Inserts were placed downstream of PλR (pFB124, pFB128, and pFB194), PBAD (pFB174), or P_lac_ (all others) control regions of appropriate plasmid/phage vectors (Table 1). LE*, in-frame CTCGAGTAA sequence appended to the end of the gene; M, start codon of mreD changed from GTG to ATG.
Plasmids pBR322 (11), pZAQ (88), pJF118EH (33), pMEL1 (85), pMLB1113 (24), pCX19 (87), pCP20 (16), pDB346 (66), pDR107 (67), pDR120 (37), pKD13 (23), pCH151 (8), pCH157 (51), and pTB59 (6) were described previously.
Unless indicated otherwise, MG1655 chromosomal DNA was used as a template in amplification reactions. Sites of interest (e.g., relevant restriction sites) are underlined in primer sequences.
To construct pCH221 (P_lac_::_gfpmut2_-T7-mrdB), mrdB (rodA) was amplified using primers 5′-TATAGAATTCATATGACGGATAATCCGAATAAAAAAACATTCTGG-3′ and 5′-CATTGTCGACTTACACGCTTTTCGACAACATTTTCC-3′. The product was treated with EcoRI and SalI, and the 1,127-bp fragment was used to replace the 13-bp EcoRI-SalI fragment of pDR107c, yielding pCH218 (PT7::_gfpmut2_-T7-mrdB). The 1,970-bp BglII-HindIII fragment of pCH218 was next used to replace the 20-bp BamHI-HindIII fragment of pMLB1113, resulting in pCH221.
For pCH222 [P_lac_::_mrdB_-_gfpmut2_], mrdB was amplified using primers 5′-TATAGAATTCATATGACGGATAATCCGAATAAAAAAACATTCTGG-3′ and 5′-GCACCTCGAGCACGCTTTTCGACAACATTTTCC-3′. The product was treated with NdeI and XhoI, and the 1,119-bp fragment was used to replace the 77-bp NdeI-XhoI fragment of pET21a, yielding pCH219 (PT7::mrdB_-His6). The 1,152-bp XbaI-XhoI fragment of pCH219 was used to replace the 1,025-bp XbaI-XhoI fragment of pCH151 (P_lac::_zipA_-gfpmut2), resulting in pCH222.
Construction of plasmid pCH235 (P_lac_::mreD_-LE) involved several steps. The annealed product of oligonucleotides 5′-TCGAGTAAGTCGACACGGTACCA-3′ (sense) and 5′-AGCTTGGTACCGTGTCGACTTAC-3′ (antisense) was used to replace the 122-bp XhoI-HindIII fragment of pCH157 (P_lac::_gfpmut2_-T7-minD minE_-His6). This resulted in pCH181 (P_lac::_gfpmut2_-T7-_minD minE_-LE), in which the His6 tag sequence in pCH157 was replaced with an XhoI site, encoding the dipeptide LE, followed by the TAA stop codon. The mreD gene was amplified using primers 5′-TATAGAATTCATATGGCGAGCTATCGTAGCCAGGGACGCTG-3′ and 5′-CGTTCTCGAGTTGCACTGCAAACTGCTGACGGAC-3′ and digested with EcoRI and XhoI. The 494-bp fragment was used to replace the 34-bp EcoRI-XhoI fragment of pDR107c, resulting in pCH217 (PT7::_gfpmut2_-T7-_mreD_-His6). Circularization of the 5,837-bp NdeI fragment of pCH217 yielded pCH223 (PT7::_mreD_-His6). pCH235 was finally obtained by replacing the 1,859-bp XbaI-XhoI fragment of pCH181 with the 512-bp XbaI-XhoI mreD fragment of pCH223.
Plasmid pCH244 (P_lac_::mreB mreC mreD yhdE) was obtained after several steps. The 3,359-bp ApoI fragment of pMEL1 was inserted in the EcoRI site of pDR107a, yielding pDB364 [PT7::_gfpmut2_-T7-mreB(5-347) mreC mreD yhdE_]. The 4,223-bp BglII-HindIII fragment of pDB364 was next used to replace the 20-bp BamHI-HindIII fragment of pMLB1113, resulting in pDB366 [P_lac::_gfpmut2_-T7-mreB(5-347) mreC mreD yhdE_]. The mreB gene was amplified using primers 5′-CGACTCTAGACAGCTTTCAGGATTATCCCTTAGTATG-3′ and 5′-GCAAAAGCTTACTCTTCGCTGAACAGGTCGCC-3′. The product was treated with XbaI and HindIII, and the 1,072-bp fragment ligated to the 7,639-bp XbaI-HindIII fragment of pCH151 (P_lac::zipA_-gfpmut2), generating pCH214 (P_lac::mreB). Finally, replacement of the 534-bp KpnI-HindIII fragment of pCH214 with the 2,879-bp KpnI-HindIII fragment of pDB366 resulted in pCH244.
To obtain pCH268 (P_lac_::gfpmut2_-T7-zapA), zapA was amplified using primers 5′-GAAGGATCCATGTCTGCACAACCCGTC-3′ and 5′-CGAGTCGACTCATTCAAAGTTTTGGTTAG-3′. The product was treated with BamHI and SalI, and the 336-bp fragment was used to replace the 1,164-bp BamHI-SalI fragment of pDR120 (P_lac::_gfpmut2_-T7-ftsZ).
For plasmid pFB112 (tet sdiA), the 1,312-bp EcoRI-PstI fragment of pCX19 was ligated to the 3,615-bp EcoRI-PstI fragment of pBR322.
For pFB118 (P_lac_::mreB), the 2,696-bp ClaI fragment of pCH244 was deleted.
To obtain pFB120 (P_lac_::_mreC_-LE), mreC was amplified using primers 5′-CTAGTCTAGAATACGAGAATACGCATAACTT-3′ and 5′-CGTTCTCGAGTTGCCCTCCCGGCGCACGCGCAGGC-3′. The product was treated with XbaI and XhoI, and the 1,128-bp fragment was used to replace the 512-bp XbaI-XhoI fragment of pCH235.
For pFB121 (P_lac_::_mreC mreD_-LE), an mreCD fragment was amplified using primers 5′-CTAGTCTAGAATACGAGAATACGCATAACTT-3′ and 5′-CGTTCTCGAGTTGCACTGCAAACTGCTGACGGAC-3′. The product was treated with XbaI and XhoI, and the 1,616-bp fragment was used to replace the 512-bp XbaI-XhoI fragment of pCH235.
Plasmid pFB124 [_c_I857(Ts) PλR::_mreC, mreD_-LE] was obtained by replacing the 1,196-bp XbaI-SalI fragment of pDB346 [_c_I857(Ts) PλR::_ftsZ_] with the 1,625-bp XbaI-SalI fragment of pFB121.
In turn, pFB128 [_c_I857(Ts) PλR::_mreD_-LE] was created by replacing the 1,625-bp XbaI-SalI fragment of pFB124 with the 521-bp XbaI-SalI fragment of pCH235.
Plasmid pFB142 (P_lac_::mreB, mreC_-LE) was created in two steps. The 1,271-bp XbaI-XhoI fragment of pCH217 was used to replace the 1,859-bp XbaI-XhoI fragment of pCH181, yielding pCH233 (P_lac::_gfpmut2_-T7-_mreD_-LE). An mreBC fragment was amplified using primers 5′-CGACTCTAGACAGCTTTCAGGATTATCCCTTAGTATG-3′ and 5′-CGTTCTCGAGTTGCCCTCCCGGCGCACGCGCAGGC-3′. The product was treated with XbaI and XhoI, and the 2,240-bp fragment was used to replace the 1,271-bp XbaI-XhoI fragment of pCH233.
For pFB149 (P_lac_::_mreB mreC mreD_-LE), the 1,033-bp BamHI-SalI fragment of pCH244 was replaced with the 359-bp BamHI-SalI fragment of pFB124.
To create pFB174 (PBAD::_mreB mreC mreD_-LE), the 1,451-bp XbaI-HindIII fragment of pLL116 (a pBAD33 derivative that will be described elsewhere) was replaced with the 2,743-bp XbaI-HindIII fragment of pFB149.
For pFB185 (P_lac_::mrdB), the 508-bp NsiI-HindIII fragment of pCH221 was used to replace the 1,252-bp NsiI-HindIII fragment of pCH222.
To construct pFB190 (P_lac_::mrdA), pTB59 (P_lac_::mrdAB) was used as a template to amplify mrdA (pbpA) with primers 5′-CTCTGAATTCCCGTGAGTGATAAGGGAGCTTTGAGTAG-3′ and 5′-GCCAAGCTTGGTCGACTTAATGGTCCTCCGCTGCGGC-3′. The product was treated with EcoRI and HindIII, and the 1,954-bp fragment was used to replace the 3,084-bp EcoRI-HindIII fragment of pTB59.
For pFB194 [_c_I857(Ts) PλR::_mrdB_], the 1,155-bp XbaI-SalI fragment of pFB185 was used to replace the 1,625-bp XbaI-SalI fragment of pFB124.
Plasmid pTB182 (ftsQAZ) was obtained in several steps. The HindIII site within ftsA on pZAQ was removed by the QuikChange procedure (Stratagene), using the mutagenic primers 5′-CAGTTGCAGGAAAAGCTCCGCCAACAAGGGG-3′ and its reverse complement, resulting in a silent change (underlined) of FtsA codon 319 (Leu). The resulting plasmid (pTB178) was next mutagenized using primers 5′-TTATGAGGCCGACGATCTAGACGGCCTCAGGCGACAG-3′ and its reverse complement, creating an XbaI site in between ftsA and ftsZ. The 4,377-bp PstI-HindIII fragment of the resulting plasmid (pTB179) was then used to replace the 12-bp PstI-HindIII fragment of pGB2, yielding pTB182. The direction of ftsQAZ transcription from this plasmid is opposite that of the aadA gene.
For pTB188 (PλR::ftsZ), pDB346 [_c_I857(Ts) PλR::_ftsZ_] was used as a template in a PCR with 5′-CGTAGGATCCGCATGCGGGATAAATATCTAACACCGTGCGTG-3′ and 5′-GCTCAAGCTTGTCGACTTAATCAGCTTGCTTACGCAGGAATG-3′. The product was treated with BamHI and HindIII, and the 1,359-bp fragment was used to replace the 20-bp BamHI-HindIII fragment of pGB2, yielding pTB188. Note that this plasmid lacks a lambda repressor and that ftsZ is constitutively transcribed in the direction opposite that of aadA.
For pYT11 (P_tac_::_relA_′), a portion of relA was amplified with primers 5′-CTTTTCTAGATTTCGGCAGGTCTGGTCCCTAAAGG-3′ and 5′-GGTCCTCGAGCTGGTAGGTGAACGGCACAATGCGCCC-3′. The product was treated with XbaI and XhoI, and the 1,401-bp fragment was used to replace an XbaI-XhoI fragment of pCH276, a plasmid whose construction will be detailed elsewhere. The 1,500-bp EcoRI-HindIII fragment of the resulting plasmid (pYT5) was next used to replace the 30-bp EcoRI-HindIII fragment of pJF118EH, yielding pYT11. The plasmid encodes the first 455 residues of RelA, followed by a glutamic acid residue and a stop codon.
Phages λCH221, λCH235, λCH268, λFB120, λFB185, λFB190, and λTB59 were obtained by crossing λNT5 with pCH221, pCH235, pCH268, pFB120, pFB185, pFB190, and pTB59, respectively, as described previously (24).
E. coli strains.
mre knockout strains were constructed by λ red recombineering, using pKD13 as a template for amplification of an aph cassette consisting of aph flanked by FLP recombinase substrate sites (frt) and appropriate mre sequences (23, 91). Knockout alleles on linear fragments were recombined with the chromosome of strain DY329 carrying plasmid pCX16 [_sdiA_]. With plating under standard conditions (LB-kanamycin [Kan] at 30°C), the number of recombinants recovered in the presence of pCX16 was, at least, 2 to 3 logs higher than in its absence.
We used the following primer sets (chromosomal sequences are underlined): for mreB<>aph, 5′-GACCTGGGTACTGCGAATACCCTCATTTATGTAAAAGGACAAGGCATCGTGTGTAGGCTGGAGCTGCTTC-3′ [primer mreB(KO)5′] and 5′-AGCCATCGGTTCTTCAATCAGGAAGACTTCACGGGCACCAGCGCCCTGCGATTCCGGGGATCCGTCGACC-3′ [mreB(KO)3′]; for mreC<>aph, 5′-ATCGGATGCAGGCAGGGGAAGTGTCTGTTTACCCTGCCTGGTCTGATACGATAAGTGTAGGCTGGAGCTGCTTC-3′ [mreC(KO)5′] and 5′-AGCGATCCCCGTTGCCGGTTCAGGTAACTTTGGCCCCATCGCGTCTGGCGAATTCCGGGGATCCGTCGACC-3′ [mreC(KO)3′]; for mreD<>aph, 5′-GTGGCGAGCTATCGTAGCCAGGGACGCTGGGTAATCTGGCTCTCTTTCCTCTAAGTGTAGGCTGGAGCTGCTTC-3′ [mreD(KO)5′] and 5′-TCAGCAAGAAAATCCACGGCCAGAGCACCCCATTGACTACACTACTCCAGAATTCCGGGGATCCGTCGACC-3′ [mreD(KO)3′]; for mreBCD<>aph, primers mreB(KO)5′ and mreD(KO)3′; for mreBC<>aph, primers mreB(KO)5′ and mreC(KO)3′; and for mreCD<>aph, primers mreC(KO)5′ and mreD(KO)3′.
Recombination yielded a set of six mre<>aph derivatives of DY329/pCX16, which all showed a spherical phenotype. The six strains were transformed with pCH244 (P_lac_::mreB mreC mreD yhdE), and transformants of each reverted to a rod shape in an IPTG (isopropyl-β-d-thiogalactopyranoside)-dependent manner. Phage P1 was grown on a transformant (containing both pCX16 and pCH244) of each strain in the presence of 250 μM IPTG, resulting in a high-titer transducing lysate for each mre<>aph allele. These lysates were then used to transduce Mre+ strains, PB103, or TB28, using various strategies to avoid selective pressure for the accumulation of undesired suppressor mutations. Generally, this was accomplished by the introduction of appropriate correcting or suppressing mre, sdiA, or ftsZ plasmids or phages into the Mre+ recipient before introduction of a chromosomal mre<>aph allele by transduction. For example, to obtain the MreBCD depletion strain FB30/pFB174 (mreBCD<>aph/cat araC PBAD::mreBCD), TB28 was transformed with pFB174 prior to transduction of mreBCD<>aph and transductants were recovered at 30°C on LB-Kan supplemented with chloramphenicol and 0.5% arabinose.
Similarly, derivatives of PB103 carrying chromosomal mre<>frt alleles (Table 1; also see Table S1 in the supplemental material) were obtained by introduction of pFB112 (tet sdiA) prior to transduction with the corresponding mre<>aph lysates. The resulting mre<>aph/pFB112 strains were then transformed with pCP20 [bla cat repA(Ts) _c_I857(Ts) PλR::flp_] (16, 23) and plated at 30°C on LB containing ampicillin (Amp) and tetracycline. Transformants were streaked on LB lacking Amp and incubated at 42°C to simultaneously induce production of Flp recombinase and block replication of pCP20. Kan- and Amp-sensitive clones were purified, resulting in the desired mre<>frt/pFB112 strains. These strains were transformed with appropriate mre plasmids and used for complementation analyses (see Table S1 in the supplemental material). The growth of some of these transformants at 37°C and in the presence of IPTG led to simultaneous correction of the rod phenotype and competitive loss of pFB112 (see Table S1 in the supplemental material), giving rise to depletion strains that lacked extra copies of sdiA, such as the MreB depletion strain FB17/pFB118/pFB124 (mreBCD<>frt/P_lac::mreB/PλR::mreCD).
For construction of mrd mutants, we used the following primer sets (chromosomal sequences are underlined): for mrdAB<>aph, 5′-CATCCTTATCACCGTGAGTGATAAGGGAGCTTTGAGTAGAAAACGCAGCGGGTGTAGGCTGGAGCTGCTTC-3′ [pbp2(KO)5′] and 5′-CGCCAGCCATGACGCGACCAAAGGTGGTTTGCGCTCTGGCGGCTATCCATTCCGGGGATCCGTCGACC-3′ [rodA(KO)3′]; and for mrdB<>aph, 5′-CGATCTGCCTGCGGAAAATCCAGCGGTTGCCGCAGCGGAGGACCATTAAGTGTAGGCTGGAGCTGCTTC-3′ [rodA(KO)5′ and rodA(KO)3′].
Recombination with the chromosome of DY329/pCX16 resulted in FB29/pCX16 (mrdAB<>aph/sdiA) and FB20/pCX16 (mrdB<>aph/sdiA), which propagated as spheres. These strains were transformed with pTB59 (P_lac_::mrdAB), which caused transformants to revert back to a rod shape in the presence of IPTG. P1 lysates were prepared on FB29/pCX16/pTB59 and FB20/pCX16/pTB59 transformants, and these were used to transduce mrdAB<>aph and mrdB<>aph into PB103 or TB28 derivatives carrying appropriate complementing plasmids and/or phages.
For the P1 transduction experiments whose results are shown in Tables 3 and 5, we used the mre<>aph and mrd<>aph lysates described above, except for the mrdB<>aph and lacIZYA<>aph transducing lysates (Table 3), which were prepared on strains FB22(λCH221) and TB12, respectively.
TABLE 3.
Suppression of Mre− and MrdB− lethality by multiple copies of sdiA
| P1 lysatea | No. of colonies for: | |
|---|---|---|
| PB103/pGB2 (wt/vector) | PB103/pCX16 (wt/sdiA) | |
| lacIZYA<>aph | 326 | 496 |
| mreBCD<>aph | 1 | 149 |
| mreB<>aph | 1b | 133 |
| mreC<>aph | 1 | 123 |
| mreD<>aph | 0 | 110 |
| mrdB<>aph | 0 | 77 |
TABLE 5.
Recovery of mreBCD<>aph and mrdAB<>aph transductants on minimal mediuma
| P1 lysate and strain | Relevant genotype | No. of colonies for indicated temp (presence of IPTG) | ||||
|---|---|---|---|---|---|---|
| 20°C (−) | 30°C (−) | 30°C (+) | 37°C (−) | 37°C (+) | ||
| mreBCD<>aph | ||||||
| PB103 | wt | ND | 103 | ND | 4 | ND |
| PB103/pFB149 | wt/P_lac_::mreBCD | ND | 134 | 149 | 0 | 87 |
| TB28 | wt | 92 | 0 | ND | ND | ND |
| mrdAB<>aph | ||||||
| PB103 | wt | ND | 96 | ND | 6 | ND |
| PB103(λTB59) | wt(P_lac_::mrdAB) | ND | 112 | 98 | 3 | 28 |
| TB28 | wt | 84 | 0 | ND | ND | ND |
Growth conditions.
Cells were routinely grown in LB (0.5% NaCl) or M9 minimal medium supplemented with 0.2% Casamino Acids, 50 μg/ml l-tryptophan, and 0.2% maltose. Low-phosphate (LP) medium contained 100 μM Tris-Cl (pH 7.5), 10 mM KCl, 17 mM NaCl, 29 mM NH4Cl, 0.3 mM KH2PO4, 2 mM MgSO4, 1 mM CaCl2, 50 μM thiamine, 20 μg/ml l-tryptophan, 0.2% Casamino Acids, and 0.2% maltose or glucose. When appropriate, medium was supplemented with 50 μg/ml Amp, 50 μg/ml spectinomycin, 25 μg/ml Kan, 25 μg/ml chloramphenicol, and/or 12.5 μg/ml tetracycline, except for cells carrying pTB63, in which case 5.0 μg/ml tetracycline was used. Other additions are specified in the text. Unless indicated otherwise, strain DY329 and derivatives were incubated at 30°C and other strains at 37°C.
Immunofluorescence and confocal microscopy.
On-slide immunofluorescence staining methods that work well with rods and filaments (1, 37) did not result in consistent labeling of FtsZ structures in large spheroids. We therefore developed a protocol for immunostaining of cellular structures in which incubations with lysozyme and antibodies are done in solution. Briefly, cells were fixed by adding 1 ml of culture directly to a mixture of formaldehyde and glutaraldehyde in NaPO4 buffer (pH 7.5), giving final concentrations of 2.4%, 0.04%, and 30 mM, respectively. The suspension was incubated for 10 min at room temperature (RT), followed by incubation for 50 min on ice. Cells were washed twice in 1 ml phosphate-buffered saline (PBS) (10.1 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), once in 1 ml GTE (20 mM Tris-Cl, 50 mM glucose,10 mM EDTA, pH 7.5), and resuspended in 0.4 ml of GTE. Aliquots (0.1 ml) of cells were treated with egg white lysozyme from a freshly prepared stock of 0.4 mg/ml in GTE to give a final concentration of 16 μg/ml and incubated for 2 to 4 min at RT. Cells were washed once in 1 ml PBS and resuspended in 1 ml PBS containing 2% bovine serum albumin (BSA). During the subsequent blocking and antibody incubation steps, samples were gently mixed in an end-over-end tube mixer. After 1 h at RT, affinity-purified rabbit polyclonal anti-FtsZ antibodies (37) were added, and incubation was continued overnight at 4°C. Cells were washed once with 1 ml PBS, resuspended in 1 ml PBS containing 2% BSA and a 1:2,500 dilution of Alexa-488 conjugated anti-rabbit immunoglobulin G (Molecular Probes), and incubated for 2 h at RT. Cells were washed twice with 1 ml PBS and resuspended in 50 μl PBS, and aliquots were spotted onto poly-l-lysine-coated coverslips. Confocal microscopy was performed with a Zeiss LSM 510 inverted laser-scanning microscope using a 100× (1.45-numerical-aperture) oil immersion objective. Images were collected using 488-nm excitation light from an argon-krypton laser, a 560-nm dichroic mirror, and a 500- to 550-nm band pass barrier filter. For all images, a z series was collected at 0.2-μm increments. Image processing, including projections and three-dimensional rotations, were performed using LSM 510 software (version 2.5).
Wide-field microscopy.
The cells in Fig. 5 and 7G were imaged on a Leica DM IRE2 microscope outfitted with a CoolSnap HQ camera (Photometrics) and a piezo-driven 100× (1.4-numerical-aperture) oil objective. Optical sections were collected at a 0.2-μm step size and with Cy3- and/or green fluorescent protein (GFP)-specific filter sets. Images were deconvolved through 40 iterations of a blind deconvolution algorithm provided in the Leica AS MDW package. As indicated, either deconvolved individual slices or maximum projections of the deconvolved image stack are shown. All other wide-field images, including time lapse series, were obtained with a Zeiss Axioplan-2 microscope setup as previously described (44). Live cells were imaged using clean but otherwise untreated microscope slides.
FIG. 5.

Aberrant FtsZ assemblies in live MreBCD-depleted spheres. The MreBCD depletion strain FB30(λCH268)/pFB174 [Δ_mreBCD_(P_lac_::_gfp_-zapA)/PBAD::_mreBCD_] was grown at 37°C in M9-maltose medium lacking arabinose and containing 50 μM IPTG. Live cells were imaged early (A and B) (OD600 = 0.2) and later (C and D) (OD600 = 0.4) during depletion of MreBCD. Cells were mixed with FM4-64 (0.5 μg/ml) immediately prior to imaging. Maximum projection GFP (2), FM4-64 (3), and merged (4) as well as corresponding DIC (1) fluorescence images are shown. Panels D5 to D8 show merged fluorescence images of individual optical slices from the top to the bottom of the cell. The arrows in panel C4 highlight some odd-looking GFP-ZapA accumulations. The arrows in panels D1 and D5 point at small vesicle-like bodies that are both visible by DIC and outlined with FM4-64 fluorescence, while the arrowheads point at much larger bodies that failed to be outlined by the dye. The arrow in panel D8 points at a membrane involution near the bottom of the cell. Bar = 2 μm.
FIG. 7.

Topological separation of internal membrane systems from the external cell membrane. Shown are live MreBCD-depleted spheroids of strain FB30/pFB174 (Δ_mreBCD_/PBAD::mreBCD) that were grown at 37°C in LB to an OD600 of 0.2. (A to C) Cells were mixed with FM4-64 and imaged immediately. Panels show DIC (1) and FM4-64 (2) fluorescence images. Arrowheads in panels A and B point to large vesicles that were not outlined by FM4-64, while the arrow in panel C points out a vesicle that was. (D to G) Cells were pulse labeled with FM4-64 for 5 min, 30 min prior to imaging. Shown are both DIC (1) and FM4-64 (2 to 6) fluorescence images. Note that all vesicles visible by DIC are now also outlined by FM4-64 fluorescence. In addition, FM4-64 stains structures not readily resolvable by DIC (e.g., arrows in panels D and F). For the cell in panel G, individual z slices from top to bottom (G2 to G5) as well as a maximum projection image (G6) are shown. Note that FM4-64-stained material is present throughout the interior of the cell. (H and I) CellTrace BTME was added 15 min, and FM1-43 immediately, before imaging. DIC (1), FM1-43 (2), and BTME (3) images are shown. Arrowheads point to material labeled with BTME but not with FM1-43. (J to L) The growth medium was supplemented with LY. Cells were gently washed in prewarmed medium lacking LY and imaged immediately. Many vesicles visible by DIC contained trapped LY (e.g., arrowhead in panel K). Some bodies that appeared as a vesicle by DIC did not retain the dye, suggesting that they were still continuous with the external CM and periplasm (arrow in panel L). Panel J illustrates that cells of the wt parent control (TB28) completely failed to retain the dye. Bar = 2 μm.
Fluorescent dyes (Molecular Probes) were visualized using Cy3-specific (FM4-64 and CellTrace BODIPY TR methyl ester [BTME]) or GFP-specific (FM1-43 and lucifer yellow [LY] CH) filter sets. FM1-43 and FM4-64 were added to live cells immediately before imaging at final concentrations of 0.25 and 0.50 μg/ml, respectively, except for the pulse-labeling experiment whose results are shown in Fig. 7D to G. For the latter, cells were concentrated 20-fold, incubated with FM4-64 (20 μg/ml) for 5 min at 37°C, washed once in prewarmed medium, resuspended in the same to the original cell density, and grown at 37°C for an additional 30 min before imaging. CellTrace BTME was used at 0.25 μM and was added to live cells 15 min before imaging. For Fig. 8A to D, cells were first chemically fixed as described above, washed once in PBS, and resuspended in PBS containing BTME. After 15 min at RT, cells were washed again in PBS and imaged. For LY internalization assays, the dye was included in the growth medium at the time of inoculation at a final concentration of 50 μg/ml. Upon reaching the desired optical density, 0.5 ml of culture was subjected to gentle centrifugation (6,000 × g for 20 s in a microcentrifuge) at RT. The supernatant was carefully removed, and cells were resuspended in 1 ml prewarmed medium by gently pipetting them up and down twice. Cells were pelleted as described above, resuspended in 50 μl prewarmed medium, and imaged immediately.
FIG. 8.

Vesicle formation does not require FtsZ polymerization. Shown are spheroids of strain FB30/pTB63/pDR144 (Δ_mreBCD_/ftsQAZ/P_lac_::sfiA). (A to D) Cells were inoculated to an OD600 of 0.05 in LP maltose containing no (A) or 0.5 mM (B) IPTG and then grown at 37°C to an OD600 of 0.3, fixed, and labeled with BTME. Shown are DIC (1) and BTME (2) fluorescence. One hundred cells of each culture were further analyzed to determine the average lengths of their long and short axes (C) and the percentages of cells containing internal membrane and/or showing signs of constriction (D). Arrowheads in panel B point to examples of vesicles visible by both DIC and BTME fluorescence. (E to F) Cells were diluted to an OD600 of 0.025 in LB supplemented with 0.5 mM IPTG and either no (E) or 50 μg/ml (F) LY and grown at 37°C to an OD600 of 0.2 to 0.3. For panel E, cells were incubated with BTME for 15 min at 37°C and then treated with FM1-43 immediately before imaging live. Shown are DIC (1), FM1-43 (2), and BTME (3) fluorescence. Note the intracytoplasmic membrane stained by BTME that was inaccessible to FM1-43. For panel F, cells were gently washed in prewarmed growth media prior to imaging. Shown are DIC (1) and LY (2) fluorescence images. Note the trapped LY in intracytoplasmic vesicles. Bar = 2 μm.
Phospholipid synthesis assay.
Strain FB21/pFB149 was grown overnight at 37°C in LP glucose medium supplemented with Amp (LP-Amp) and 200 μM IPTG. Cells were washed once in LP-Amp and then used to inoculate two cultures containing 50 ml LP-Amp and either no or 1 mM IPTG to an optical density at 600 nm (OD600) of 0.001. Cultures were shaken at 37°C, and both the increases in optical densities and the changes in cell morphologies were monitored. At the indicated time points (T1 and T2), three aliquots were removed from each culture. A 1.0-ml aliquot was used to chemically fix cells for later determination of average cell dimensions, while a 5.0-ml aliquot was used to prepare a whole-cell extract for determination of total cell protein per ml culture. To measure incorporation of 32Pi in total phospholipid, 0.8 ml of culture was mixed with prewarmed 32Pi (8,000 Ci/mmol) to 50 μCi/ml, and incubation was continued for 15 min at 37°C. Phospholipids were extracted essentially as described previously (10). Briefly, 3 ml of a 1:2 chloroform-methanol mixture was added, and the mixture was vortexed vigorously for 30 s, followed by incubation on ice for 15 min. The mixture was vortexed briefly after sequential addition of 1 ml each of chloroform and water and then subjected to centrifugation at 5,800 × g for 15 min at 4°C. A 1.4-ml aliquot of the lower phase was washed by sequential addition of 1.40 ml methanol and 1.26 ml water, followed by centrifugation as described above. A 0.2-ml aliquot of the lower phase was then mixed with 5.0 ml Econosafe cocktail (RPI), and radioactivity was measured with a Beckman scintillation counter. Values were converted to numbers of disintegrations per minute/μg whole-cell protein.
Cell dimensions and geometrical considerations.
Cell dimensions were measured from differential interference contrast (DIC) images by using Object-Image 2.15 (83). The position of the long axis in spheroids was judged by eye, and the short axis was measured perpendicular to the long one. The volume (V), surface (S), and circumference (C) of a sphere were calculated using _V_s = 4/3π_r_3, _S_s = 4π_r_2, and C_s = 2π_r and those of a rod (capsule) with V_c = 4/3π_r_3 + π_r_2_h, S_c = 4π_r_2 + 2π_rh, and C_c = 2π_r, with r representing radius and h cylinder length. The volume of a prolate spheroid (c > a) was obtained using V_sph = 4/3π_a_2_c, with c representing polar radius and a equatorial radius.
Other methods.
Whole-cell extracts were prepared as described previously (37). Protein concentrations were measured using the noninterfering protein assay (NI; G-Biosciences), with BSA as a standard. Quantitative Western analyses were done essentially as before (45).
RESULTS
Construction of unsuppressed _mre_- and _mrd_-null and depletion strains of E. coli.
To study the physiological relevance of the Mre proteins in E. coli, we used λ red-mediated recombineering to create sets of strains carrying chromosomal mre mutations. Careful construction and maintenance of these strains was prompted by our initial observations, consistent with those by others (49, 70), that propagation of mre mutants under common growth conditions appears to select for progeny that produce an elevated level of the division protein FtsZ. Thus, our first attempts to create mre<>aph lesions (Fig. 1A) in the recombineering strain DY329 [λ_c_I857 Δ(_cro_-bioA)] yielded no or very few recombinants (data not shown), suggesting the lesions caused death. This result was not in agreement with the viability of strains carrying the classical mreB129 or _mre_-678 [Δ(_mreB_-rng)] alleles (85).
A clue to what hindered the construction of mre knockouts came from observing strain PA340-678/pCH244 (Δ_mreBCD_/P_lac_::mreBCD), which carries the classical chromosomal _mre_-678 allele (84) and a complementing plasmid containing mreBCD downstream of the lac promoter (Fig. 1A). Cells grew as spheres in the absence of IPTG and as rods in its presence. In the latter case, however, a small but significant fraction of rods contained polar septa, leading to the production of minicells. Minicells were not observed in strain PA340/pCH244, indicating that their formation by the shape-corrected mutant was a property neither shared by its Mre+ parent nor induced by expression of the mre genes from the plasmid (data not shown). Because cooverexpression of the ftsQ, -A, and -Z genes both suppresses the lethality of mrd lesions (59, 82) and induces minicell formation (88), we hypothesized that similar to what occurs with mrd, (i) the mre genes in E. coli might be essential, explaining our difficulties in obtaining null alleles; (ii) existing mre mutants may have accumulated second-site mutations that lead to an elevated level of FtsQ, FtsA, and/or FtsZ; and (iii) elevated levels of the division proteins restore viability and allow mre mutants to propagate as spheres.
Accordingly, quantitative Western analyses showed that the classical mreB129 and _mre_-678 derivatives of strain PA340 (85) contained about two times more FtsZ than the parent (Table 2). Moreover, introduction of plasmid pCX16 (sdiA) in the recombineering strain DY329 now allowed the recovery of viable mre knockout derivatives at frequencies that were at least 2 logs higher than without the plasmid (data not shown). SdiA positively regulates a promoter (ftsQ_2_p) upstream of ftsQAZ, and cells carrying pCX16 contained three- to fourfold the normal level of FtsZ (87) (Table 2). The use of DY329/pCX16 for recombineering allowed for viable mrd knockout derivatives (Fig. 1B) to be readily obtained as well.
TABLE 2.
Relative FtsZ levels in viable mre mutants
| Straina | Relevant genotype | IPTG concn (μM) | Relative FtsZ levelb | SD |
|---|---|---|---|---|
| PA340 | wt | 0 | 1.0 | |
| PA340-129 | mreB129 | 0 | 2.0 | 0.5 |
| PA340-678 | _mre_-678 | 0 | 2.4 | 0.8 |
| PB103 | wt | 0 | 1.0 | |
| PB103/pCX16 | wt/sdiA | 0 | 4.2 | 1.6 |
| FB2/pCX16 | mreB<>aph/sdiA | 0 | 3.0 | 0.7 |
| FB2/pCH244 | mreB<>aph/P_lac_::mreBCD | 500 | 1.0 | 0.2 |
| FB2sup/pGB2 | mreB<>aph sup?c/vector | 0 | 2.6 |
Suppression of mre and mrd lethality by pCX16 (sdiA) was also evident when knockout alleles were transferred by P1-mediated transduction from the original DY329/pCX16 recombinants to strains of different backgrounds (Table 3 and results not shown). For example, the presence of pCX16 in the acceptor strain PB103 had little effect on the transduction frequency of a lacIZYA<>aph allele, but it increased the number of viable mre<>aph or mrdB<>aph transductants by about 2 logs. Thus, the mre<>aph and mrdB<>aph alleles were all detrimental to survival, but viability was enhanced by increased SdiA activity in all cases.
The presence of the sdiA plasmid was not absolutely required for allelic transfer, as transduction of mre<>aph alleles to PB103/pGB2 did yield rare spherical transductants (Table 3). Further analyses of one of these (FB2sup [mreB<>_aph_]) showed that its FtsZ level was over twofold higher than normal (Table 2), indicating it had undergone a second alteration, leading to increased production of the division protein. To avoid a selective advantage for such undefined (and undesired) suppressors of Mre− or Mrd− lethality, care was taken to provide all knockout strains with complementing (Mre+ or Mrd+) and/or lethality-suppressing (SdiA+ or Fts+) plasmids or phages during subsequent strain manipulations.
Polarity of mreB and mreC lesions.
Initial complementation experiments indicated that the mre<>aph alleles were polar on expression of downstream genes, precluding a firm conclusion as to the role of each gene (not shown). We proceeded with complementation experiments using strains in which the aph gene had been evicted by FLP recombinase, leaving only the 82-bp frt scar sequence on the chromosome (23). To maintain viability, these strains also carried the sdiA plasmid pFB112 (tetA sdiA), a ColE1 derivative conferring tetracycline resistance. As expected, each of the mre<>frt/pFB112 strains grew as spheres. Complementation was studied with a set of six ColE1 derivatives which confer resistance to Amp and carry one or more of the mre genes downstream of the lac promoter (Table 1 and Fig. 1A). The mre<>frt/pFB112 strains were transformed with each one of the P_lac_::mre plasmids. Cells were plated on LB agar containing Amp and IPTG, and transformants were examined for both cell morphology and loss of the incompatible pFB112 plasmid.
As summarized in Table S1 in the supplemental material, plasmid pCH244 (P_lac_::mreBCD) was capable of restoring a rod shape to each of the mre<>frt strains. In addition, pCH235 (P_lac_::mreD) restored the rod shape in the mreD<>frt strain. However, the mreB<>frt lesion failed to be restored by pFB118 (P_lac_::mreB) unless cells also harbored pFB124 (PλR::mreCD), a compatible plasmid carrying mreC and mreD downstream of a temperature-inducible λ PR promoter. Similarly, the mreC<>frt allele could be corrected only by P_lac_::mreC plasmids that carry mreD in cis (pCH244 or pFB121) or when mreD was coexpressed in trans from pFB128 (PλR::mreD). Others previously noted that frameshift or frt deletion-replacement lesions in mreB are polar on the expression of mreC and mreD (61, 86). Our complementation results are consistent with this and further show that the chromosomal mreC<>frt lesion (Fig. 1A) is similarly polar on the expression of mreD.
Whereas each transformant in which the rod shape was restored had lost pFB112, all transformants that remained spherical had retained this sdiA plasmid, even though the antibiotic in the medium (Amp) favored maintenance of the incompatible P_lac_::mre competitors (see Table S1 in the supplemental material). Apparently, pFB112 provided all spherical Mre− cells with a selective advantage, supporting the conclusion that extra copies of sdiA allowed them to propagate.
Each mre gene is required for both maintenance of rod shape and normal viability.
To study unsuppressed Mre− phenotypes, we used strains that lack an sdiA plasmid and in which transcription of one or all of the mre genes can be shut off by omitting IPTG or arabinose from the growth medium. Specific depletion of MreB or MreC was accomplished by supplying cells with an appropriate source of MreC and/or MreD to compensate for the polarities associated with the chromosomal mreB and mreC lesions described above.
As shown in Fig. 2, cells of strain FB17/pCH244 (mreBCD<>frt/P_lac_::mreBCD) (row 8) grew about as well as the control strain PB103/pCH244 (wt/P_lac_::mreBCD) (row 7) on LB at 37°C in the presence of IPTG (columns A to C). Growth of FB17/pCH244 was negligible in the absence of the inducer (columns D to F), however, confirming that depletion of all three Mre proteins severely limits the ability of cells to propagate. Identical results were obtained upon the specific depletion of MreB, MreC, or MreD separately, using strains FB17/pFB118/pFB124 (mreB<>frt/P_lac_::mreB/PλR::mreCD) (row 2), FB10(λFB120)/pFB128 [mreC<>aph(P_lac_::mreC)/PλR::mreD_] (row 4), or FB11(λCH235) [mreD<>aph(P_lac::mreD)] (row 6), respectively. As detailed further below, depletion of any of the Mre proteins caused cells to grow into large spheres that failed to divide properly (see Table S2 in the supplemental material).
FIG. 2.

Mre− lethality and suppression of MreBCD− lethality by extra SdiA. Even-numbered rows show that depletion of MreB (row 2), MreC (row 4), MreD (row 6), or MreBCD (row 8) causes a severe growth defect and that the latter can be suppressed by extra SdiA (row 10). Odd-numbered rows show appropriate wt controls. Cultures were grown to density at 37°C in LB supplemented with appropriate antibiotics and IPTG and were diluted 104 (columns A and D), 105 (B and E), or 106 (C and F)-fold. Aliquots (10 μl) were spotted on LB plates containing either 0.1% glucose (D to F) or IPTG (A to C) at 100 μM (rows 3 to 6) or 250 μM (other rows). Plates were incubated overnight at 37°C and photographed. The strains used were PB103/pFB118/pFB124 and FB17/pFB118/pFB124 (rows 1 and 2), PB103(λFB120)/pFB128 and FB10(λFB120)/pFB128 (rows 3 and 4), PB103(λCH235) and FB11(λCH235) (rows 5 and 6), PB103/pCH244 and FB17/pCH244 (rows 7 and 8), and PB103/pCH244/pCX16 and FB17/pCH244/pCX16 (rows 9 and 10).
These results confirmed that all three mre genes are required to maintain the rod shape of E. coli cells (84). Others recently concluded that all three mre genes are also essential for viability (49, 86). However, Kruse et al. did not consider _frt_-associated polarity in the mre operon, leaving the possibility that only the loss of MreD affected viability (49). The transduction results obtained by Wachi et al. are more convincing, although it is unclear whether each of the mre plasmids used in their study would be capable of correcting the corresponding mre lesion(s) in the chromosome (86). Our complementation and depletion results are comprehensive and show quite conclusively that each of the three Mre proteins is indeed required for normal viability. Note, however, that the proteins are only conditionally essential, as shown by subsequent experiments discussed below.
Overexpression of FtsZ is sufficient to restore viability to Mre− cells.
In addition to stimulating transcription of the ftsQAZ division genes (87), SdiA affects the expression of many other genes as well (89).
To test whether an increased level of just FtsZ is sufficient to suppress Mre− lethality, we used strain FB30/pFB174 (mreBCD<>aph/PBAD::mreBCD) carrying either pDR3 (P_lac_::ftsZ) or the vector control pMLB1113. Aliquots of serially diluted cultures were spotted on LB agar supplemented with either 0.5% arabinose, 0.1% glucose, or 0.1% glucose plus 100 μM IPTG. Both strains grew well in the presence of arabinose (MreBCD+) but failed to form colonies in the presence of glucose (MreBCD−) when IPTG was absent. The presence of IPTG, however, specifically allowed pDR3-carrying cells to grow in the presence of glucose (MreBCD− FtsZ++), showing that overproduction of FtsZ is indeed sufficient to overcome the growth defect of Mre− cells (Fig. 3A).
FIG. 3.

Conditional viability of Mre− cells. (A) The MreBCD depletion strain FB30/pFB174 (Δ_mreBCD_/PBAD::mreBCD) carrying either plasmid pDR3 (P_lac_::ftsZ) (row 2) or a vector control (row 1) was grown to density at 37°C in LB with 0.5% arabinose. Cultures were diluted and spotted on LB plates containing 0.5% arabinose (columns A to C), 0.1% glucose (columns D to F), or 0.1% glucose plus 100 μM IPTG (columns G to I). Plates were incubated at 37°C. (B) Overnight cultures of strain TB28 (wt) carrying either pDR3 (P_lac_::ftsZ) (row 2) or a vector control (row 1) were diluted and spotted on LB plates containing the A22-stock solvent methanol (columns A to C), 10 μg/ml A22 (columns D to F), or 10 μg/ml A22 plus 50 μM IPTG (columns G to I). Plates were incubated at 37°C. (C) The MreBCD depletion strain FB21/pFB149 (Δ_mreB_/P_lac_::mreBCD) (rows 2 and 4) and its wt parent TB28/pFB149 (rows 1 and 3) were grown overnight at 37°C in LB with 250 μM IPTG. Cultures were diluted and spotted on M9-maltose plates containing 250 μM IPTG (columns A to C) or 0.1% glucose (columns D to F). Plates were incubated at 30°C (rows 1 and 2) or 20°C (RT; rows 3 and 4). (D) An overnight culture of strain TB28 (wt) was diluted and spotted on M9-maltose plates containing methanol (row 1) or 5 μg/ml A22 (row 2). Plates were incubated at RT or 37°C, as indicated. (E) The MreBCD depletion strain FB30/pFB174 (Δ_mreBCD_/PBAD::mreBCD) (rows 1 and 2) and its wt parent TB28/pFB174 (rows 3 and 4) carrying either plasmid pYT11 (P_tac_::_relA_′) (rows 2 and 4) or pJF118EH (vector) (rows 1 and 3) were grown to density at 30°C in M9-maltose with 0.5% arabinose. Cultures were diluted in LB and spotted on an LB plate with 50 μM IPTG, which was incubated at 30°C. Overnight cultures were serially diluted 104 (columns A, D, and G)-, 105 (columns B, E, and H)-, and 106 (columns C, F, and I)-fold (A to D) or 103 (column A)-, 104 (column B)-, 105 (column C)-, and 106 (column D)-fold (E) in LB, and 10-μl aliquots were spotted in each case. Plates were incubated for 2 days (M9 at RT) or overnight (all others).
The antibacterial compound S_-(3,4-dichlorobenzyl)isothiourea (A22) causes a rod-to-sphere shape conversion in E. coli and other gram-negative species and likely interferes directly with the activities of the MreB protein (35, 43, 48). Therefore, the results mentioned above predicted that ftsZ overexpression might also alleviate A22 toxicity. This was indeed the case as determined by spot titer analyses of wt strain TB28 carrying either pDR3 (wt/P_lac::ftsZ) or the vector control pMLB1113. Cells were spotted on LB agar containing either no A22, 10 μg/ml A22, or 10 μg/ml A22 plus 50 μM IPTG. A22 prevented colony formation in the absence of IPTG, but overexpression of ftsZ in the pDR3-carrying cells overcame the toxicity of A22 to a significant degree (Fig. 3B). Cells in the latter case propagated as spheres (not shown), indicating that ftsZ overexpression did not interfere with the effect of A22 on cell shape but rather allowed resultant spheres to survive and grow.
The Mre proteins are dispensable for viability at low growth rates.
In the experiments described thus far, cells were cultured with rich (LB) medium at 30°C or 37°C. The growth phenotypes of Mre− cells were characterized more rigorously by depleting MreBCD from derivatives of two parent strains, PB103 and TB28, on both rich (LB) and poor (M9) media and at three different temperatures (37°C, 30°C, and 20°C). The parent strains have distinct backgrounds, and TB28 grows significantly faster than PB103, especially on minimal medium (Table 4). Depletion strains FB17/pFB149 and FB21/pFB149 (Δ_mreBCD_/P_lac_::mreBCD) and their respective parent controls, PB103/pFB149 and TB28/pFB149 (wt/P_lac_::mreBCD), were subjected to spot titer analyses on medium containing either 0.1% glucose or 250 μM IPTG. The results (shown in Fig. S1 in the supplemental material) are summarized in Fig. 3C and Table 4. As anticipated, FB21/pFB149 failed to grow in the presence of glucose (MreBCD−) under most conditions (row 2 in Fig. 3C and even rows in Fig. S1B in the supplemental material). Strikingly, however, it grew almost as well as the parent control on minimal medium at RT (∼20°C) (row 4 in Fig. 3C and row 12 in Fig. S1B in the supplemental material). Strain FB17/pFB149 (see Fig. S1A in the supplemental material) similarly failed to grow in the presence of glucose on LB at 37°C and 30°C (rows 2 and 4) but grew about as well as its parent control on M9 at both 30°C and 20°C (rows 10 and 12) and even formed some tiny colonies on LB at 20°C and on M9 at 37°C (rows 6 and 8). When growing in the presence of glucose under permissive conditions, cells of each depletion strain propagated as spheres (not shown), indicating that expression of mreBCD from the plasmid was sufficiently repressed. Compared to the doubling times of the parent strains in liquid medium under comparable conditions (Table 4), these results indicated that while Mre functions are essential for viability at moderate-to-high growth rates (approximate mass doubling time [∼_T_d] <150 min), they become dispensable during slow growth (∼_T_d > 300 min).
TABLE 4.
Growth rates, FtsZ levels, and viabilities of spherical derivatives
| Medium and temp (°C) | Result for PB103 | Result for TB28 | ||||||
|---|---|---|---|---|---|---|---|---|
| _T_d (min)a | [FtsZ]b | Growthc | _T_d (min)a | [FtsZ]b | Growthc | |||
| MreBCD− | MrdAB− | MreBCD− | MrdAB− | |||||
| LB | ||||||||
| 37 | 34 | 1.0 | − − − | − − − | 27 | 1.1 | − − − | − − − |
| 30 | 60 | 1.0 | − − − | − − − | 43 | 1.0 | − − − | − − − |
| 25 | 100 | ND | + − − | + + + | 75 | ND | − − − | + + + |
| M9 | ||||||||
| 37 | 151 | ND | + − − | + + + | 60 | ND | − − − | + + + |
| 30 | 302 | 0.9 | + + + | + + + | 75 | 0.8 | − − − | + + + |
| 25 | >500 | ND | + + + | + + + | 301 | ND | + + + | + + + |
These observations were further supported by P1 transduction experiments in which wt cells were mixed with a mreBCD<>aph lysate and incubated at different temperatures on selective M9 plates. The results paralleled those of the depletion experiments in that normal transduction frequencies of TB28 and PB103 could be attained, but only at sufficiently low temperatures (Table 5). The resulting mreBCD<>aph derivatives of TB28 and PB103 were spherical and grew about as well as their wt parents on minimal medium at low temperatures. Moreover, even after several rounds of purification under permissive conditions, they failed to grow on richer medium and/or at higher temperatures (not shown).These results indicate that conditions of slow growth relieve the selective pressure on Mre− cells to acquire alterations that lead to increased FtsZ production. One possibility is that slow growth itself leads to an increased FtsZ level. We detected no significant changes in FtsZ levels between fast- and slow-growing cells of PB103 or TB28 (Table 4), however, arguing against this possibility.
The ability of unsuppressed Mre− cells to propagate under slow-growth conditions suggested that slow growth might also reduce the toxicity of A22. Figure 3D shows that while A22 prevented growth of TB28 on LB agar at 37°C, the drug was indeed markedly less effective at inhibiting colony formation at 20°C.
Suppression of Mrd− lethality by ftsZ or slow growth.
Cooverexpression of ftsQ and ftsA with ftsZ was previously found to be required to restore viability to amdinocillin-treated cells on rich medium (59). This predicted that, in contrast to what was found for Mre-depleted spheres (see above), overexpression of ftsZ by itself might not be sufficient to rescue Mrd-depleted ones. We addressed this issue by using strain FB40(λTB59) [mrdAB<>frt (P_lac_::mrdAB)] carrying either pTB182 (PQAZ::ftsQAZ), pTB188 (PλR::ftsZ), or a vector control (pGB2). Aliquots of serially diluted cultures, including the Mrd+ parent controls, were spotted on LB agar containing either 100 μM IPTG or 0.1% glucose. All strains grew well in the presence of IPTG (MrdAB+). MrdAB-depleted cells carrying the vector control failed to grow, but those that carried either plasmid pTB182 or pTB188 (MrdAB− FtsQAZ++ or MrdAB− FtsZ++, respectively) plated efficiently (see Fig. S2 in the supplemental material). Moreover, pTB188 (PλR::ftsZ) rescued MrdAB-depleted cells about as well as cells that were depleted for MreBCD in a parallel control experiment (see Fig. S2 in the supplemental material). We conclude that elevated expression of ftsZ is sufficient to alleviate the growth defects of both Mre− and Mrd− cells.
wt cells resist killing by amdinocillin on poor medium (4, 47), and a mrdB (rodA)-null mutant was reported to survive on poor medium as well (28). Hence, it was likely that slow-growth conditions would also allow unsuppressed MrdAB− spheres to propagate. To verify this, spot titer analyses were performed with the MrdAB depletion strains FB39(λTB59) and FB40(λTB59) [Δ_mrdAB_(P_lac_::mrdAB)] and their parent controls, PB103(λTB59) and TB28(λTB59) [wt(P_lac_::mrdAB)], respectively. As summarized in Table 4, the results (provided in Fig. S3 in the supplemental material) were similar to those obtained with the MreBCD depletion strains. Although the depletion strains failed to grow in the presence of glucose (MrdAB−) on LB agar at 37°C and 30°C, they grew about as well as the parent controls under conditions favoring slower mass increase. Transduction experiments again supported these observations in that stable mrdAB<>aph derivatives of both PB103 and TB28 could be obtained at normal frequencies as long as they were selected for and maintained under conditions avoiding too-rapid growth (Table 5, and data not shown).
Suppression of MreBCD− lethality by RelA′.
Cells become resistant to killing by amdinocillin when concentrations of the stringent alarmone ppGpp rise above a threshold that is still well below that needed to stop growth altogether (47, 81). Given the similar growth requirements of Mrd− and Mre− spheres noted above, we suspected that the lethality associated with depletion of the MreBCD proteins on rich medium might be suppressed by increased ppGpp as well. To test this, we stimulated alarmone synthesis in wt and Mre-depleted cells by IPTG-induced expression of an overactive form of (p)ppGpp synthase (RelA′, lacking residues 456 to 744) (47, 69) from plasmid pYT11 (P_tac_::_relA_′). As expected, the inducer caused a reduction in the growth rate of pYT11-carrying cells, and growth ceased completely at 250 μM IPTG. In LB medium without arabinose and containing only 50 μM IPTG, _T_ds increased from 59 to103 min in wt rods (TB28) and from 76 to 108 min in MreBCD-depleted spheres (FB30/pFB174) (Table 6). Spot-titer analyses showed that expression of RelA′ under these conditions was sufficient to suppress the lethality of MreBCD-depleted spheres (Fig. 3E). Although the modestly reduced growth rate might have contributed to the ability of Mre− spheres to survive in this experiment, this is unlikely to be the sole explanation, as spheres in which _relA_′ is not artificially induced fail to survive unless the _T_d value surpasses 150 min, at least (Table 4).
TABLE 6.
Effects of RelA′ production on doubling times, FtsZ levels, and cell sizesa
| Strain | Genotype | IPTG concn (μM) | _T_db (min) | [FtsZ]c,f | Long axisd,f (μm) | Short axisd,f (μm) | Vold,e,f (μm3) |
|---|---|---|---|---|---|---|---|
| TB28/pJF118EH | wt/vector | 0 | 61 | ND | ND | ND | ND |
| TB28/pJF118EH | wt/vector | 50 | 59 | 1.0 | 4.1 (0.9) | 1.1 (0.1) | 3.45 (1.1) |
| TB28/pYT11 | wt/P_tac_::_relA_′ | 0 | 62 | ND | ND | ND | ND |
| TB28/pYT11 | wt/P_tac_::_relA_′ | 50 | 103 | 1.3 (0.0) | 1.9 (0.4) | 0.9 (0.1) | 1.0 (0.3) |
| FB30/pFB174/pJF118EH | Δ_mreBCD_/PBAD::mreBCD/vector | 50 | 76 | 1.1 (0.1) | 4.0 (0.1) | 3.0 (0.6) | 20.2 (10.9) |
| FB30/pFB174/pYT11 | Δ_mreBCD_/PBAD::mreBCD/P_tac_::_relA_′ | 0 | 68 | ND | ND | ND | ND |
| FB30/pFB174/pYT11 | Δ_mreBCD_/PBAD::mreBCD/P_tac_::_relA_′ | 50 | 108 | 1.6 (0.1) | 2.3 (0.6) | 1.7 (0.3) | 3.9 (2.6) |
Production of RelA′ was accompanied by substantial reductions in the average sizes of both wt rods and MreBCD− spheres (Table 6), indicating an elevated division frequency. Quantitative Western analyses indicated modest increases (∼30 to 60%) in relative FtsZ levels in the exponentially growing, RelA′-producing rods and spheres (Table 6). Whether the combination of a reduced mass doubling rate with this small increase in FtsZ is sufficient to explain the significantly smaller size of RelA′-producing cells, and/or to explain the ability of the spheres to propagate under these conditions, is presently unclear. Either way, these results further emphasize the similarities between amdinocillin-induced (PBP2−) and Mre− spheres.
We conclude that the growth requirements of genetically unsuppressed Mre− and Mrd− spheres are quite similar. Both are viable at low growth rates, and their deaths at high growth rates can be prevented by increases in ppGpp levels and/or an extra supply of just the FtsZ division protein.
Conditional lethality of Mre− cells is associated with a division defect and aberrant assembly of FtsZ.
The finding that ftsZ overexpression allows propagation of Mre− cells under nonpermissive growth conditions suggested that, as what was inferred for spherical mrd mutants (82), the lethality associated with loss of mre might be primarily caused by a division defect. The phenotype of Mre− cells supported this possibility. Strains completely lacking one or more of the Mre proteins, but carrying an sdiA plasmid, grew as spheres of various sizes. Many of these appeared to be in the process of constriction, and immunostaining with anti-FtsZ antibodies showed the protein associated with these sites. In most of the smaller spheres, FtsZ had accumulated in well-defined rings, although some rings showed atypical branches (see Fig. S4A in the supplemental material). About 10 to 20% of these populations consisted of distinctly larger cells, likely due to unequal inheritance of the suppressing sdiA plasmid. In these cells, FtsZ invariably appeared assembled in more-complex patterns that often included isolated patches and foci as well as more-extended structures that failed to span the girth of the cell but appeared branched and/or folded back on themselves (see Fig. S4B in the supplemental material).
Depletion of each (or all) of the three Mre proteins under nonsuppressing conditions resulted in a uniform giant-sphere phenotype (see Table S2 in the supplemental material). For example, when cells of the MreB depletion strain FB17/pFB118/pFB124 (mreB<>frt/P_lac_::mreB/PλR::mreCD) were shifted from LB medium containing IPTG to medium lacking the inducer, cells initially grew and divided as rods but then lost the rod shape and ultimately formed very large spherical cells. FtsZ assembled in typical rings early during depletion (Fig. 4A and B), but the nondividing large cells that formed later on again contained the protein in more-complex patterns as described above (Fig. 4C to E).
FIG. 4.
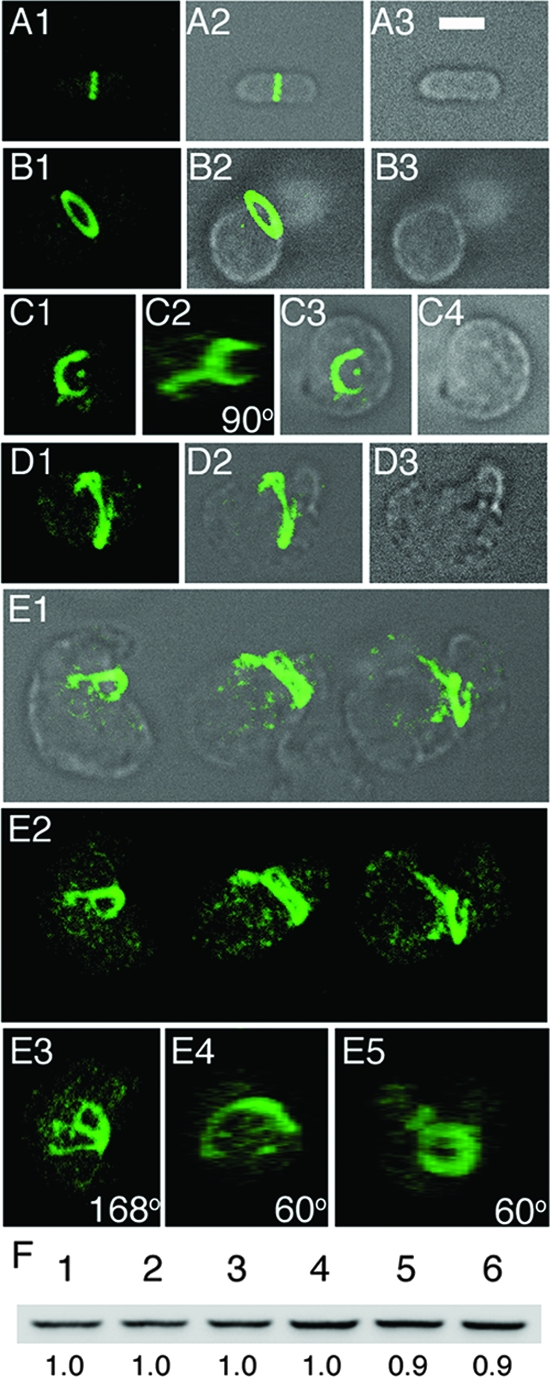
MreB-depleted cells contain normal levels of FtsZ but form aberrant FtsZ structures. (A to E) Immunofluorescence confocal microscopy of MreB-depleted cells with anti-FtsZ antibody, Strain FB17/pFB124/pFB118 (Δ_mreBCD_/PλR::mreCD/P_lac_::mreB) was grown in the presence of either 250 μM IPTG (A) or 0.1% glucose (B to E). Samples for staining were taken both early (B) (OD600 = 0.2) and late (A and C to E) (OD600 = 0.6) during growth/depletion. Maximum projection (A1, B1, C1, D1, and E2), DIC (A3, B3, and C4), and merged (A2, B2, C3, D2, and E1) fluorescence images are shown. In panel C2, the image in panel C1 is rotated 90° about the y axis. Panels E3, E4, and E5 show y axis rotations of the left-hand (168°), middle (60°), and right-hand (60°) cell in panel E2, respectively. Bar = 2 μm. (F) Anti-FtsZ immunoblot of whole-cell extracts of MreB-depleted spheres (lanes 3 and 6) and rod-shaped controls (other lanes). Extracts were prepared on strain PB103 (wt) (lanes 1 and 4), and its MreB depletion derivative FB17/pFB124/pFB118 was grown in the presence of either 250 μM IPTG (lanes 2 and 5) or 0.1% glucose (lanes 3 and 6). Cells were harvested both early (OD600 = 0.2) (lanes 1 to 3) and late (OD600 = 0.4 to 0.5) (lanes 4 to 6) during growth/depletion, and each lane received 10 μg total protein. Measured intensity values of FtsZ bands in lanes 2 and 3 were normalized to that in lane 1 and those in lanes 5 and 6 to that in 4. Resulting relative values are shown below the lanes. Cells were grown in LB at 37°C in each case (A to F).
To ensure that the odd FtsZ assembly patterns were not an immunostaining artifact, we also localized FtsZ in live Mre− spheres, using a GFP fusion to the cytoplasmic FtsZ-binding protein ZapA as a convenient marker for FtsZ assembly. Figure 5 shows cells of the MreBCD depletion strain FB30(λCH268)/pFB174 [Δ_mreBCD_(P_lac_::_gfp_-zapA)/PBAD::_mreBCD_] inoculated in M9-maltose medium at 37°C. The medium lacked arabinose to shut down mreBCD expression and contained IPTG to induce expression of _gfp_-zapA. In addition, CM was visualized by treatment with the membrane dye FM4-64 immediately before microscopy. Early during depletion, cells appeared as fat rods, and GFP-ZapA (i.e., FtsZ) formed a ring or arc across the long axes of the rods (Fig. 5A and B). Later on, cells again had become large spheres with complex patterns of FtsZ accumulation (Fig. 5C and D), corroborating the results obtained with immunostaining.
Figure 5D highlights features of a large Mre− spheroid that appears in the process of division. It contains a shallow constriction perpendicular to the middle of its long axis, and the bulk of FtsZ appears to have assembled in a zone around the constriction. The constriction is asymmetric, however, in that the bottom part of the cell shows a clear invagination that is associated with a fairly well-defined arc of FtsZ (D7 and D8), whereas invagination is less obvious in the top part, where FtsZ seems present in ill-defined clusters scattered about midcell (D5 and D6). As Mre− cells grow into very large spheres under these nonpermissive conditions, we imagine that many such constriction attempts eventually abort.
One possible explanation for the failure of Mre− spheres to divide properly under nonpermissive growth conditions, and for the fact that extra FtsZ can restore division, is that the absence of the Mre proteins somehow caused a drop in the level of FtsZ. This was not supported by Western analyses, however, as we detected no significant change in the level of FtsZ in nondividing MreB-depleted spheres (Fig. 4F, lane 6) compared to that in dividing rod-shaped control cells (lanes 4 and 5).
Vesicle-like bodies in E. coli spheres.
The cell in Fig. 5D also shows another striking feature of Mre− spheres under nonpermissive conditions, which is the presence of vesicle-like bodies in their interiors. Imaging by DIC indicated the presence of a large vesicle-like body in the left-hand half of the cell and that of smaller ones elsewhere (D1). Most of these were not stained by the membrane-impermeable FM4-64 dye, suggesting that if these compartments were surrounded by membrane, it was discontinuous with the externally accessible CM. One of the optical slices shows a clear small circle of FM4-64 staining near the cell center, however, suggesting the presence of a finger-like involution of the CM at this site that reaches well into the body of the cell. Some FtsZ clusters surrounding this FM4-64-stained material can be seen as well (D5). Another projection of FM4-64 stain that appears continuous with the CM is visible in a plane near the cell bottom (D8).
Vacuole-like inclusions were previously noted upon inactivation of PBP2 (MrdA) by amdinocillin in E. coli (54) and in a Salmonella enterica serovar Typhimurium rodA (mrdB) mutant grown on soft agar (20). In addition, they were observed in a number of E. coli shape mutants with ill-characterized lesions (2, 3, 38, 54, 63). Two of these older studies included thin section transmission electron microscopy analyses of the mutant cells. Allison (3) performed these studies on a mon (envB) mutant (2) that may have been allelic with one of the mre genes (54, 85), while Henning et al. (38) studied a temperature-sensitive shape mutant (lss12) that may have been allelic to mrdA as it produced a thermolabile PBP2 protein (75). Both studies showed the presence of CM involutions, stacked cisternae, and vesicle-like compartments traversing the cytoplasmic space of large spherical cells. These compartments appeared lined by a unit membrane, and their lumens lacked ribosomes, suggesting that they formed by involutions of the CM. Whether all intracytoplasmic membrane was continuous with the CM was not assessed (3, 38).
To better define the genetic requirements for vacuolization in shape mutants, we depleted each of the Mre and Mrd proteins separately under nonsuppressing conditions and observed cells by both membrane staining and DIC. In each case, cells formed large spheres with readily apparent vesicle-like inclusions (see Table S2 in the supplemental material). Therefore, the phenomenon is not provoked by the absence of any of the shape proteins specifically but is more likely a general consequence of growth as a nondividing sphere per se. As this phenotype is inherently interesting and correlates with the failure of spheres to divide properly, we studied the formation of vesicle-like inclusions in Mre− spheres in more detail.
Involution of the CM and endocytosis in E. coli spheres.
To ensure that the vesicle-like bodies that we observed in spheres were bounded by CM, we visualized the latter in live MreBCD-depleted cells with a fusion of GFP to the N-terminal transmembrane domain of the bitopic CM protein ZipA (GFP) (45). The fluorescent fusion accumulated around each vesicle that was visible by DIC, suggesting that they were indeed surrounded by CM (Fig. 6B and C). Topologically, the lumens of these bodies are expected to correspond to extracytoplasmic space. If they are bounded by CM only, this space should correspond to the periplasm. Though unlikely, it is also conceivable that they are lined with both CM and outer membrane (OM), in which case the lumen is expected to be compartmentalized further. To probe these possibilities, we used a GFP fusion that is targeted to the periplasm via the twin arginine transport system (TTGFP) (6). Panels G to I of Fig. 6 illustrate that TTGFP indeed accumulated in the lumen of each vesicle. In addition, the fusion distributed evenly within vesicular space, indicating that it was not compartmentalized further.
FIG. 6.

Intracytoplasmic membrane compartments in MreBCD-depleted spheroids. Shown are live cells of the MreBCD depletion strain FB30/pFB174 (Δ_mreBCD_/PBAD::mreBCD) producing either transmembrane GFP from lysogenic phage λCH178 [P_lac_::zipA(1-183)-gfp_] (A to C) or periplasmic TTGFP from plasmid pTB6 (P_lac::ss_torA_-gfp) (D to J). Cells were grown to an OD600 of 0.2 at 37°C in LB with 50 μM IPTG and either with (A and D) or without (other panels) 0.5% arabinose. Both DIC (1) and GFP (2) fluorescence images are shown in panels A to I. Panels J show a time lapse series of a spheroid exposed to lysozyme. For this experiment, 2 μl of culture was applied to a slide and covered with a coverslip. A 1-μl aliquot of egg white lysozyme (100 μg/ml in GTE) was then pipetted against the edge of the coverslip, where it was drawn under by capillary action. GFP fluorescence was recorded immediately at 1-s intervals (J1 to J6). The arrowhead in panel J1 points to a compartment that quickly released TTGFP into the medium, in contrast to other compartments that retained the fusion throughout the procedure (arrows). Panel J7 shows a DIC image of the lysozyme-treated sphere a few seconds after the image in panel J6 was taken. Bar = 2 μm.
In most MreBCD-depleted spheres, both GFP and TTGFP also accumulated in intracytoplasmic patterns that were not or poorly distinguishable by DIC (e.g., Fig. 6B2 and F2). These patterns likely corresponded to membrane compartments, such as small vesicles and/or stacked forms, with lumens too small to detect by DIC.
As mentioned above, only a subset of vesicle-like bodies showed accumulation of fluorescence at their peripheries when the membrane-impermeable CM dye FM4-64 was added to Mre− spheres immediately before microscopy (Fig. 7A to C). In contrast, when spheres were pulse labeled with the dye 30 min prior to observation, virtually all bodies that were visible by DIC were now also clearly outlined by a fluorescent border (Fig. 7D to G). In addition, as was observed with the GFP and TTGFP probes, the dye accumulated in various other patterns that traversed the sphere's interior and that were often quite extensive. At the time of observation, the spheres in this experiment (Fig. 7D to G) had an average diameter of 3.7 μm (n = 102) and 90% contained vesicle-like and/or other fluorescent patterns within their interiors. Thus, the interiors of the majority of Mre− spheres contained a considerable amount of membrane originating, at least in part, from externally accessible CM. Moreover, the fact that a substantial subset of vesicular bodies were not immediately accessible to FM4-64 (e.g., Fig. 7A and B) suggested that they were discontinuous with the external CM and periplasm and, thus, represented “true” vesicles.
The latter inference was confirmed by three additional experiments. In the first, spheres were incubated with the membrane-permeable membrane dye CellTrace BTME for 15 min and then mixed with the membrane-impermeable CM dye FM1-43 just prior to microscopy. As expected, BTME stained the membrane of each vacuole that was visible by DIC as well as additional internal membrane structures, whereas FM1-43 stained only a subset of these structures (Fig. 7H and I). In the second, spheres producing TTGFP were resuspended in isotonic buffer and observed by time lapse microscopy immediately after addition of lysozyme. Upon disintegration of the murein wall, a subset of vesicle-like bodies released TTGFP into the medium, implying that their lumens were continuous with the exterior periplasm. Over half remained intact, however, showing that they were topologically separate from the cell's exterior CM and periplasm (Fig. 6J and data not shown). In the third experiment, wt rods or Mre− spheres were grown in the presence of LY, a water-soluble, membrane-impermeant fluorescent compound that is excluded from the cytoplasm by the CM barrier but should have access to the periplasm as it is sufficiently small (522 Da) to pass OM porins (60). Cells were then washed in medium lacking LY and immediately imaged live. Cells of the wt parent TB28 completely failed to retain LY, indicating that if the dye entered the periplasm it readily washed out again (Fig. 7J). Similarly, the dye appeared absent from the external periplasms of Mre− spheres, as well as from a subset of vesicle-like bodies, suggesting that these were still continuous compartments. However, the dye remained trapped in the majority of vesicle-like bodies that were visible by DIC, indicating that this subset had become topologically separate from the external CM and periplasm and, thus, represented “true” endosomes (Fig. 7K and L).
We conclude that under nonpermissive growth conditions, spherical Mre− cells form extensive involutions of the CM into the cytoplasm, resulting in elaborate membranous compartments in the cell interior. Moreover, many of the involutions must be subject to an endocytic-like membrane fission event that releases them as a closed compartment into the cytoplasm.
Membrane involution and scission do not require FtsZ assembly.
What causes the involution of CM and the generation of internal vesicles in spherical cells? We considered the possibility that unsuccessful attempts at cell division by spherical cells under nonpermissive conditions might be a primary cause of CM invasion into the cytoplasm. Therefore, we examined the ability of Δ_mre_ cells to accumulate internal membrane while FtsZ polymerization in the cells was inhibited by the SfiA (SulA) protein. Strain FB30/pTB63/pDR144 (Δ_mreBCD_/PQAZ::ftsQAZ/P_lac_::sfiA) was grown in the absence or presence of IPTG, and cells were stained with BTME to visualize membrane. In the absence of inducer (SfiA−), the extra supply of FtsZ from pTB63 allowed these Δ_mreBCD_ cells to grow and divide as small spheres (Fig. 8A and C). Over 50% of the spheres were in the process of constriction (Fig. 8D), and no obvious internal membrane accumulations were detectible by DIC or fluorescence microscopy under these conditions (Fig. 8A). In the presence of IPTG (SfiA+), cells ceased constriction and increased in size as expected. After only about 2.6 mass doublings, over 60% of these nondividing spheres contained internal membrane systems that were readily detectible by DIC and/or fluorescence microscopy (Fig. 8B to D). In addition, double staining with permeable (BTME) and nonpermeable (FM1-43) membrane dyes (Fig. 8E), as well as LY-internalization assays (Fig. 8F), again indicated that many of the internal membrane structures in the nondividing FB30/pTB63/pDR144 spheres were discontinuous with the external CM.
We conclude that FtsZ polymerization is not required for generation of the internal membrane compartments in spherical cells of E. coli.
Spherical cells fail to adjust the rate of phospholipid synthesis.
Though no data were shown and no implications were discussed, Henning et al. mentioned that lss12 [mrd(Ts)?] spheres synthesized phospholipids at the same rate as wt rods (38). This is notable as a rod-to-sphere conversion is accompanied by an increase in the volume-to-surface ratio. Hence, a failure to properly reduce the net rate of phospholipid synthesis after conversion to spheres would lead to excess membrane, which, in turn, could well be the primary cause of the extensive internal membrane systems that develop in E. coli mre and mrd mutants under nonpermissive growth conditions. To address this possibility, we compared the incorporation of radiolabeled orthophosphate (32Pi) into total phospholipid in MreBCD+ rods and MreBCD-depleted spheres of strain FB21/pFB149 [mreB<>aph/P_lac_::_mreBCD_]. Cells were inoculated in LP medium at 37°C in the presence (MreBCD+) or absence (MreBCD−) of 1 mM IPTG. Relative rates of phospholipid synthesis were determined using aliquots of cells taken at both mid (T1)- and late (T2)-logarithmic growth (Fig. 9). At both time points, the cells that were growing without IPTG were both distinctly spherical and maintained mass doubling rates similar to that of their rod-shaped counterparts (Fig. 9A and C). Figure 9B shows that whereas the rate of phospholipid synthesis declined as cells of either shape entered stationary phase, the rates were virtually identical for both cell types at both T1 and T2.
FIG. 9.
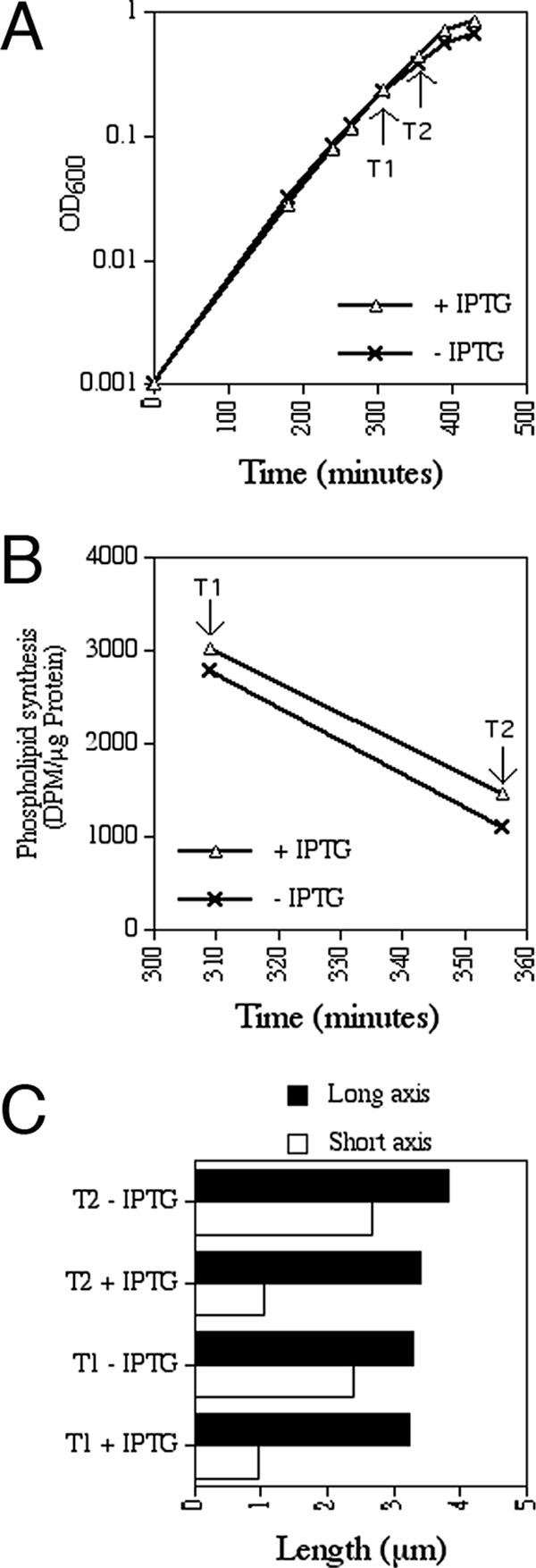
Phospholipid synthesis rates in rods and MreBCD-depleted spheres. The MreBCD depletion strain FB21/pFB149 (Δ_mreB_/P_lac_::mreBCD) was grown at 37°C in LP glucose with no or 1 mM IPTG, and the increase in optical density was monitored over time (A). At time points T1 and T2, aliquots were removed to determine phospholipid synthesis rates (B) and cell shape parameters (C).
It thus appears that upon a rod-to-sphere conversion, E. coli cells indeed fail to respond to their new surface requirements and continue to produce phospholipid membrane at a rate suitable for rod-shaped cells.
Mislocalization of division proteins to internal membrane systems in Mre− spheres.
The finding that depletion of any of the five shape proteins from fast-growing cells resulted in large nondividing spheres with internal membrane systems raised the possibility that the division block in the spheres is causally related to the invasion of CM into their interiors (see Table S2 in the supplemental material). This idea was supported by the distribution of GFP-ZapA in Mre-depleted spheres. The fusion accumulated not only in various patterns on the exterior CM but also in clusters and patches surrounding interior membrane compartments, indicating nonproductive assembly of FtsZ on the latter (Fig. 10B to E).
FIG. 10.

Assembly of division proteins on intracytoplasmic membrane. (A to E) Shown are cells of the MreBCD depletion strain FB30(λCH268)/pFB174 [Δ_mreBCD_ (P_lac_::_gfp_-zapA)/PBAD::mreBCD_]. Cells were grown at 37°C to an OD600 of 0.2 in M9-maltose supplemented with 50 μM IPTG and either no (B to E) or 0.5% (A) arabinose. DIC (1) and GFP-ZapA (2) fluorescence images are shown. Note the accumulation of fluorescence in foci/patches at the cell exteriors as well as surrounding internal vesicles in panels B to E. (F to H) λDR122 (P_lac::gfp_-minDE) lysogens of TB28 (wt) (F) and the MreBCD depletion strain FB30/pFB174 (Δ_mreBCD/PBAD::mreBCD) (G and H) were grown at 37°C to an OD600 of 0.2 to 0.4 in LB supplemented with 50 μM IPTG but lacking arabinose. DIC (1) and GFP-MinD time lapse (2 to 4) fluorescence images at 10-s intervals are shown in panels F and G. Note the transient assembly of GFP-MinD on both the CMs and the surfaces of nearby internal vesicles as it oscillates about the short axis of the spheroid in panel G. The large sphere in panel H was treated with BTME prior to imaging. Shown are DIC fluorescence (1), BTME fluorescence (5), GFP-MinD time-lapse fluorescence at 15 s intervals (2 to 4), and merged GFP-MinD and BTME fluorescence (6 to 8) images. Note the periodic movement of GFP-MinD between BTME-stained sites in the interior of the sphere (H2, H4, H6, and H8) and sites on the CM (H3 and H7).
The oscillating Min system normally controls placement of the division machinery by antagonizing FtsZ assembly on the CM at off-center sites. In E. coli, proper Min function relies on alternating bulk assembly and disassembly phases of the MinD ATPase on the CM at either cell end (Fig. 10F) (67, 68). We observed that MinD readily assembles on the intracytoplasmic membrane systems in spheroids as well. GFP-MinD and MinE were produced from a lysogenic phage during depletion of MreBCD in strain FB30(λDR122)/pFB174 [Δ_mreBCD_(P_lac_::_gfp_-minDE)/PBAD::_mreBCD_]. Consistent with other studies in which Min proteins were observed in spheroids (19, 70, 77), GFP-MinD was seen to oscillate between opposite sites of the exterior CM, most commonly along the longest axes of the spheroids. In addition, however, GFP-MinD also transiently decorated interior vesicles that were visible by DIC. Decoration by the fusion was most obvious on any vesicle that resided near one of the MinD landing sites on the exterior CM (Fig. 10G). The fusion also associated transiently with internal structures not readily resolvable by DIC (Fig. 10H). Panels H2 to H4 show oscillation of the fusion in a large spheroid that contained extensive internal membrane systems as visualized with the BTME dye. GFP-MinD moved from assembly sites in the spheroid's interior (H2) to two opposite landing sites on the external CM (H3), back again to interior sites (H4), and so on. Overlays of the GFP and BTME channels indicated that GFP-MinD accumulation sites in the sphere's interior invariably coincided with the presence of internal membrane at these sites (H5 to H8).
In a previous study, a yellow fluorescent protein-MinD fusion was observed to sometimes assemble into subsurface loops and filaments in spheres of an mreB mutant (70). The membrane was not visualized in this study, and the significance of these puzzling MinD patterns remained unclear. Our results suggest that the ability of MinD to assemble on intracytoplasmic vesicles and membrane systems of more-complicated shapes may also have been responsible for the subsurface loops and filaments, respectively, observed in that study.
We propose that the assembly of FtsZ and other division proteins on interior membrane systems contributes significantly to the lethal division defect in rapidly growing Mre− and Mrd− spheres. Nonproductive assemblies on interior membrane will compete with potentially productive ones on the exterior CM for pools of unassembled FtsZ and other septal ring components. As the amount of FtsZ, in particular, is normally limiting the number of functional septal rings that a cell produces (88), this competition must lower the division rate. Moreover, disturbances of Min dynamics by interior membrane are expected to compound the problem by allowing FtsZ assembly at too many inappropriate sites.
DISCUSSION
Avoiding selective pressure for secondary genetic alterations, we created comprehensive sets of mre and mrd cell shape mutants of E. coli and used these to study the importance of rod shape to the viability of cells under various conditions. One notable general conclusion of our results is that a lack of one or more of each of the five Mre and Mrd shape proteins results in a very similar phenotype. In each case, mutant cells (i) grew and divided as small spheres that appeared genetically stable, under conditions of slow mass increase; (ii) grew into giant nondividing and nonpropagating spheres with elaborate intracytoplasmic membrane systems, under more-common conditions favoring moderate to fast mass increase; and (iii) could be rescued by a modest increase in the production of FtsZ, which allowed them to grow and divide as small spheres at any growth rate. It is evident that, like the Mrd proteins (4, 5, 28, 47, 82), MreB, -C, and -D are only conditionally essential. This result helps explain incongruous conclusions in the literature on the matter of mre essentiality in E. coli (49, 50, 70, 84-86). In addition, these similarities in mutant phenotypes lend further support to the notion that the Mre and Mrd proteins cooperate in the same complex and/or pathway to effect maintenance of rod shape (14).
One striking feature of all shape mutants examined was the presence of elaborate membrane systems in the interiors of the large nondividing spheroids that formed under nonpermissive conditions. Although de novo formation cannot be excluded, our results indicate that many, if not all, of these interior membrane systems originated from FtsZ-independent involution and elaboration of the CM. Importantly, MreBCD-depleted spheres failed to adjust phospholipid synthesis to their new surface requirements but rather continued synthesis at about the same rate as rod-shaped control cells. This has interesting implications for the regulation of phospholipid synthesis (21, 65), as it suggests the absence of a mechanism that couples membrane synthesis to actual cell surface requirements. One intriguing possibility is that allowing or mediating such coupling is actually one of the functions of the shape proteins. If so, the mechanism seems to require all five Mre and Mrd proteins, as depletion of each resulted in excess internal membrane. Alternatively, a mechanism that “senses” the surface and transmits its state to the phospholipid synthesis machinery does not exist in E. coli. Rather, the rate of phospholipid synthesis may be primarily, or solely, coupled to the rate of increase in cell mass (64), and being rod-shaped is somehow hard-wired in the system to arrive at a proper membrane/mass ratio. In any event, the failure to reduce phospholipid synthesis upon becoming spherical provides a ready rationale for the “excess” membrane in the interiors of Mre− and Mrd− spheroids under nonpermissive conditions.
It is also remarkable that while some of the internal membrane systems in nondividing spheroids were continuous with the external CM, others were clearly not. In addition, several assays indicated that many, if not all, of the discontinuous systems had at some point been continuous with externally accessible CM. This implies that the discontinuous intracytoplasmic compartments resulted from involution of the CM followed by a membrane scission event that released them in the cytoplasm. We are unaware of any other well-documented example of membrane endocytosis in E. coli or related bacterial species. Though the present example concerns cells that are not in a physiologically “normal” state, it does raise the question of what is responsible for the scission event that separates the “endosomes” from the external CM. Membrane fusion or fission rarely occurs spontaneously (17, 57), suggesting the possibility that some enzymatic activity in E. coli is capable of stimulating endosome formation.
Given the failure to adjust phospholipid synthesis rate, one might also expect an excess of OM in MreBCD-depleted spheroids under nonpermissive conditions. Indeed, we did observe an occasional TTGFP-filled bleb emanating from the surfaces of FB30/pFB174/pTB6 [Δ_mreBCD_/PBAD::mreBCD/P_lac_::ss_torA_-_gfp_] spheroids grown in the absence of arabinose and presence of IPTG. However, the occurrence of such blebs seemed rather low (about 1 out of every 10 spheres), and the culture medium seemed similarly barren of free fluorescent vesicles (data not shown). It is possible that excess OM was shed as vesicles that were too small to readily detect by fluorescence microscopy in this fashion (<300 nm) (34). Alternatively, perhaps phospholipid transport to the OM does slow down to a rate that is more appropriate to the surface requirement of a spheroid, and almost all of the excess phospholipid that the cell produces is incorporated in the intracytoplasmic systems. More work will be needed to distinguish between these possibilities.
This and previous studies indicate that the inability of shape mutants to propagate under nonpermissive conditions is primarily due to a cell division defect. A popular explanation for this is that the amount of FtsZ that is normally available is insufficient to accommodate the increased circumference of a cell after it converts from rod to sphere (49, 82). However, following through on this geometrical argument indicates that it is incomplete, at best. For example (Fig. 11), consider that FtsZ concentration remains constant and, thus, that its amount increases with cell mass (volume). Further assume a rod-shaped cell with a diameter of 1.4 μm that is about ready to begin Z-ring assembly/constriction when it reaches 3.6 μm from tip to tip. This cell then has a volume of 4.82 μm3 (line b in Fig. 11), a surface of 15.83 μm2, a circumference of 4.40 μm, a volume-to-surface ratio of 0.31 μm, and a volume-to-circumference (V/C) ratio of 1.10 μm2 (line a in Fig. 11). If this cell were to morph instantly to a sphere with a diameter of 2.10 μm, volume remains at 4.82 μm3, but V/C ratio decreases to 0.73 μm2 as circumference increases to 6.59 μm. Thus, to divide at the same mass as a rod, this sphere might indeed require an additional 50% [100 × (6.59 − 4.40)/4.40] FtsZ molecules to be able to assemble a complete ring. As mass increases, however, the V/C ratio rises quickly (Fig. 11, upper), and we calculate that if the sphere increased in volume by only a factor of 1.83 to 8.84 μm3, it would once again attain a V/C ratio of 1.10 μm2 (intersection of lines a and c in Fig. 11). In other words, the sphere would once again contain a sufficient amount of FtsZ to form a complete ring after less than one additional mass doubling. Why then do unsuppressed mre and mrd mutants not simply propagate as dividing spheres with a moderately increased average mass at any growth rate?
FIG. 11.
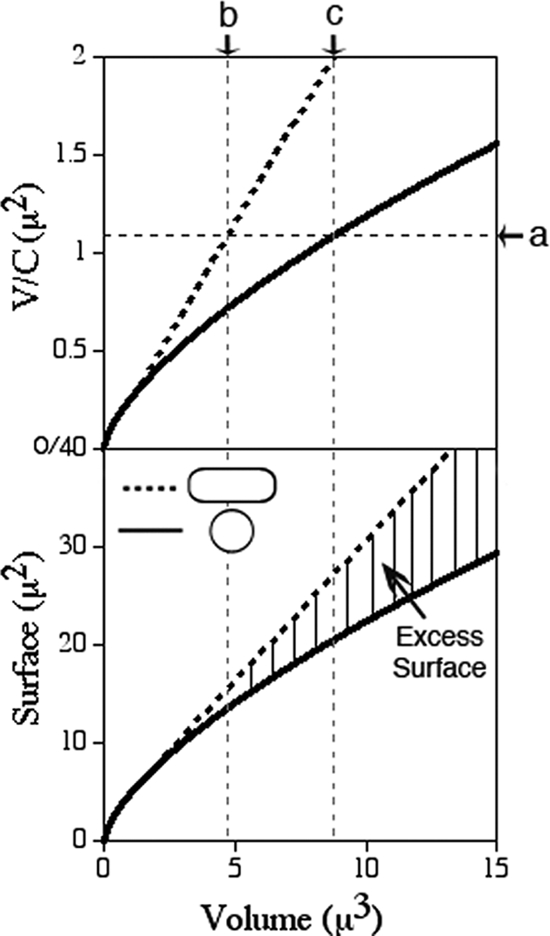
Relationships between volume, surface, and circumference in rods and spheres. The upper panel shows the increase of V/C ratio with increasing volumes of rod-shaped (stippled line) and spherical (solid line) cells. The lower panel shows the increase in surface of rod-shaped and spherical cells with increasing volume. The hatched area highlights the difference in surface requirements between the two cell shapes. See text for details.
Based on the results of this study, we now think it likely that the presence of intracytoplasmic membrane in fast-growing spheres contributes directly to the accompanying division block by diverting FtsZ and other division proteins into nonproductive assemblies on their surfaces. The failure of spheres to adjust phospholipid synthesis to their shape can lead to large excesses of membrane, and this problem becomes worse as spheres increase in mass (Fig. 11, lower). As an example, we consider the same cell as that mentioned above. Upon instantly morphing into a sphere with a volume of 4.82 μm3, this cell would face only a moderate excess of CM of 2.02 (15.83 − 13.81) μm2 (∼15%). In principle, it would again accumulate a sufficient amount of FtsZ (V/C ratio = 1.10 μm2) upon further growth to a volume of 8.84 μm3 (line c in Fig. 11). However, it would now also have accumulated an excess of 6.63 (27.30 − 20.67) μm2 membrane, which is over 30% more than needed to cover its surface and by itself sufficient to cover a sphere with a diameter of 1.45 μm. Note that this amount of excess intracytoplasmic membrane would be even higher if spheres were to reduce the fraction of total phospholipid (∼1/3) that is normally transported to the OM (see above). In our model, this excess intracytoplasmic membrane competes effectively with the external CM for FtsZ, delaying division further. As a result, cell mass keeps increasing, but so does the proportion of excess membrane (Fig. 11, bottom). Meanwhile, the chance of noncanonical patterning of the Min system increases, and the cell circumference becomes several times that of a normal rod. This combination of factors is likely to render assembly of a functional division apparatus increasingly rare as spheroids increase in mass, which is consistent with the dearth of typical Z “rings” in large spheroids (Fig. 4, 5, and 10; also see Fig. S4 in the supplemental material).
Assuming that membrane synthesis is indeed solely coupled to mass increase and that excess intracytoplasmic membrane interferes with division, small spheres might have the best chance to divide because they contain the least excess membrane. In fact, a sphere with a diameter of 1.83 μm (volume = 3.21 μm3, surface = 10.52 μm2, circumference = 5.75 μm) would have no excess membrane at all in our example, as its volume-to-surface ratio of 0.31 μm matches that of our sample rod-shaped cell mentioned above. However, as both its mass is smaller (by 33%) and its circumference larger (by 31%), its V/C ratio of 0.56 μm2 is only about half that required for division. Put differently, this sphere has not yet accumulated a sufficient amount of FtsZ to accommodate its circumference. It is important to note, however, that a mere doubling in FtsZ concentration would provide this small sphere with enough FtsZ to assemble a complete ring before excess membrane becomes a problem. The absence of discernible intracytoplasmic membrane in FtsZ-overproducing Δ_mreBCD_ spheres (e.g., Fig. 8A) is consistent with this scenario.
Although this model renders it easy to appreciate why a modest increase in FtsZ production would allow mre and mrd mutants to propagate as small spheres under otherwise nonpermissive conditions, the innate ability of the mutants to propagate as spheres under permissive conditions cannot be readily explained by geometrical arguments alone. About half of MreBCD-depleted spheres of strain FB30/pFB149 (Δ_mreBCD_/P_lac_::mreBCD) propagating with native levels of FtsZ under permissive conditions (in liquid M9 at 22°C) still appeared to contain some amount of excess intracytoplasmic membrane, as judged by the presence of small BTME-stained foci that were mostly distributed along their peripheries (see Fig. S5 in the supplemental material). This suggests that, at least under slow-growth conditions, cells can tolerate a certain amount of excess CM. We imagine that cells manage to build up sufficient pools of FtsZ and other division proteins to assemble a functional division apparatus as long as the ratio of “excess” to total available membrane surface remains below some critical value at the time of assembly. Based on the division phenotypes of min slmA double mutants, which are similarly medium dependent, we previously suggested that assembly of a constriction-competent septal ring in a slow-growing cell may be accomplished at a lower minimum concentration of FtsZ than that in a fast-growing one (7). The same suggestion can be offered here, but what might be responsible for a more efficient use of available FtsZ during slow mass increase remains unclear.
Supplementary Material
[Supplemental material]
Acknowledgments
We thank Thomas Bernhardt, Cynthia Hale, and Yu-Ting Su for plasmid construction and advice; David McDonald, Patrick Viollier, and Masaaki Wachi for materials; Patricia Conrad and Minh Lam for advice on confocal and deconvolution microscopy; and Arne Rietsch and Patrick Viollier for comments on the manuscript.
This work was supported by a Human Frontiers Science Program award (RGP0001/2003) and NIH GM57059 (to P.D.) and NIH NRSA Institutional Training Grant T32GM08056 (to F.B.). Some of the images were prepared at the Confocal Microscopy Core Facility of the Comprehensive Cancer Center of CWRU/UHC, supported by NIH P30 CA43703.
Footnotes
▿
Published ahead of print on 9 November 2007.
REFERENCES
- 1.Addinall, S. G., and J. Lutkenhaus. 1996. FtsA is localized to the septum in an FtsZ-dependent manner. J. Bacteriol. 1787167-7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adler, H. I., C. E. Terry, and A. A. Hardigree. 1968. Giant cells of Escherichia coli. J. Bacteriol. 95139-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allison, D. P. 1971. Giant cells of Escherichia coli: a morphological study. J. Bacteriol. 1081390-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbour, A. G., L. W. Mayer, and B. G. Spratt. 1981. Mecillinam resistance in Escherichia coli: dissociation of growth inhibition and morphologic change. J. Infect. Dis. 143114-121. [DOI] [PubMed] [Google Scholar]
- 5.Begg, K. J., and W. D. Donachie. 1998. Division planes alternate in spherical cells of Escherichia coli. J. Bacteriol. 1802564-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernhardt, T. G., and P. A. de Boer. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 521255-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernhardt, T. G., and P. A. de Boer. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol. Cell 18555-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernhardt, T. G., and P. A. J. de Boer. 2003. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 481171-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhavsar, A. P., and E. D. Brown. 2006. Cell wall assembly in Bacillus subtilis: how spirals and spaces challenge paradigms. Mol. Microbiol. 601077-1090. [DOI] [PubMed] [Google Scholar]
- 10.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37911-917. [DOI] [PubMed] [Google Scholar]
- 11.Bolivar, F., R. L. Rodriguez, P. J. Greene, M. C. Betlach, H. L. Heyneker, and H. W. Boyer. 1977. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 295-113. [PubMed] [Google Scholar]
- 12.Bouloc, P., D. Vinella, and R. D'Ari. 1992. Leucine and serine induce mecillinam resistance in Escherichia coli. Mol. Gen. Genet. 235242-246. [DOI] [PubMed] [Google Scholar]
- 13.Cabeen, M. T., and C. Jacobs-Wagner. 2005. Bacterial cell shape. Nat. Rev. Microbiol. 3601-610. [DOI] [PubMed] [Google Scholar]
- 14.Carballido-López, R. 2006. The bacterial actin-like cytoskeleton. Microbiol. Mol. Biol. Rev. 70888-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carballido-Lopez, R., A. Formstone, Y. Li, S. D. Ehrlich, P. Noirot, and J. Errington. 2006. Actin homolog MreBH governs cell morphogenesis by localization of the cell wall hydrolase LytE. Dev. Cell 11399-409. [DOI] [PubMed] [Google Scholar]
- 16.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1589-14. [DOI] [PubMed] [Google Scholar]
- 17.Chernomordik, L. V., and M. M. Kozlov. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72175-207. [DOI] [PubMed] [Google Scholar]
- 18.Churchward, G., D. Belin, and Y. Nagamine. 1984. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 31165-171. [DOI] [PubMed] [Google Scholar]
- 19.Corbin, B. D., X. C. Yu, and W. Margolin. 2002. Exploring intracellular space: function of the Min system in round-shaped Escherichia coli. EMBO J. 211998-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa, C. S., and D. N. Anton. 1999. Conditional lethality of cell shape mutations of Salmonella typhimurium: rodA and mre mutants are lethal on solid but not in liquid medium. Curr. Microbiol. 38137-142. [DOI] [PubMed] [Google Scholar]
- 21.Cronan, J. E. 2003. Bacterial membrane lipids: where do we stand? Annu. Rev. Microbiol. 57203-224. [DOI] [PubMed] [Google Scholar]
- 22.Daniel, R. A., and J. Errington. 2003. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell 113767-776. [DOI] [PubMed] [Google Scholar]
- 23.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Boer, P. A. J., R. E. Crossley, and L. I. Rothfield. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56641-649. [DOI] [PubMed] [Google Scholar]
- 25.de Boer, P. A. J., R. E. Crossley, and L. I. Rothfield. 1988. Isolation and properties of minB, a complex genetic locus involved in correct placement of the division site in Escherichia coli. J. Bacteriol. 1702106-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Defeu Soufo, H. J., and P. L. Graumann. 2005. Bacillus subtilis actin-like protein MreB influences the positioning of the replication machinery and requires membrane proteins MreC/D and other actin-like proteins for proper localization. BMC Cell Biol. 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Den Blaauwen, T., M. E. Aarsman, N. O. Vischer, and N. Nanninga. 2003. Penicillin-binding protein PBP2 of Escherichia coli localizes preferentially in the lateral wall and at mid-cell in comparison with the old cell pole. Mol. Microbiol. 47539-547. [DOI] [PubMed] [Google Scholar]
- 28.de Pedro, M. A., W. D. Donachie, J. V. Höltje, and H. Schwarz. 2001. Constitutive septal murein synthesis in Escherichia coli with impaired activity of the morphogenetic proteins RodA and penicillin-binding protein 2. J. Bacteriol. 1834115-4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Divakaruni, A. V., R. R. Loo, Y. Xie, J. A. Loo, and J. W. Gober. 2005. The cell-shape protein MreC interacts with extracytoplasmic proteins including cell wall assembly complexes in Caulobacter crescentus. Proc. Natl. Acad. Sci. USA 10218602-18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dye, N. A., Z. Pincus, J. A. Theriot, L. Shapiro, and Z. Gitai. 2005. Two independent spiral structures control cell shape in Caulobacter. Proc. Natl. Acad. Sci. USA 10218608-18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ehlert, K., and J. V. Höltje. 1996. Role of precursor translocation in coordination of murein and phospholipid synthesis in Escherichia coli. J. Bacteriol. 1786766-6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figge, R. M., A. V. Divakaruni, and J. W. Gober. 2004. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol. Microbiol. 511321-1332. [DOI] [PubMed] [Google Scholar]
- 33.Furste, J. P., W. Pansegrau, R. Frank, H. Blocker, P. Scholz, M. Bagdasarian, and E. Lanka. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48119-131. [DOI] [PubMed] [Google Scholar]
- 34.Gerding, M. A., Y. Ogata, N. D. Pecora, H. Niki, and P. A. de Boer. 2007. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol. Microbiol. 631008-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gitai, Z., N. A. Dye, A. Reisenauer, M. Wachi, and L. Shapiro. 2005. MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120329-341. [DOI] [PubMed] [Google Scholar]
- 36.Guyer, M. S., R. R. Reed, J. A. Steitz, and K. B. Low. 1981. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harbor Symp. Quant. Biol. 45135-140. [DOI] [PubMed] [Google Scholar]
- 37.Hale, C. A., and P. A. J. de Boer. 1999. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ and independent of FtsA. J. Bacteriol. 181167-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henning, U., K. Rehn, V. Braun, B. Hohn, and U. Schwarz. 1972. Cell envelope and shape of Escherichia coli K12. Properties of a temperature-sensitive rod mutant. Eur. J. Biochem. 26570-586. [DOI] [PubMed] [Google Scholar]
- 39.Henriques, A. O., P. Glaser, P. J. Piggot, and C. P. Moran. 1998. Control of cell shape and elongation by the rodA gene in Bacillus subtilis. Mol. Microbiol. 28235-247. [DOI] [PubMed] [Google Scholar]
- 40.Höltje, J. V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62181-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikeda, M., T. Sato, M. Wachi, H. K. Jung, F. Ishino, Y. Kobayashi, and M. Matsuhashi. 1989. Structural similarity among Escherichia coli FtsW and RodA proteins and Bacillus subtilis SpoVE protein, which function in cell division, cell elongation, and spore formation, respectively. J. Bacteriol. 1716375-6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishino, F., W. Park, S. Tomioka, S. Tamaki, I. Takase, K. Kunugita, H. Matsuzawa, S. Asoh, T. Ohta, B. G. Spratt, and M. Matsuhashi. 1986. Peptidoglycan synthetic activities in membranes of Escherichia coli caused by overproduction of penicillin-binding protein 2 and RodA protein. J. Biol. Chem. 2617024-7031. [PubMed] [Google Scholar]
- 43.Iwai, N., K. Nagai, and M. Wachi. 2002. Novel S-benzylisothiourea compound that induces spherical cells in Escherichia coli probably by acting on a rod-shape-determining protein(s) other than penicillin-binding protein 2. Biosci. Biotechnol. Biochem. 662658-2662. [DOI] [PubMed] [Google Scholar]
- 44.Johnson, J. E., L. L. Lackner, and P. A. J. de Boer. 2002. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ-rings. J. Bacteriol. 1842951-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson, J. E., L. L. Lackner, C. A. Hale, and P. A. De Boer. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J. Bacteriol. 1862418-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones, L. J., R. Carballido-Lopez, and J. Errington. 2001. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell 104913-922. [DOI] [PubMed] [Google Scholar]
- 47.Joseleau-Petit, D., D. Thevenet, and R. D'Ari. 1994. ppGpp concentration, growth without PBP2 activity, and growth-rate control in Escherichia coli. Mol. Microbiol. 13911-917. [DOI] [PubMed] [Google Scholar]
- 48.Kruse, T., B. Blagoev, A. Lobner-Olesen, M. Wachi, K. Sasaki, N. Iwai, M. Mann, and K. Gerdes. 2006. Actin homolog MreB and RNA polymerase interact and are both required for chromosome segregation in Escherichia coli. Genes Dev. 20113-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kruse, T., J. Bork-Jensen, and K. Gerdes. 2005. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 5578-89. [DOI] [PubMed] [Google Scholar]
- 50.Kruse, T., J. Moller-Jensen, A. Lobner-Olesen, and K. Gerdes. 2003. Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 225283-5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lackner, L. L., D. M. Raskin, and P. A. de Boer. 2003. ATP-dependent interactions between Escherichia coli Min proteins and the phospholipid membrane in vitro. J. Bacteriol. 185735-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leaver, M., and J. Errington. 2005. Roles for MreC and MreD proteins in helical growth of the cylindrical cell wall in Bacillus subtilis. Mol. Microbiol. 571196-11209. [DOI] [PubMed] [Google Scholar]
- 53.Lee, J. C., J. H. Cha, D. B. Zerbv, and G. C. Stewart. 2003. Heterospecific expression of the Bacillus subtilis cell shape determination genes mreBCD in Escherichia coli. Curr. Microbiol. 47146-152. [DOI] [PubMed] [Google Scholar]
- 54.Long, W. S., C. L. Slayman, and K. B. Low. 1978. Production of giant cells of Escherichia coli. J. Bacteriol. 133995-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowe, J., F. van den Ent, and L. A. Amos. 2004. Molecules of the bacterial cytoskeleton. Annu. Rev. Biophys. Biomol. Struct. 33177-198. [DOI] [PubMed] [Google Scholar]
- 56.Matsuzawa, H., S. Asoh, K. Kunai, K. Muraiso, A. Takasuga, and T. Ohta. 1989. Nucleotide sequence of the rodA gene, responsible for the rod shape of Escherichia coli: rodA and the pbpA gene, encoding penicillin-binding protein 2, constitute the rodA operon. J. Bacteriol. 171558-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMahon, H. T., and J. L. Gallop. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438590-596. [DOI] [PubMed] [Google Scholar]
- 58.Mercer, K. L., and D. S. Weiss. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J. Bacteriol. 184904-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navarro, F., A. Robin, R. D'Ari, and D. Joseleau-Petit. 1998. Analysis of the effect of ppGpp on the ftsQAZ operon in Escherichia coli. Mol. Microbiol. 29815-823. [DOI] [PubMed] [Google Scholar]
- 60.Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67593-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nilsen, T., A. W. Yan, G. Gale, and M. B. Goldberg. 2005. Presence of multiple sites containing polar material in spherical Escherichia coli cells that lack MreB. J. Bacteriol. 1876187-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogura, T., P. Bouloc, H. Niki, R. D'Ari, S. Hiraga, and A. Jaffe. 1989. Penicillin-binding protein 2 is essential in wild-type Escherichia coli but not in lov or cya mutants. J. Bacteriol. 1713025-3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Onoda, T., J. Enokizono, H. Kaya, A. Oshima, P. Freestone, and V. Norris. 2000. Effects of calcium and calcium chelators on growth and morphology of Escherichia coli L-form NC-7. J. Bacteriol. 1821419-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Podkovyrov, S., and T. J. Larson. 1995. Lipid biosynthetic genes and a ribosomal protein gene are cotranscribed. FEBS Lett. 368429-431. [DOI] [PubMed] [Google Scholar]
- 65.Raetz, C. R. 1978. Enzymology, genetics, and regulation of membrane phospholipid synthesis in Escherichia coli. Microbiol. Rev. 42614-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raskin, D. M., and P. A. J. de Boer. 1997. The MinE ring: an FtsZ-independent cell structure required for selection of the correct division site in E. coli. Cell 91685-694. [DOI] [PubMed] [Google Scholar]
- 67.Raskin, D. M., and P. A. J. de Boer. 1999. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc. Natl. Acad. Sci. USA 964971-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothfield, L., A. Taghbalout, and Y. L. Shih. 2005. Spatial control of bacterial division-site placement. Nat. Rev. Microbiol. 3959-968. [DOI] [PubMed] [Google Scholar]
- 69.Schreiber, G., S. Metzger, E. Aizenman, S. Roza, M. Cashel, and G. Glaser. 1991. Overexpression of the relA gene in Escherichia coli. J. Biol. Chem. 2663760-3767. [PubMed] [Google Scholar]
- 70.Shih, Y. L., I. Kawagishi, and L. Rothfield. 2005. The MreB and Min cytoskeletal-like systems play independent roles in prokaryotic polar differentiation. Mol. Microbiol. 58917-928. [DOI] [PubMed] [Google Scholar]
- 71.Shih, Y. L., T. Le, and L. Rothfield. 2003. Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc. Natl. Acad. Sci. USA 1007865-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Slovak, P. M., S. L. Porter, and J. P. Armitage. 2006. Differential localization of Mre proteins with PBP2 in Rhodobacter sphaeroides. J. Bacteriol. 1881691-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soufo, H. J., and P. L. Graumann. 2003. Actin-like proteins MreB and Mbl from Bacillus subtilis are required for bipolar positioning of replication origins. Curr. Biol. 131916-1920. [DOI] [PubMed] [Google Scholar]
- 74.Spratt, B. G. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 722999-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spratt, B. G., A. Boyd, and N. Stoker. 1980. Defective and plaque-forming lambda transducing bacteriophage carrying penicillin-binding protein-cell shape genes: genetic and physical mapping and identification of gene products from the _lip_-_dacA_-_rodA_-_pbpA_-leuS region of the Escherichia coli chromosome. J. Bacteriol. 143569-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tamaki, S., H. Matsuzawa, and M. Matsuhashi. 1980. Cluster of mrdA and mrdB genes responsible for the rod shape and mecillinam sensitivity of Escherichia coli. J. Bacteriol. 14152-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thanedar, S., and W. Margolin. 2004. FtsZ exhibits rapid movement and oscillation waves in helix-like patterns in Escherichia coli. Curr. Biol. 141167-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tiyanont, K., T. Doan, M. B. Lazarus, X. Fang, D. Z. Rudner, and S. Walker. 2006. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc. Natl. Acad. Sci. USA 10311033-11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van den Ent, F., L. A. Amos, and J. Lowe. 2001. Prokaryotic origin of the actin cytoskeleton. Nature 41339-44. [DOI] [PubMed] [Google Scholar]
- 80.van den Ent, F., M. Leaver, F. Bendezu, J. Errington, P. de Boer, and J. Lowe. 2006. Dimeric structure of the cell shape protein MreC and its functional implications. Mol. Microbiol. 621631-1642. [DOI] [PubMed] [Google Scholar]
- 81.Vinella, D., R. D'Ari, A. Jaffe, and P. Bouloc. 1992. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J. 111493-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vinella, D., D. Joseleau-Petit, D. Thevenet, P. Bouloc, and R. D'Ari. 1993. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J. Bacteriol. 1756704-6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vischer, N. O. E., P. G. Huls, and C. L. Woldringh. 1994. Object-Image: an interactive image analysis program using structured point collection. Binary 6160-166. [Google Scholar]
- 84.Wachi, M., M. Doi, Y. Okada, and M. Matsuhashi. 1989. New mre genes mreC and mreD, responsible for formation of the rod shape of Escherichia coli cells. J. Bacteriol. 1716511-6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wachi, M., M. Doi, S. Tamaki, W. Park, S. Nakajima-Iijima, and M. Matsuhashi. 1987. Mutant isolation and molecular cloning of mre genes, which determine cell shape, sensitivity to mecillinam, and amount of penicillin-binding proteins in Escherichia coli. J. Bacteriol. 1694935-4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wachi, M., K. Osaka, T. Kohama, K. Sasaki, I. Ohtsu, N. Iwai, A. Takada, and K. Nagai. 2006. Transcriptional analysis of the Escherichia coli mreBCD genes responsible for morphogenesis and chromosome segregation. Biosci. Biotechnol. Biochem. 702712-2719. [DOI] [PubMed] [Google Scholar]
- 87.Wang, X., P. A. J. de Boer, and L. I. Rothfield. 1991. A factor that positively regulates cell division by activating transcription of the major cluster of essential cell division genes of Escherichia coli. EMBO J. 103363-3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ward, J. E., and J. Lutkenhaus. 1985. Overproduction of FtsZ induces minicell formation in E. coli. Cell 42941-949. [DOI] [PubMed] [Google Scholar]
- 89.Wei, Y., J. M. Lee, D. R. Smulski, and R. A. LaRossa. 2001. Global impact of sdiA amplification revealed by comprehensive gene expression profiling of Escherichia coli. J. Bacteriol. 1832265-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Young, K. D. 2003. Bacterial shape. Mol. Microbiol. 49571-580. [DOI] [PubMed] [Google Scholar]
- 91.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 975978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]