Phospho-regulation of β-Catenin Adhesion and Signaling Functions (original) (raw)
. Author manuscript; available in PMC: 2008 Mar 31.
Published in final edited form as: Physiology (Bethesda). 2007 Oct;22:303–309. doi: 10.1152/physiol.00020.2007
Abstract
β-Catenin plays a critical structural role in cadherin-based adhesions and is also an essential co-activator of Wnt-mediated gene expression. The degree to which β-catenin participates in these two functions is dictated by the availability of β-catenin binding partners, and an emerging theme is that these binding interactions are regulated by phosphorylation. Inputs from various cell-signaling events can therefore impact β-catenin function, which may be necessary for the finely tuned adhesive and signaling responses required for tissue morphogenesis.
The ability of cells to adhere and differentiate into distinct tissues is a defining feature of multicellular organisms. The cadherin/catenin adhesion system is the major means by which cells adhere to one another. Without this protein complex, the cell-cell rearrangements that drive morphogenetic processes, such as gastrulation and dorsal closure, are completely arrested (73). β-Catenin, a central, structural component of this adhesion complex, is also required for mediating differentiation processes initiated by Wnt signals. Wnts are secreted lipoglycoproteins that act as morphogens to pattern tissues and are used reiteratively throughout development to instruct cells to adopt particular fates. These cell fate decisions are directed by the expression of various genes that are cell type and context dependent (reviewed in Ref. 12). For example, in intestine, the Wnt pathway maintains progenitor and stem cell populations (75), whereas in liver Wnts control nitrogen metabolism and ammonia detoxification along the periportal axis (4). Wnt-mediated gene expression is ultimately controlled by a transcriptional complex that contains a DNA-binding factor known as lymphocyte enhancer factor (LEF)/T-cell factor (TCF) and a cadherin-free form of β-catenin. In this complex, β-catenin serves as an obligate co-activator through its ability to recruit components that promote chromatin remodeling and transcriptional initiation/elongation (review in Ref. 80).
There has been much interest in understanding how adhesion and Wnt signaling are coordinated through the use of this common component, β-catenin. Forced overexpression of cadherins can compete for and sequester the signaling pool of β-catenin (21, 33). Conversely, reductions in cadherin protein levels can enhance β-catenin signaling (14), suggesting that the absolute level of cadherins may set thresholds for Wnt signals. More recent studies, however, imply that the extent to which β-catenin is used for signaling and adhesion can be strongly influenced by the phosphorylation of β-catenin binding partners. This review will highlight recent evidence showing the importance of phosphorylation in the regulation of β-catenin nuclear signaling and adhesive functions.
β-Catenin Structure
β-Catenin belongs to the armadillo family of proteins, which are characterized by a central domain consisting of a repeating 42 amino acid motif termed the “arm repeat.” These repeats were originally identified in the Drosophila segment polarity gene product and β-catenin ortholog Armadillo (60). This repeat has since been found in other proteins, such as the adhesion proteins plakoglobin (also known as γ-catenin) and p120ctn, and is considered to be a versatile protein-binding interface (13). X-ray crystallographic analysis of the central armadillo domain of β-catenin shows that its 12 arm repeats form a superhelix of helices that create a long, positively charged groove (39). β-Catenin uses this single binding surface to interact with many of its negatively charged ligands, such as the cadherin adhesion receptor, the Axin/APC degradation complex, and the LEF/TCF transcription factors.
Since β-catenin engages these various binding partners in a mutually exclusive fashion, β-catenin function is dictated by which factor associates with the arm domain. Although not universal, an emerging theme is that phosphorylation of these binding partners introduces negative charges that enhance interactions along β-catenin’s positively charged groove and, therefore, increases binding affinity. Thus ligand phosphorylation can influence the extent to which β-catenin is used for cell-cell adhesion or transcription. Phosphorylation of β-catenin itself can affect some of these ligand interactions, and therefore these cases will also be discussed. The NH2 and COOH termini of β-catenin are unstructured regulatory regions that largely recruit essential co-factors for adhesion and signaling (see below). In certain circumstances, the COOH terminus may limit the availability of the β-catenin arm repeat region for cadherins or APC (8, 11, 26).
β-Catenin and the Cadherin Adhesive Complex
β-Catenin is a central component of the cadherin/catenin adhesive complex (FIGURE 1). Cadherins are type I, single-pass transmembrane glycoproteins that mediate Ca2+-dependent intercellular adhesion. Specific adhesive binding is conferred by the cadherin ectodomain, which engages an identical molecule on the surface of an adjacent cell (5, 45), whereas the cadherin cytoplasmic domain mediates the structural and signaling activities required for adhesion. In addition to an interaction with β-catenin, cadherins associate with two other catenin proteins, termed α- and p120-catenin. α-Catenin is an actin binding protein that dynamically links the cadherin complex to the actin cytoskeleton (19, 61, 85). α-Catenin lacks an armadillo domain and is, therefore, structurally unrelated to β-catenin. p120ctn belongs to a subfamily of armadillo proteins and regulates cadherin surface levels by antagonizing endocytosis (17, 83) and promoting cadherin clustering (87), possibly through its ability to inhibit Rho (79). Although α-catenin and p120ctn are important regulators of cell-cell adhesion (reviewed in Refs. 24, 67), β-catenin binding to cadherin remains a prerequisite for adhesion due to its role in protecting the cadherin cytoplasmic domain from rapid degradation (40), enhancing the efficiency of endoplasmic reticulum to cell surface transport (10) and recruiting α-catenin to sites of cell-cell contact (19, 85). Therefore, posttranslation modifications that regulate the β-catenin/cadherin interaction will have important consequences for cell-cell adhesion.
FIGURE 1. Summary of β-catenin-mediated adhesion and signaling.
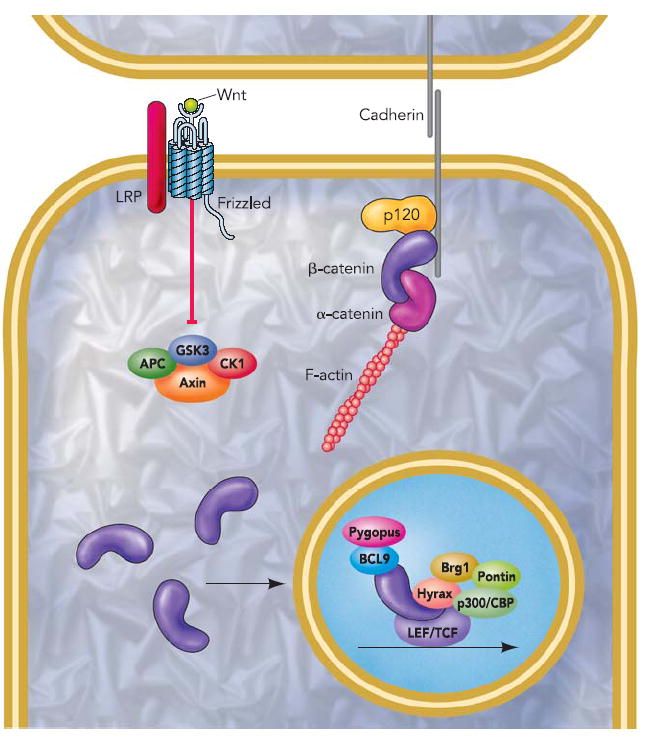
See text and http://www.stanford.edu/~rnusse/wntwindow.html for details.
Phospho-regulation of the β-Catenin/Cadherin Interaction
Although it has been long appreciated that cadherins are phosphorylated within a serine-rich stretch that comprises the β-catenin binding domain (70), the contribution of distinct phosphorylations has been only recently revealed. Specifically, in vitro phosphorylation of cadherin serine residues 834, 836, and 842 significantly enhances the affinity with which β-catenin binds cadherins by 300-fold (Refs. 11, 41; note that numbering for E-cadherin is based on the sequence of Rimm and Morrow, Ref. 62). Comparison of co-crystals comprised of phospho-cadherin/β-catenin and non-phospho-cadherin/β-catenin complexes reveals a molecular explanation for this affinity difference, since phosphorylation generates more molecular contacts at the cadherin/β-catenin interface. Although GSK3β and CK2 have been shown to robustly phosphorylate these sites in vitro (41, 48), the endogenous cell-signaling events, kinases, and in vivo phospho-sites have remained poorly defined.
Not all cadherin phosphorylation sites enhance β-catenin binding affinity. For example, phosphorylation of cadherin at S-846 by CK1 (S-840 in the Rimm and Morrow E-cadherin sequence) can reduce β-catenin binding to cadherin, and a mutant E-cadherin that mimics phosphorylation at this site (S846D) exhibits decreased adhesive activity due to enhanced internalization (20). Similarly, tyrosine phosphorylation of N-cadherin by src at Y860 (corresponding to Y831 in E-cadherin) antagonizes β-catenin binding. Expression of a mutant N-cadherin that cannot be phosphorylated at this site (Y860F) prevents the transmigration of cells across an endothelial monolayer (59). Since both CK1 and src sites are in close proximity to the CK2 sites described by Huber and Weis (41), it will be important to see whether these phosphorylation-dependent reductions in β-catenin binding to cadherin can now be rationalized at the molecular level. Notwithstanding these rationalizations, it certainly appears that the role of cadherin phosphorylation in modulating β-catenin binding affinity will be both kinase and site dependent, and may even depend on the precise order of these phosphorylations (9).
Although this review is mostly focused on the phosphorylation of β-catenin binding partners, it is important to mention that the ligand-binding region of β-catenin is itself a substrate of src and EGFR family kinases, and phosphorylation of tyrosine 654 (which sits in the 12th arm repeat of β-catenin) can reduce cadherin binding and adhesive functions (55, 63). Importantly, the consequences of phosphorylating this residue have been rationalized at the molecular level, since pY-654 clashes with a key aspartate residue in the cadherin (41). Although the aforementioned studies present clear evidence that the β-catenin/cadherin binding interaction can be regulated, whether these phosphorylations control β-catenin binding like a simple on/off switch or in a more graded fashion is currently under debate. In this regard, a number of physiological situations are known to exist where cadherin adhesive activity is altered (e.g., during embryo compaction or convergence/extension movements required for gastrulation); however, evidence for wide-scale perturbation of the β-catenin/cadherin binding is lacking (reviewed in Ref. 28). Thus it will be important to learn more about the various ways that this binding interface can be regulated. Despite these complexities, it certainly seems that phosphorylation events that modulate the cadherin/β-catenin binding interaction can affect the abundance and/or function of cadherin adhesive complexes (FIGURE 2A). Moreover, evidence that cadherin/β-catenin binding can be influenced by CK1, GSK3β, and CK2 kinases, which play key roles in regulating the nuclear signaling form of β-catenin (discussed below), opens up the possibility that this interaction may be even modulated by Wnt signals.
FIGURE 2. Phospho-regulation of β-catenin protein interactions.
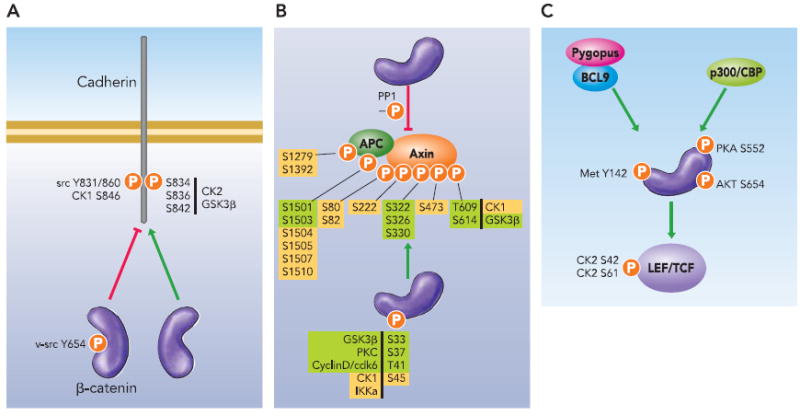
A: phosphorylation of cadherin residue, Y860, and β-catenin residue, Y654, by src prevents the cadherin/β catenin interaction. Also inhibitory to the cadherin/catenin interaction is phosphorylation of cadherin S846 (S840 in the human E-cadherin sequence) by CK1. In contrast, GSK3β and CK2-mediated phosphorylation of cadherin S834, S836, and S842 enhances associations with β -catenin. B: β catenin preferentially binds to phosphorylated APC/axin complex. CK1 and GSK3β hyperphosphorylate the degradation complex and increase the affinity of APC/axin for β-catenin. Once bound to APC/axin, β-catenin becomes phosphorylated at S45 by CK1 (or IKKα and cyclinD/cdk6). Subsequent phosphorylations at S33, S37, and T41 by GSK3β (or PKC) lead to ubiquitination and degradation of β-catenin. Phosphatase PP1 reverses CK1-mediated phosphorylation of axin and allows β-catenin to escape the degradation complex. Corresponding kinases and phosphorylations are highlighted in the same color. C: phosphorylation of LEF/TCF by CK2 enhances interaction with β-catenin and promotes the recruitment of β-catenin to Wnt-regulated promoters. Met-mediated phosphorylation of Y142 allows BCL9/pygopus to bind the NH2 terminus of β-catenin, whereas phosphorylation of S552 and S675 by Akt and PKA promote interactions with additional transcriptional coactivators, such as CBP. See text for references. Arrows reflect enhanced interactions. P, phosphorylation site.
Phospho-regulation of β-Catenin Stability
Although cadherin-bound β-catenin is relatively stable, the cytosolic pool of β-catenin is continually flagged for degradation by an elaborate, phosphorylation-based mechanism. The NH2 terminus of cytosolic β-catenin is constitutively phosphorylated by a dual-kinase mechanism. CK1α phosphorylates β-catenin at serine 45, and this priming phosphorylation results in subsequent phosphorylation by GSK3β at residues 41, 37, and 33 (49, 88) (FIGURE 2B). These phosphorylation events are coordinated by the scaffold protein axin, which has binding sites for β-catenin, CK1, GSK3β, as well as other factors required for Wnt-dependent and -independent signaling events (50). β-Catenin that is phosphorylated at residues 37 and 33 is ultimately recognized by the β-TrCP E3-ligase complex, ubiquitylated, and rapidly degraded by the 26S proteasome (31). The adenomatous polyposis coli (APC) tumor suppressor gene product is also an axin-binding partner and is thought to remove NH2 terminally phosphorylated β-catenin from the axin complex for transfer to the degradation machinery (84). Thus phosphorylation-dependent degradation of β-catenin is a key step in turning off Wnt signals. Importance for this mechanism is further supported by the existence of loss-of-function mutations in APC and axin as well as activating point mutations within β-catenin’s NH2 terminal phosphorylation sites, which play causal roles in various cancers (57).
“A key consequence of Wnt-mediated axin dephosphorylation and GSK3β inhibition is that residues 41, 37, and 33 of β-catenin remain unphosphorylated, and bTrCP-mediated degradation is prevented.”
Phospho-regulation of the β-Catenin Degradation Complex
We are beginning to appreciate that the recruitment and progression of β catenin through the axin/APC degradation complex is regulated by a series of ordered phosphorylation events. Phosphorylation of axin by CK1 and GSK3β can increase axin binding to β-catenin (42, 51, 81). This allows β-catenin to be a more efficient substrate of CK1α and GSK3β, enhancing NH2 terminal phosphorylation of β-catenin by >20,000-fold (15). Axin also promotes phosphorylation of APC by CK1ε and GSK3β (64, 65), increasing the affinity of APC for β-catenin from 3 μM to 10 nM (30). Since the affinity of phospho-APC for β-catenin is stronger than the affinity of phospho-axin for β-catenin, APC can compete with axin for binding to β-catenin. This phosphorylation-dependent competition, therefore, may displace axin from β-catenin, allowing Axin to bind another molecule of β-catenin. Although it remains unclear how APC coordinates phospho-β-catenin recognition by β-TrCP and the proteasome, it is currently appreciated that APC inhibits β-catenin signaling by promoting the flux of β-catenin molecules through the axin complex (30).
Wnt signals inhibit the axin degradation complex by acting through a receptor complex that contains a seven-pass transmembrane protein known as Frizzled (Fz), along with its co-receptor, low-density receptor-related protein (LRP 5 or 6) (7, 77). Co-receptor engagement activates a signaling “relay” protein known as Disheveled (Dsh), and together, Fz/LRP and Dsh can antagonize the axin complex at multiple levels. Activated LRP can bind axin (16, 89), which may disrupt the axin scaffold complex (25). In addition, Dsh can inhibit GSK3β activity (47, 88), resulting in decreased axin phosphorylation and binding to β-catenin (42, 51, 81). Importantly, a recent study has found that Wnt-induced axin dephosphorylation can be mediated by a specific phosphatase, PP1 (51). Thus controlled phosphorylation and dephosphorylation of axin (by CK1/GSK3β and PP1, respectively) appear to be major means by which β-catenin levels are regulated.
Given this central role for phosphorylation in the regulation of β-catenin signaling, this multi-protein degradation complex is poised to receive inputs from other signaling pathways. In this regard, the NH2 terminal GSK3β sites of β-catenin can be also phosphorylated by PKC, which may explain how noncanonical Wnts antagonize β-catenin signaling (29). Additionally, β-catenin S45 can be phosphorylated by IKKα (58). β-Catenin S45 and axin CK1 sites can also be cross-regulated by CyclinD/Cdk6 and cyclinA/Cdk2, respectively, which may enable cell cycle regulators to attenuate Wnt signals (43, 56) (FIGURE 2B). Other examples include the cell fate regulator, Notch, and the cell metabolism regulator, TSC1, which can lower cytosolic β-catenin protein levels (32, 52). Conversely, PKA activation by trimeric Gα protein (37), cdc42 (82), and the p68RNA helicase (86) can enhance β-catenin protein levels. The mechanisms by which these very different molecular components affect axin-complex structure and function remain to be determined. Nonetheless, these data indicate that Wnt/β-catenin signals are subject to fine tuning by diverse signaling inputs, integrating cell shape, cycling, metabolism, and fate decisions.
NH2 Terminally Unphosphorylated β-Catenin and Signaling
A key consequence of Wnt-mediated axin dephosphorylation and GSK3β inhibition is that residues 41, 37, and 33 of β-catenin remain unphosphorylated, and βTrCP-mediated degradation is prevented. Although Wnt-induced accumulation of cytosolic β-catenin is a key feature of β-catenin signaling, studies have indicated that elevated levels alone cannot explain β-catenin/TCF transcriptional activation (27). Indeed, it is now appreciated that Wnt signaling is specifically mediated through molecular forms of β-catenin that remain unphosphorylated at residues 37 and 41 (69). Although there is currently no molecular explanation for the superior signaling activity of NH2 terminally unphosphorylated β-catenin, some studies observe that NH2 terminally unphosphorylated β-catenin accumulates more readily in nuclei (36, 69, 76), suggesting that phosphorylation of β-catenin at residues 33, 37, and 41 may recruit a factor that primarily promotes nuclear export. Alternatively, phosphorylation of β-catenin may restrict access to LEF-/TCF-regulated promoters, since phospho-β-catenin can interact with LEF, but ternary phospho-β-catenin/LEF/DNA complexes cannot be detected in gel-shift assays (66). Regardless of the molecular explanation, it is clear that a better understanding of the components that act on these phosphorylation sites will be required.
Phospho-regulation of β-Catenin-Mediated Transcription
After Wnt activation, the cytosolic form of β-catenin gains access to the nuclear compartment by a constitutive shuttling mechanism that is independent of classical nuclear localization sequences (22, 44). Since the armadillo repeat region of β-catenin resembles the HEAT repeats found in the nuclear import factor importin-β, it is postulated that β-catenin can mediate its own nuclear import through direct interactions with nucleoporins (71). Thus β-catenin is thought to continually shuttle in/out of the nucleus, and interactions with either cytosolic or nuclear proteins ultimately influence β-catenin’s distribution. For example, when β-catenin is co-expressed with TCF, which contains a classic lysine-rich NLS, β-catenin is preferentially localized to the nucleus (3). Conversely, when β-catenin is co-expressed with axin, it remains cytoplasmic (74). What controls whether β-catenin will be incorporated into axin (cytoplasmic) or TCF (nuclear) complexes? As discussed above, phosphorylation of axin enhances binding to β-catenin, and evidence also suggests that phosphorylation of TCF family members can influence binding to β-catenin. Specifically, phosphorylation of TCF3 by CK1 enhances, whereas GSK3β inhibits, TCF3 binding to β-catenin (46). Moreover, phosphorylation of LEF by CK2 at S42 and 61 increases the affinity of β-catenin for LEF-bound chromatinized templates and enhances gene transcription (78). Thus it appears that local activation of kinases on LEF/TCF-regulated promoters may not only drive gene-specific β-catenin recruitment but may also influence nuclear retention.
Once β-catenin binds to TCF, the NH2- and COOH-terminal regions of β-catenin recruit complexes that promote transcriptional activation. The NH2 terminus of β-catenin binds to a complex consisting of the adaptor legless/Bcl9 and pygopus, which promote transcriptional activation in a manner that is not fully understood (18, 38). The COOH-terminal region binds proteins involved in chromatin remodeling, such as CBP/p300 histone acetylases (35), Brg-1 (1), and TTRAP/TIP60 and mixed-lineage-leukemia (MLL1/MLL2) SET1-type complexes (68), as well as components that influence transcription initiation by RNA polymerase II, such as parafibromin/hyrax of the histone ubiquitination polymerase associated factor-1 (PAF1) complex (54), the helicase pontin52 (2), and TATA binding protein (34). Recent studies suggest that phosphorylation of β-catenin may modulate the recruitment of some of these factors. For example, phosphorylation of β-catenin at NH2-terminal residue Y142 by the Met receptor promotes Bcl9-2 binding (6). Moreover, phosphorylation of β-catenin at COOH-terminal residues S552 and S675 by AKT and PKA can enhance β-catenin/TCF reporter activation, possibly through association with histone acetylases (23, 72). Lastly, recent work by Miyabayashi and colleagues (53) suggests that the particular co-activators recruited to β-catenin may dictate which target genes are activated and that this differential recruitment can be regulated by phosphorylation. Specifically, the general protein phosphatase 2A (PP2A) controls binding of CBP vs. p300 to β-catenin/TCF-regulated promoters to influence the expression of growth- vs. differentiation-promoting genes (53). Thus it appears that local modulation of β-catenin transcriptional activation, either at the level of TCF/β-catenin binding or β-catenin/co-factor interactions, may be used to control the multiple nuclear targets required for Wnt-mediated tissue patterning (FIGURE 2C).
Summary
Phosphorylation and the coordination of β-catenin signaling and adhesive functions
To many, β-catenin is a curious protein: it has no obvious intrinsic catalytic activity, i.e., it is not a nucleotide binding protein that harnesses the energy of hydrolysis to move ions or exchange binding partners. Rather, β-catenin is a protein whose core armadillo repeat structure allows it to bind many partners, and it is through this binding that structural and functional information is conveyed. Phosphorylation of β-catenin and, in particular, its adhesive (cadherin), regulatory complex (axin and APC), and transcription factor (TCF) ligands has emerged as a major means of regulating the relative strength of these interactions. Better understanding of the signaling inputs that regulate these phosphorylation events will not only be important for understanding how adhesion and Wnt signaling are coordinated during tissue morphogenesis but will be important in designing strategies to attenuate the nuclear signaling/oncogenic function of β-catenin, while sparing or promoting the cadherin-based tumor suppressor activities of β-catenin.
Acknowledgments
We thank Annette Flozak, Meghan Thorne, and Rigen Mo for suggestions.
References
- 1.Barker N, Hurlstone A, Musisi H, Miles A, Bienz M, Clevers H. The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 2001;20:4935–4943. doi: 10.1093/emboj/20.17.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer A, Chauvet S, Huber O, Usseglio F, Rothbacher U, Aragnol D, Kemler R, Pradel J. Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J. 2000;19:6121–6130. doi: 10.1093/emboj/19.22.6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 4.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, Colnot S. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 5.Boller K, Vestweber D, Kemler R. Cell-adhesion molecule uvo-morulin is localized in the intermediate junctions of adult intestinal epithelial cells. J Cell Biol. 1985;100:327–332. doi: 10.1083/jcb.100.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between beta-catenin’s adhesive and transcriptional functions. Genes Dev. 2004;18:2225–2230. doi: 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 8.Castano J, Raurell I, Piedra JA, Miravet S, Dunach M, Garcia de Herreros A. Beta-catenin N- and C-terminal tails modulate the coordinated binding of adherens junction proteins to beta-catenin. J Biol Chem. 2002;277:31541–31550. doi: 10.1074/jbc.M204376200. [DOI] [PubMed] [Google Scholar]
- 9.Catimel B, Layton M, Church N, Ross J, Condron M, Faux M, Simpson RJ, Burgess AW, Nice EC. In situ phosphorylation of immobilized receptors on biosensor surfaces: application to E-cadherin/beta-catenin interactions. Anal Biochem. 2006;357:277–288. doi: 10.1016/j.ab.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 10.Chen YT, Stewart DB, Nelson WJ. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J Cell Biol. 1999;144:687–699. doi: 10.1083/jcb.144.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi HJ, Huber AH, Weis WI. Thermodynamics of beta-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J Biol Chem. 2006;281:1027–1038. doi: 10.1074/jbc.M511338200. [DOI] [PubMed] [Google Scholar]
- 12.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 2003;13:463–471. doi: 10.1016/s0962-8924(03)00167-3. [DOI] [PubMed] [Google Scholar]
- 14.Cox RT, Kirkpatrick C, Peifer M. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J Cell Biol. 1996;134:133–148. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, Dale TC, Pearl LH. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003;22:494–501. doi: 10.1093/emboj/cdg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- 17.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de la Roche M, Bienz M. Wingless-independent association of Pygopus with dTCF target genes. Curr Biol. 2007;17:556–561. doi: 10.1016/j.cub.2007.01.063. [DOI] [PubMed] [Google Scholar]
- 19.Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dupre-Crochet S, Figueroa A, Hogan C, Ferber EC, Bialucha CU, Adams J, Richardson EC, Fujita Y. Casein kinase 1 is a novel negative regulator of e-cadherin-based cell-cell contacts. Mol Cell Biol. 2007;27:3804–3816. doi: 10.1128/MCB.01590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fagotto F, Funayama N, Gluck U, Gumbiner B. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol. 1996;132:1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr Biol. 1998;8:181–190. doi: 10.1016/s0960-9822(98)70082-x. [DOI] [PubMed] [Google Scholar]
- 23.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox DT, Peifer M. Cell adhesion: separation of p120’s powers? Curr Biol. 2007;17:R24–R27. doi: 10.1016/j.cub.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 25.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci USA. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottardi CJ, Gumbiner BM. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J Cell Biol. 2004;167:339–349. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guger KA, Gumbiner BM. A mode of regulation of beta-catenin signaling activity in Xenopus embryos independent of its levels. Dev Biol. 2000;223:441–448. doi: 10.1006/dbio.2000.9770. [DOI] [PubMed] [Google Scholar]
- 28.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 29.Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim EY, Lee SS, Kim M, Kim TK, Shin JG, Oh S. Protein-kinase-C-mediated beta-catenin phosphorylation negatively regulates the Wnt/beta-catenin pathway. J Cell Sci. 2006;119:4702–4709. doi: 10.1242/jcs.03256. [DOI] [PubMed] [Google Scholar]
- 30.Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9:207–210. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 32.Hayward P, Brennan K, Sanders P, Balayo T, DasGupta R, Perrimon N, Martinez Arias A. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development. 2005;132:1819–1830. doi: 10.1242/dev.01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, Kintner C, Noro C, Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 34.Hecht A, Litterst CM, Huber O, Kemler R. Functional characterization of multiple transactivating elements in beta-catenin, some of which interact with the TATA-binding protein in vitro. J Biol Chem. 1999;274:18017–18025. doi: 10.1074/jbc.274.25.18017. [DOI] [PubMed] [Google Scholar]
- 35.Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000;19:1839–1850. doi: 10.1093/emboj/19.8.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendriksen J, Fagotto F, van der Velde H, van Schie M, Noordermeer J, Fornerod M. RanBP3 enhances nuclear export of active (beta)-catenin independently of CRM1. J Cell Biol. 2005;171:785–797. doi: 10.1083/jcb.200502141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005;25:9063–9072. doi: 10.1128/MCB.25.20.9063-9072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmans R, Stadeli R, Basler K. Pygopus and legless provide essential transcriptional coactivator functions to armadillo/beta-catenin. Curr Biol. 2005;15:1207–1211. doi: 10.1016/j.cub.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 39.Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 40.Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI. The cadherin cytoplasmic domain is unstructured in the absence of beta-catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem. 2001;276:12301–12309. doi: 10.1074/jbc.M010377200. [DOI] [PubMed] [Google Scholar]
- 41.Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- 42.Jho E, Lomvardas S, Costantini F. A GSK3beta phosphorylation site in axin modulates interaction with beta-catenin and Tcf-mediated gene expression. Biochem Biophys Res Commun. 1999;266:28–35. doi: 10.1006/bbrc.1999.1760. [DOI] [PubMed] [Google Scholar]
- 43.Kim SI, Park CS, Lee MS, Kwon MS, Jho EH, Song WK. Cyclin-dependent kinase 2 regulates the interaction of Axin with beta-catenin. Biochem Biophys Res Commun. 2004;317:478–483. doi: 10.1016/j.bbrc.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 44.Krieghoff E, Behrens J, Mayr B. Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J Cell Sci. 2006;119:1453–1463. doi: 10.1242/jcs.02864. [DOI] [PubMed] [Google Scholar]
- 45.Leckband D, Sivasankar S. Mechanism of homophilic cadherin adhesion. Curr Opin Cell Biol. 2000;12:587–592. doi: 10.1016/s0955-0674(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 46.Lee E, Salic A, Kirschner MW. Physiological regulation of beta-catenin stability by Tcf3 and CK1epsilon. J Cell Biol. 2001;154:983–993. doi: 10.1083/jcb.200102074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Yuan H, Weaver CD, Mao J, Farr GH, 3rd, Sussman DJ, Jonkers J, Kimelman D, Wu D. Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1. EMBO J. 1999;18:4233–4240. doi: 10.1093/emboj/18.15.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lickert H, Bauer A, Kemler R, Stappert J. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/beta-catenin interaction and strengthens cell-cell adhesion. J Biol Chem. 2000;275:5090–5095. doi: 10.1074/jbc.275.7.5090. [DOI] [PubMed] [Google Scholar]
- 49.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 50.Luo W, Lin SC. Axin: a master scaffold for multiple signaling pathways. Neurosignals. 2004;13:99–113. doi: 10.1159/000076563. [DOI] [PubMed] [Google Scholar]
- 51.Luo W, Peterson A, Garcia BA, Coombs G, Kofahl B, Heinrich R, Shabanowitz J, Hunt DF, Yost HJ, Virshup DM. Protein phosphatase 1 regulates assembly and function of the beta-catenin degradation complex. EMBO J. 2007;26:1511–1521. doi: 10.1038/sj.emboj.7601607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates beta-catenin signaling activity. J Biol Chem. 2003;278:5947–5951. doi: 10.1074/jbc.C200473200. [DOI] [PubMed] [Google Scholar]
- 53.Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668–5673. doi: 10.1073/pnas.0701331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mosimann C, Hausmann G, Basler K. Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/armadillo. Cell. 2006;125:327–341. doi: 10.1016/j.cell.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 55.Murase S, Mosser E, Schuman EM. Depolarization drives beta-catenin into neuronal spines promoting changes in synaptic structure and function. Neuron. 2002;35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- 56.Park CS, Lee MS, Oh HJ, Choi KY, Yeo MG, Chun JS, Song WK. Modulation of beta-catenin by cyclin-dependent kinase 6 in Wnt-stimulated cells. Eur J Cell Biol. 2007;86:111–123. doi: 10.1016/j.ejcb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 58.Provost E, Yamamoto Y, Lizardi I, Stern J, D’Aquila TG, Gaynor RB, Rimm DL. Functional correlates of mutations in beta-catenin exon 3 phosphorylation sites. J Biol Chem. 2003;278:31781–31789. doi: 10.1074/jbc.M304953200. [DOI] [PubMed] [Google Scholar]
- 59.Qi J, Wang J, Romanyuk O, Siu CH. Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol Biol Cell. 2006;17:1261–1272. doi: 10.1091/mbc.E05-10-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riggleman B, Wieschaus E, Schedl P. Molecular analysis of the armadillo locus: uniformly distributed transcripts and a protein with novel internal repeats are associated with a Drosophila segment polarity gene. Genes Dev. 1989;3:96–113. doi: 10.1101/gad.3.1.96. [DOI] [PubMed] [Google Scholar]
- 61.Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rimm DL, Morrow JS. Molecular cloning of human E-cadherin suggests a novel subdivision of the cadherin superfamily. Biochem Biophys Res Commun. 1994;200:1754–1761. doi: 10.1006/bbrc.1994.1656. [DOI] [PubMed] [Google Scholar]
- 63.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 64.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 65.Rubinfeld B, Tice DA, Polakis P. Axin-dependent phosphorylation of the adenomatous polyposis coli protein mediated by casein kinase 1epsilon. J Biol Chem. 2001;276:39037–39045. doi: 10.1074/jbc.M105148200. [DOI] [PubMed] [Google Scholar]
- 66.Sadot E, Conacci-Sorrell M, Zhurinsky J, Shnizer D, Lando Z, Zharhary D, Kam Z, Ben-Ze’ev A, Geiger B. Regulation of S33/S37 phosphorylated beta-catenin in normal and transformed cells. J Cell Sci. 2002;115:2771–2780. doi: 10.1242/jcs.115.13.2771. [DOI] [PubMed] [Google Scholar]
- 67.Scott JA, Yap AS. Cinderella no longer: alpha-catenin steps out of cadherin’s shadow. J Cell Sci. 2006;119:4599–4605. doi: 10.1242/jcs.03267. [DOI] [PubMed] [Google Scholar]
- 68.Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20:586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Staal FJ, Noort Mv M, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep. 2002;3:63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stappert J, Kemler R. A short core region of E-cadherin is essential for catenin binding and is highly phosphorylated. Cell Adhes Commun. 1994;2:319–327. doi: 10.3109/15419069409014207. [DOI] [PubMed] [Google Scholar]
- 71.Suh EK, Gumbiner BM. Translocation of beta-catenin into the nucleus independent of interactions with FG-rich nucleoporins. Exp Cell Res. 2003;290:447–456. doi: 10.1016/s0014-4827(03)00370-7. [DOI] [PubMed] [Google Scholar]
- 72.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 73.Tepass U, Gruszynski-DeFeo E, Haag TA, Omatyar L, Torok T, Hartenstein V. Shotgun encodes Drosophila E-cadherin and is preferentially required during cell rearrangement in the neurectoderm and other morphogenetically active epithelia. Genes Dev. 1996;10:672–685. doi: 10.1101/gad.10.6.672. [DOI] [PubMed] [Google Scholar]
- 74.Tolwinski NS, Wieschaus E. Armadillo nuclear import is regulated by cytoplasmic anchor Axin and nuclear anchor dTCF/Pan. Development. 2001;128:2107–2117. doi: 10.1242/dev.128.11.2107. [DOI] [PubMed] [Google Scholar]
- 75.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 76.van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- 77.Wang HY, Liu T, Malbon CC. Structure-function analysis of Frizzleds. Cell Signal. 2006;18:934–941. doi: 10.1016/j.cellsig.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 78.Wang S, Jones KA. CK2 controls the recruitment of Wnt regulators to target genes in vivo. Curr Biol. 2006;16:2239–2244. doi: 10.1016/j.cub.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 79.Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 80.Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20:1394–1404. doi: 10.1101/gad.1424006. [DOI] [PubMed] [Google Scholar]
- 81.Willert K, Shibamoto S, Nusse R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 1999;13:1768–1773. doi: 10.1101/gad.13.14.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu X, Quondamatteo F, Lefever T, Czuchra A, Meyer H, Chrostek A, Paus R, Langbein L, Brakebusch C. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006;20:571–585. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xing Y, Clements WK, Kimelman D, Xu W. Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex. Genes Dev. 2003;17:2753–2764. doi: 10.1101/gad.1142603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127:139–155. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 87.Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 89.Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]