TNFα blockade in human diseases: Mechanisms and future directions (original) (raw)
. Author manuscript; available in PMC: 2009 Feb 1.
Published in final edited form as: Clin Immunol. 2007 Oct 3;126(2):121–136. doi: 10.1016/j.clim.2007.08.013
Abstract
Tumor necrosis factor-alpha (TNFα) antagonists have shown remarkable efficacy in a variety of immune-mediated inflammatory diseases (IMIDs). Therapeutic scope and limitations of these agents are reviewed in a recently published article in the Journal. In spite of their therapeutic popularity, little is known about their modes of action in vivo and factors that limit their scope of therapeutic use. Intriguingly, while all TNFα antagonists including blocking antibodies and soluble receptors are effective in certain IMIDs, only anti-TNFα antibodies are effective in other IMIDs. Early efforts at understanding how TNFα antagonists act in IMIDs centered on their ability to neutralize soluble TNFα or to block TNF receptors from binding to their ligands. Subsequent studies suggested a role of complement-mediated lysis or antibody-dependent cell cytotoxicity in their therapeutic effects. More recent models postulate that TNFα blockers may act by affecting intracellular signaling, with the end result being a hastened cell cycle arrest, apoptosis, suppression of cytokine production, or improved Treg cell function. TNFα antagonists can also modulate the functions of myofibroblasts and osteoclasts, which might explain how TNFα antagonists reduce tissue damage in chronic IMIDs. Focusing on the human therapeutic experience, this analytical review will review the biology of mechanisms of action, the limiting factors contributing to disease restriction in therapeutic efficacy, and the mechanism and frequency of treatment-limiting adverse responses of TNFα antagonists. It is hoped that the overview will address the needs of clinicians to decide on optimal use, spur clinical innovation, and incite translational researchers to set priorities for in vivo human investigations.
Keywords: Adalimumab, Antibody dependent cytoxicity, Apoptosis, Autoantibody, Autoimmune diseases, Biologic therapies, Caspase, CDP-571, Certolizumab, Complement mediated cytolysis, Crohn's disease, Cytokine, Dendritic cells, Etancerceot, Golimumab, Graft-versus-host disease, Immune regulation, Immunotherapy, Infection, Inflammatory bowel disease, Inflammatory diseases, Infliximab, Myofibroblast, Onercept, Osteoclast, Pegsunercept, Regulatory T cells, Rheumatoid arthritis, Small molecule inhibitors, Tuberculosis, Tumor necrosis factor-alpha, Tumor necrosis factor receptor, Tumor necrosis factor signalling
Introduction
Immune-mediated inflammatory diseases (IMID) such as rheumatoid arthritis (RA), inflammatory bowel disease (IBD), psoriatic arthritis (PsA), vasculitis, ankylosing spondylitis (AS), and juvenile chronic arthritis (JCA) incur substantial costs to patients and society at large [1]. New treatments targeting disease mechanisms, popularly referred to as biological modifier therapies, are revolutionizing the treatment of such diseases. Tumor necrosis factor-alpha (TNFα) antagonism is one such arsenal in this expanding armamentarium of biological therapies. Since their first license for clinical use in 1998, the three approved TNFα antagonists have shown clear benefits in a series of randomized, controlled trials enrolling over 8000 patients with inflammatory diseases. Efficacy and safety profiles of these agents in IMIDs have been reviewed in a recently published article in the Journal [116].
Despite the therapeutic popularity of these three agents, much remains to be learned about their modes of action, the limiting factors for scope and efficacy of therapeutic use, and the mechanism and frequency of adverse responses. Here, we review the biology of mechanisms of action, the limiting factors contributing to disease restriction in therapeutic efficacy, and the mechanism and frequency of treatment-limiting adverse responses.
TNFα antagonists
TNFα is a prototype member of the TNF family of ligands that bind the corresponding TNF receptor (TNFR) family [2]. TNFα is generated and expressed by immune cells. It mediates numerous inflammatory and immunoregulatory activities [3]. Several lines of evidence suggest that TNFα contributes to tissue inflammation in IMIDs.
Two monoclonal antibodies (mAb) and a soluble receptor, which bind to soluble and membrane forms of TNFα and can neutralize the pathological effects of TNFα in IMIDs, are licensed for clinical use (reviewed in [116]).
Infliximab (INF)
INF is a chimeric mAb, composed of human constant regions of IgG1κ, with murine variable regions. It binds to both soluble and transmembrane TNFα with high affinity and specificity, but not to TNFβ. The binding of INF to membrane TNFα can mediate programmed cell death. The high specificity of the INF mAb decreases the potential for nonspecific effects on other biological pathways. However, the murine variable region segment of the mAb can lead to production of human anti-mouse Abs potentially limiting therapeutic efficacy [4]. INF is administered by intravenous (i.v.) infusion at a dose of 3 to 10 mg/kg every 8 weeks. It is indicated for use in RA, AS, CD, ulcerative colitis, PsA and chronic severe plaque psoriasis.
Etanercept (ETA)
Soluble TNF receptors bind to and inactivate TNFα, effectively lowering the amount of TNFα that is available for binding to membrane-bound receptors [5]. ETA is a dimeric fusion protein consisting of two extracellular domains of the human p75 (75 kDa) TNF receptor (sTNFRII), linked to the Fc portion of a type 1 human immunoglobulin (IgG1). The Fc portion helps to retain the molecule in the circulation [6]. By competitive inhibition, the two sTNFRII arms bind two of the three receptor-binding sites on the TNF trimer. TNF binding to the cell surface receptors is prevented [7], signal transduction is checked, and hence TNFα-induced proinflammatory activity is inhibited [6]. Of note, low concentrations of soluble TNF receptors stabilize the structure of TNFα, but higher concentrations inhibit TNFα binding to TNFRI and TNFRII on target cells [8]. ETA is administered as subcutaneous (s.c.) injections of 25 mg twice a week or 50 mg s.c. once a week. Usual dose for psoriasis is 50 mg twice weekly for 3 months, followed by a maintenance regimen dose of 50 mg per week. It is indicated for use in RA, JCA, PsA, AS, and moderate to severe psoriasis.
Adalimumab (ADA)
ADA (D2E7) is a complete human IgG1 anti-TNFα mAb that has been generated through repertoire cloning. It binds to the soluble and transmembrane forms of TNFα with high affinity, thereby preventing TNFα from binding to its receptors. In vitro studies have also demonstrated its effect on the induction of cell lysis and apoptosis [9]. It is generally administered at a dose of 40 mg s.c. every 2 weeks, or at higher doses administered once a week. It is indicated for use in RA, PsA, AS, and moderate to severe Crohn's disease.
How does TNFα blockade work in disease states?
Early efforts at understanding how anti-TNF biologics work in immune-mediated disease centered on the ability of anti-TNF Abs to neutralize soluble TNFα or to block TNF receptors from binding to their ligands [10]. Subsequent studies suggested that complement-mediated lysis or Ab-dependent cytotoxicity (ADCC) played a role in the effects of these biologics [11]. More recent models postulate that anti-TNF biologics may work by affecting intracellular signaling, with the end result being a hastened cell cycle arrest, apoptosis, or suppression of cytokine production [12]. These in vitro and a few in vivo studies that have given some insight into the possible mechanisms by which anti-TNF agents may improve diseases are described in the following sections and summarized in Tables 1 and 2 and Figs. 1 and 2.
Table 1.
Effects of TNFα blockade in RA
| Target | Mechanism of action | Evidence | Reference |
|---|---|---|---|
| Cytokine production | Decreases production of proinflammatory cytokines | In vitro | [114] |
| Cytokine production | Decreases production of proinflammatory cytokines | In vivo | [28,29] |
| Infiltrating immune cells | Reduce the number of infiltrating synovial granulocytes, CD3+ T cells, CD22+ B cells, and CD68+ macrophages, and neutrophilsReduction in the expression of the chemokines IL-8 and MCP-1 | In vivo | [32,115] |
| Monocytes and Macrophages | Induces apoptosis both in the synovial fluid and peripheral blood | In vivo | [35] |
| Peripheral blood T cells | Induces apoptosis via caspase 3 | In vitro | [17] |
| Anemia of chronic disease | Increase haemoglobin levelsReduce erythropoietin levels | In vivo | [44] |
| Endothelial cells and atherosclerosis | Decreases serum E-selectin, ICAM-1 levels and VEGFImprove endothelial function | In vivo | [24,25,37,38] |
| T regulatory cells and dendritic cells (DC) | Increase in circulating regulatory T cellsDecrease the expression of DC activation markers and prevention of DC maturation | In vivo | [39,41] |
Table 2.
Effects of infliximab in IBD
| Target | Mechanism of action | Evidence | References |
|---|---|---|---|
| Lamina propria T cells | Induces apoptosis by binding to tmTNF, inducing caspase 3, caspase 8, Bax and Bak | In vitro | [12,17,19,47] |
| Lamina propria mononuclear cells | Decreases production of proinflammatory cytokines | In vitro | [48] |
| (Jurkat) T cells | Upregulate E-selectin and IL-10, arrest G0/G1 Antibody-dependent cell cytotoxicityComplement-mediated cytotoxicity | In vitro | [12,15] |
| Peripheral blood mononuclear cells | Induces apoptosis | In vitro | [20] |
| Peripheral blood T cells | Induces apoptosis via caspase 3 | In vitro | [17] |
| Epithelial cells | Restores colonic epithelial barrier measured by decreasing number of apoptotic cells and increasing resistance | In vitro | [53] |
| Endothelial cells | Decreases CD40 and VCAM-1 expression on intestinal microvasculature | In vivo | [51] |
| Soluble TNF | Neutralizes soluble TNF | In vitro | [17] |
Figure 1.

Extracellular molecular targets of TNF antagonists in human diseases. Potential common targets in various diseases are shown on the left. Targets that have been specifically reported in rheumatoid arthritis (RA) and Crohn's disease (CD) are highlighted in the boxes on the right. Pathways in which TNF-antagonists block are shown by red symbol [2,23,113].
Figure 2.
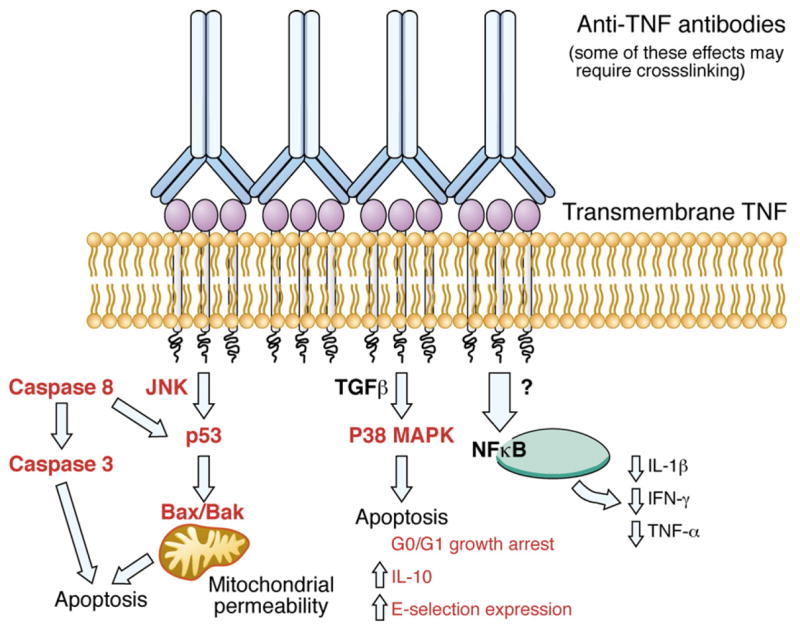
Reverse intracellular signaling cascade induced by the binding of anti-TNFAbs to trans-membrane TNF (tmTNF). This is one potential mechanism accounting for the in vitro effect of anti-TNF agents as distilled from several sources. See text for further details. Red=induced by anti-TNF binding to tmTNF.
Neutralization of soluble TNFα by anti-TNFα agents
Anti-TNFα Abs and soluble TNF receptors can reduce inflammation by directly neutralizing the activity of TNFα (Fig. 1). The TNF receptor exists as one of two isomers, a p55 (TNFR I) and a p75 receptor (TNFR II). These two isoforms of TNF receptor may be found on the membrane of monocytes and T lymphocytes (mTNFR), or circulating in the serum as a soluble receptor (sTNFR). The binding of TNF to a circulating receptor essentially neutralizes its action and prevents patients against overwhelming shock in the face of infection or inflammation. Patients with CD have increased levels of both soluble TNFRs [13]. Gustot et al. found that INF decreases serum levels of sTNFRII in patients with active CD. However, it has no effect on the levels of sTNFRII. This decrease may be secondary to a feedback mechanism whereby serum TNFα induces the expression of sTNFRII, which, in turn, neutralizes TNF and its proinflammatory effects.
A recent study sought to address the specificity of anti-TNF binding to the p55 or p75 receptor [14]. They used a human lung carcinoma cell line A549 transfected with a luciferase reporter system for binding to p55 receptor and the Jurkat cell line transfected with a chimeric p75 construct with a similar luciferase reporter. This system allowed them to measure the ability of INF, ADA, or certolizumab (see Looking to the future: research and innovation priorities to improve and extend therapeutic use and safety) to neutralize soluble TNF or membrane-bound TNF signaling in vitro specifically via the p55 or p75 receptor. Interestingly, all three agents equally neutralized membrane-bound TNF via the p55 or p75 receptors, but certolizumab pegol was 2-fold more potent in neutralizing soluble TNF signaling via either receptor than INF or ADA.
Complement-mediated lysis and ADCC
Fossati et al. used a system of NS0 myeloma cells expressing high levels of tmTNF to show that ETA, INF, ADA, and certolizumab all bound to tmTNF, although ETA bound fewer TNF molecules [15]. However, only ETA, INF, and ADA were able to affect complement-dependent lysis and ADCC. Certolizumab exhibits neither ADCC nor complement-mediated lysis because it is a Fab' fragment without an Fc portion.
Intracellular signaling mediated by anti-TNFα agents
Binding of an anti-TNF molecule to tmTNF can potentially elicit one or more effects, including apoptosis mediated by reverse signaling, the downregulation of cytokine production, or growth arrest. Mechanistic studies of anti-TNF agents have yielded variable results due to differences in experimental conditions. The consequences of membrane binding depend largely on the systems in which these mechanisms are tested. Several of these studies have been performed using cell lines into which a gene encoding non-cleavable mTNF has been transfected. As discussed later in the next subsection, investigators have found differences in various anti-TNF agents in their ability to bind tmTNF [16]. Newer data suggest that the cross-linking of anti-TNF Ab on the cell is responsible for its reverse intracellular signaling, inhibiting the production of inflammatory cytokines, inducing apoptosis, and causing growth arrest. A few studies have shown differential binding of different anti-TNF agents to soluble versus membrane TNF with ensuing differences in ability to induce apoptosis in cells, although the results have been variable [17,18].
Explanation of TNF signaling
Antibody binding to tmTNF induces a reverse (outside to inside) intracellular signaling cascade (Fig. 2). The resulting release of intracellular calcium, cytokine production, and E-selectin expression is associated with caspase-dependent apoptosis [12]. This apoptosis does not seem to require Fas–Fas ligand interaction, however [19].
The consequences of TNF binding to its membrane-bound receptor are several. Human Jurkat T cells stably transfected with membrane-bound TNF upregulate levels of E-selectin as well as IL-10 when incubated in the presence of INF [18]. These cells are also arrested in the G0/G1 phase of the cell cycle. The three transmembrane serine residue domains on mTNF are essential to its function. It is also likely that p53, a transcription factor that regulates apoptosis and cell cycle arrest, acting through intracellular signaling proteins Bax and Bak, is responsible in part for the INF-induced apoptotic effect [12]. Caspase 8 acts to promote apoptosis through its downstream effects on caspase 3, as well as on proteins that influence mitochondrial permeability, Bcl-2, Bc-xL (anti-apoptotic), Bax and Bak (pro-apoptotic). It is the balance of these proteins that determines the cell's fate. It has been postulated that INF may work to induce apoptosis through these caspase 8-mitochondrial permeability pathways [20].
Notably, binding of TNF to TNFRI causes upregulation of pro-apoptotic signals but it also leads to the translocation of NF-κB into the nucleus, protecting the cell from apoptosis. Whether the end result of TNF binding is apoptosis of the cell or protection from apoptosis depends on the cell type and its state of activation.
ADA appears to have a similar mechanism of action as INF in inducing caspase-dependent apoptosis [21]. This study found that ADA induced caspase 3-dependent apoptosis of a monocyte cell line in vitro as well as in vivo in severe combined immunodeficient (SCID) mice reconstituted with human monocytes (THP-1), and that its effects could be reversed by administering a caspase inhibitor to the mice contemporaneously.
Functional modulation of myofibroblasts
Anti-TNF agents, INF, ETA and ADA, enhance the production of tissue inhibitor of metalloproteinases (TIMP)-1 [22]. INF can also enhance the migration of myofibroblasts, which may reduce MMP activity and facilitate the wound healing. Interestingly, INF reduced collagen production by CD myofibroblasts. These effects offer a new mechanism by which anti-TNF agents may reduce tissue damage in chronic IMIDs.
Mechanistic studies in individual diseases are discussed in the following sections.
Evidence from inflammatory joint disease
Normally, TNFα and other proinflammatory cytokines are maintained in balance by anti-inflammatory factors. This balance is shifted in favor of the proinflammatory cytokines in inflammatory diseases. TNFα upregulates adhesion molecules on the endothelium, stimulates fibroblast proliferation, and recruits leukocytes from the circulation into the synovial fluid [23]. TNFα stimulates production of other cytokines and chemokines, reactive oxygen intermediates, nitric oxide and prostaglandins, and increases the rate of tissue remodeling by matrix-degrading proteases [24,25]. TNFα promotes angiogenesis and osteoclast differentiation and activates osteoclasts to resorb bone, leading to joint erosions particularly at the marginal surfaces [26,27]. It also increases the rate of tissue remodeling by matrix-degrading proteases [24,25] and directly mediates pain, fever, and cachexia.
Effect on cytokine production
Therapy with TNFα inhibitors leads to an overall reduction in proinflammatory cytokine levels in patients with RA. Longitudinal studies showed levels of serum IL-6 falling, followed by the reduction in the acute-phase proteins amyloid A, haptoglobin and fibrinogen [28]. Synovial synthesis of TNFα, IL-1α and IL-1β were also reduced in RA patients following a single infusion of INF [29]. Reduced IL-1 levels result in reduced synthesis of MMP and other degradative enzymes, such as proMMP-1 and proMMP-3 [28,30,31].
Effect on infiltrating cells
The inflamed joint contains synovial macrophages, and fibroblasts known as synoviocytes. Treatment with INF, ETA, and ADA in RA patients reduces the numbers of infiltrating synovial granulocyte, CD3+ T cells, CD22+ B cells, and CD68+ macrophages and a concomitant reduction in the expression of the chemokines IL-8 and MCP-1 [32]. Exposure to ADA also restored normal chemotactic neutrophil activity [33], decreased influx of leukocytes to the inflamed joints [34], and reduced activation marker CD69 on neutrophils in patients [33].
Increased apoptosis is another mechanism by which anti-TNF agents can reduce infiltrating cells. For example, treatment of RA patients with either ETA or INF increased synovial apoptosis of monocyte/macrophage populations. However, no changes in the rate of apoptosis were noticed in lymphocytes from synovial fluid or peripheral blood [35].
Effect on endothelial cell activation and atherosclerosis
Expression of angiogenic factor vascular endothelial growth factor (VEGF) is reduced in synovial membrane from patients after TNF blockade therapy. This suggests that these treatments might reduce neovascularization in the pannus characteristic within rheumatoid joint [36]. In fact, analysis of markers of endothelium (e.g. von Willibrand factor, CD31) and neovasculature (αvβ3) does show reduced vascularity after anti-TNF treatment (Table 1 and Fig. 1).
RA is associated with accelerated atherosclerosis. TNFα might play a role in the development of RA-associated cardiovascular disease by increasing endothelial cell expression of adhesion molecules in the synovial tissue [24,25]. Treatment with anti-TNFα agents results in a dose-dependent reduction in the expression of endothelial adhesion molecules in the synovial tissue [25] and decreases in soluble forms of intracellular adhesion molecule-1 (ICAM-1) and E-selection [37]. A later study showed an improvement in endothelium-dependent vasodilatation of the brachial artery after 12 weeks of INF treatment [38].
Effect on immunoregulation
Impaired function of a subset of T cells called regulatory T (Treg) cells has been implicated in the pathogenesis of RA. Indeed, Treg cells from patients with active RA are defective in their ability to suppress proinflammatory cytokine production by activated Tcells and monocytes in vitro. Anti-TNFα Ab treatment significantly boosts the numbers of circulating Treg cells; the increase in Treg cells correlated with a reduction in CRP [39]. Further, TNFα blockade inhibits the expansion of human CD4+VLA-1+ effector T cells in vitro, while augmenting the percentage of Treg cells in cultures [40]. These data suggest modulation of Treg cells as a potential mechanism of action of TNF blockers in IMIDs.
Effect on dendritic cells (DC)
As early as 24 h following treatment with INF, there was a decrease in the percentages of circulating CD11c+ and CD123+ DC; expression of activation marker CD83 was also reduced on DC in the treated patients. After 6 months of treatment, clinical improvement was associated with a less active phenotype of DC. This study supports the role of anti-TNFα in preventing the maturation and activation of DC [41].
Effect on osteoclasts
The formation and activation of osteoclasts at the cartilage–pannus junction is an essential component of RA-associated bone loss. TNFα promotes osteoclast differentiation and activates osteoclasts to resorb bone, leading to joint erosions [26] (Fig. 1). These processes involve an interaction between receptor activator of NF-κB ligand (RANKL) and its decoy receptor, osteoprotegerin (OPG) [26]. Anti-TNFα therapy can modulate the OPG/RANKL system by reducing the RANKL/OPG ratio [42]. This might be a potential mechanism by which anti-TNFα therapy retards radiographic damage in patients with RA.
Effect on bone marrow
TNFα can affect the bone marrow, contributing to the anemia of chronic disease in patients with RA [43]. Although the mechanism is unclear, administration of anti-TNFα mAb cA2 to RA patients with anemia of chronic disease resulted in increased hemoglobin levels in a dose-dependent manner. These changes were accompanied by a reduction in both erythropoietin and IL-6 levels [44].
In summary, TNFα inhibitors can affect various cell types and mediators involved in the pathogenesis of RA. These agents can influence antigen presentation and self-reactive Tcell activation via their effects on DC; increase the numbers and functional capacity of Treg cells to suppress effector T cells; reduce inflammatory cell infiltration at the sites of inflammation by modulating cell migration and inducing their apoptosis; reduce proinflammatory cytokine production; modulate the functions of endothelial cells and osteoclasts; and even affect bone marrow microenvironment (Table 1).
Evidence from inflammatory bowel disease
Myriad derangements in innate and acquired immunity characterize IBD. Among these are resistance of activated Th1-polarized effector T cells to apoptosis [45], deficiencies in local IL-10 production, and increased production of TNFα by local monocytes/macrophages. Following evidences support that anti-TNF biologics might correct some of these impairments.
Effect on apoptosis
One of the best-described mucosal immune derangements in IBD is a resistance of lamina propria T cells to apoptosis [45,46]. INF's ability to induce apoptosis of activated lymphocytes in the inflamed biopsies or peripheral blood may underlie its therapeutic efficacy in IBD (Table 2 and Fig. 2). One study performed colonoscopy with biopsy in 10 IBD patients before, and 10 weeks after, a course of INF (administered at weeks 0, 2, and 6) and in 10 control subjects with functional diarrhea [19]. They found that patients treated with INF had increasing numbers of TUNEL-staining apoptotic lamina propria T cells, approaching the number of apoptotic lamina propria Tcells of those in the control group. Another study noted a significantly increased percentage of TUNEL-positive cells, mostly CD3+ T cells, in colonic biopsies from CD patients 24 h after INF infusion (5.4 cells/HPF to 22.9 cells/HPF) [47]. Furthermore, lamina propria Tcells isolated from the colonic biopsies were more sensitive to apoptosis than peripheral blood T cells when cultured in the presence of INF [19]. In resonance with this finding, another study showed that the activated, but not resting, T cells were susceptible to apoptosis in the presence of INF. However, Lugering et al. demonstrated increased apoptosis in peripheral blood mononuclear cells (PBMCs), as measured by annexin V staining by flow cytometry, before and 4 h after INF infusion in patients with CD. The apoptotic cells showed induction of caspase 3. These changes also correlated with an absolute decrease in the percentage of monocytes in the peripheral blood after INF infusion [20].
One study compared the abilities of ETA and INF to neutralize TNF and to induce apoptosis in vitro [17]. Both INF and ETA neutralized the effects of TNFα in HeLa cells transfected with a NF-κB reporter GFP construct. However, whereas INF bound to the activated peripheral blood lymphocytes (PBL) and lamina propria monocytes isolated from the colonic biopsies from patients with CD, there was no detectable level of ETA binding to those same cells. They further ascribed this binding of INF to transmembrane TNF rather than to receptor-bound TNF. Furthermore, INF, but not ETA, induced apoptosis as measured by annexin V and 7-AAD staining in PBLs stimulated by mixed lymphocyte reaction. Caspase 3, but not cell-mediated immunity or complement-mediated cytolysis, played a role in this apoptosis.
Whether INF exerts its therapeutic effect by inducing apoptosis in PBMCs is a point of contention. For example, one study found no increase in the number of apoptotic cells when PBMCs isolated from patients with CD were cultured overnight in the presence of INF [48]. In fact, the researchers believed that these cells were stabilized against apoptosis. Stimulation of monocytes with LPS did not increase their susceptibility to apoptosis in the presence of INF. It remains to be determined whether increased apoptosis of activated lymphocytes represents a direct effect of TNF inhibitors. It is also unclear whether increased apoptosis of activated mucosal lymphocytes is in fact the mechanism of therapeutic efficacy of anti-TNF agents.
Effect on intracellular signaling
INF seems to act on several cellular targets, including proteins that regulate apoptosis, progression through the cell cycle, and expression of cytokines. INF binding transiently stimulates p38 MAP kinase both in vivo in patients with CD, as well as in vitro. Both INF and ETA are able to reduce STAT3 phosphorylation to a similar degree in cultured T cells taken from patients with CD. It may well be the neutralization of sTNF that may be important in decreasing STAT3 signaling as sTNF acts in an autocrine fashion to regulate STAT3 [49].
Effect on cytokine production
The immunopathogenesis of CD culminates in inflammation via the interplay of several cytokines including not only TNFα but also others including IL-1β and IFNγ. INF exerts its anti-inflammatory effects by acting on multiple constituents in these cytokine networks (Table 2 and Fig. 1). Ringheanu et al. [48] found that INF induced “reverse signaling” in isolated PBMCs in vitro to downregulate the production of inflammatory cytokines. They showed that monocytes produced less TNFα after they had been exposed to LPS or SEA when cultured with INF. PBMC isolated from patients before and 1 h after INF infusion and stimulated with LPS or SEA had fewer copies of TNFα, IL-1β , IL-6, and IL-8 mRNA as measured by RT–PCR. Activated T cells isolated from colonic biopsies of patients with CD cultured in the presence of INF were found to decrease their expression of IFNγ [50].
Effects on CD40–CD40L and lymphocyte homing
INF can also decrease lymphocyte homing. CD40, also a member of the TNF receptor family, is found on the surface of B cells as well as on some epithelial (and endothelial) cells. Recent fascinating results point to modulation of levels of CD40 in the microvasculature of intestinal tissue in patients with CD as a possible therapeutic mechanism of INF. CD40–CD40L ligation resulted in the upregulation of cell surface markers and inflammatory signals. Endothelial cells express CD40 that recognizes the CD40L on the surface of platelets and lamina propria T cells. Once engaged, CD40 causes endothelial cells to upregulate surface markers responsible for lymphocyte recruitment as well as produce proinflammatory cytokines and chemokines (Table 2 and Fig. 1). Danese et al. [51] showed that INF significantly reduced levels of intestinal endothelial expressed CD40 and VCAM-1 in treated patients with CD, as well as their levels of soluble CD40L.
Effect on barrier function
The epithelial cells are prime players in the innate immune response and they have been implicated as playing a major role in the dysregulated immune homeostasis of CD. Foci of epithelial cell apoptosis may be the cause of increased ionic permeability in IBD [52]. Zeissig et al. [53] showed that patients with CD had a higher index of apoptotic epithelial cell foci than healthy controls. Patients treated with INF had epithelial barriers that were restored to normal. These findings correlated functionally with epithelial resistance, but had no relationship to the expression of tight junction proteins.
In summary, anti-TNF drug INF appears to promote apoptosis of inflammatory cells infiltrating the inflamed mucosa, modulate intracellular signaling and inhibit pro-inflammatory cytokine and chemokine production, and assist with the repair of epithelial barrier. Which of these mechanisms are critical for INF-induced protection in patients with IBD remain to be determined.
Mechanistic lessons from animal models
Availability of animal models that mimic human RA or IBD permits in vivo mechanistic studies that would otherwise be difficult to perform in patients. In fact, direct evidence for the role of TNFα in the pathogenesis of RA came from the Tg 197 transgenic mice, which carry a human TNF transgene with a modified 3'-untranslated region. This mutation results in constitutive production of human TNFα in these mice. By the age of 4 weeks, all human Tg 197 mice spontaneously develop a severe bilateral, symmetric, erosive, and disabling polyarthritis similar to RA [54]. Treatment of these arthritic mice with a mAb against TNF completely prevents the development of the disease [54,55].
Studies in early 1990s also implicated TNFα in the development of collagen-induced arthritis (CIA) that is recognized as the industry standard for studying potential therapeutic targets for treatment of RA. CIA is elicited through intradermal injection of chick or bovine type II collagen (CII) in the presence of Freund's complete adjuvant into genetically susceptible mice [56]. In initial studies, i.p. injections of TN3-19.12, a hamster IgG1 mAb to murine TNFα/β, into CIA mice either before or after the onset of clinical arthritis significantly reduced paw swelling and histological severity of arthritis without reducing the incidence of arthritis or the level of the circulating antitype II collagen IgG [57]. Co-administration of anti-CD4 and a sub-optimal dose of anti-TNFα/β to the CIA mice not only reduced paw-swelling and prevented limb involvement and joint erosion, it was also effective in preventing an Ab response to the hamster anti-TNF Abs [58]. These findings may have implications for a long-term therapy in human disease, as they suggest a possibility for treatment that may help eliminate the adverse outcome of autoimmunity following anti-TNFα treatment. These findings were also confirmed using the human p55 TNF receptor–IgG fusion protein (p55–sf2), co-administered with anti-CD4 [59]. Combined anti-TNFα/anti CD4 therapy suppressed both TNFα and IL-1β expression in the DBA/1 CIA mouse model. In addition, this combined therapy also caused the normalization of serum amyloid P levels [60]. In a separate study, anti-TNFα blockade, but not anti-CD4 Abs, significantly reduced the frequency of cells expressing TNFα, IL-1β, VLA-4, VCAM-1 and CD4+ Tcells and macrophages in the joint of CIA-induced mice model [61].
In another, the motheaten arthritis mouse model, treatment with the soluble TNFRI reduced serum TNFα levels and improved the life span of the treated mice. It did not, however, reduce the serum levels of IgM and IgM anti-dsDNA Abs in comparison to control mice [62].
A variety of animal models of both acute and chronic intestinal inflammation have been developed to study CD [63]. These include chemically induced, genetically manipulated and immune-mediated models of gut inflammation, each of which addresses specific aspects of the disease pathogenesis. In addition, the TNF ARE and SAP1/YitFc mouse strains spontaneously develop chronic ileal inflammation that closely resembles human CD, with regard to disease location, histologic features and clinical responses to therapy, and thus have provided essential insights into the pathogenesis of human CD. These mouse models have been utilized for testing the efficacy of potential therapies such as anti-TNF blockades. For example, in the SAMP1/YitFc model [64], a single injection of a chimeric antimurine TNF Ab suppressed intestinal inflammation and epithelial cell damage [65], which was associated with reduced apoptosis of intestinal epithelial cells, while the lamina propria mononuclear cells showed increased cell apoptosis. These data demonstrate that one of the mechanisms of action of anti-TNF therapy involves homeostatic regulation of apoptosis in mucosal and lamina propria mononuclear cells, resulting in a net decrease of chronic inflammation.
To study the in vivo effects of TNF inhibition on human cells, some investigators have used a humanized mouse model where SCID/beige mice were reconstituted with human monocytic cell line THP-1 or the human T cell line Jurkat. In these mice, administration of a mouse–human IgG1 chimeric anti-TNF Ab induced apoptosis of monocytes and T lymphocytes and decreased the production of human cytokines IL-10 and IL-12 [66]. Cell death in these mice was independent of FcγR binding or complement activation and resulted from apoptosis induction in a caspase-dependent pathway. This mouse model was also utilized to study the mechanism of apoptosis induction by the fully humanized mAb ADA, as well as the chimeric mAb INF. Following treatment with either ADA or INF, a high percentage of the human THP-1 cells were apoptotic [21]. Moreover, apoptosis induction by ADA could be abrogated by treatment of these mice with a pancaspase inhibitor. Theses data indicated that caspase activation is involved in TNFα-induced T cell apoptosis in vivo.
In summary, the use of animal models has not only demonstrated the role of TNFα in the development of chronic joint and intestinal inflammation, it has paved the way for TNFα-based therapies. Ongoing and future animal studies will be critical to unravel new mechanisms by which anti-TNFα therapies act in IMIDs.
Limiting factors contributing to disease restriction in therapeutic efficacy
Anti-TNF agents have clear therapeutic benefit in some IMIDs while they have relatively weaker or no effect in other diseases. Even in diseases where anti-TNF treatments have been shown to be successful, more than one third of patients do not respond to treatment. This lack of responsiveness may be due to a different mechanism(s) of disease in some patients. In other words, chronic joint or intestinal inflammation clinically diagnosed as RA or IBD, respectively, actually represent different pathological disease entities. Alternatively, TNFα may play a pathogenetic role only in certain stages of disease.
Another important area is to understand why and how certain anti-TNF agents have better outcomes in one disease and not in another. For instance, ETA is an important drug in the treatment of patients with RA, but a large clinical trial showed that it had no effect at the dose regimen used on decreasing the gut inflammation in IBD. In vitro studies attempted to explore differences in binding to tmTNF between ETA and INF. One must take care, however, to interpret the results of in vitro studies carefully, as they are not always generalizable to patients with complex chronic diseases. Furthermore, the results of many mechanistic studies of anti-TNF biologics vary because of the conditions under which they are run. The consequences of membrane binding depend largely on the systems in which these mechanisms are tested. Several of these studies have been done using cell lines into which a gene encoding non-cleavable mTNF has been transfected. When tmTNF is transfected into cells, investigators take care to protect it from an enzyme that would otherwise cleave the TNF molecule, called TNFα-converting enzyme (TACE) [67]. At first the gene encoding tmTNF was transfected as a deletion mutant, but more recently, Harashima et al. [68] developed a method of transfecting cells with a tmTNF rendered non-cleavable by site-directed mutagenesis. Using these methods, investigators assessed the number of anti-TNF molecules that can bind tmTNF either by flow cytometry or by radioassay [16].
TNF exists in monomeric and biologically active trimeric forms. INF is able to bind to both monomeric as well as trimeric forms of TNF, but because of its binding site on TNF occurs in the cleft between subunits, ETA can only bind to the trimeric form [16]. This binding configuration may explain why the number of mTNF molecules bound by INF is 1.5 to 3 times as many as ETA, while their affinities in monovalent interactions are similar. Scallon et al. [16] performed pulse–chase experiments of radiolabeled INF and ETA and found that while both bound tmTNF, ETA had a higher off-rate, signifying that the rate of exchange of ETA with tmTNF is higher than that of INF. A recent study from the Abbott Laboratories compared the binding properties of the three FAD-approved anti-TNF agents [69]. Using a BIAcore 3000 instrument, they found similar affinities of the three antagonists for soluble TNF, but with differing on-rates and off-rates. ETA had a faster binding on-rate and a faster dissociation off-rate to soluble TNF than INF or ADA. All three TNFα antagonists also bound to membrane TNF with similar affinities. Other factors, such as the ability of ETA to bind both TNFα and TNFβ and the ability of mAbs to bind only to TNFα, might also explain some differences in treatment outcomes using these agents.
The cross-linking of anti-TNF agent on the cell may elicit reverse cell signaling, causing reduced production of inflammatory cytokines, and increased apoptosis and growth arrest. If the surface density of mTNF on the cell is high, it would be expected that there is more opportunity for Ab cross-linking [66]. ETA may have greater efficacy in the treatment of patients with RA because circulating rheumatoid factor as well as complement component C1q may act to cross-link this Ab when bound to lymphocytes in vivo.
Some authors argue that INF binds to both tmTNF as well as soluble TNFα, while ETA binds primarily to soluble TNF, and so cannot mediate caspase-dependent apoptosis [17]. Others claim that both INF and ETA bind to the uncleavable form of membrane bound TNF, but INF binds with higher avidity [18]. By binding to cells bearing membrane-bound TNF, INF is able to induce apoptosis of those cells [70]. Monocytes isolated from the peripheral blood of patients with CD undergo apoptosis after binding of INF in vitro, i.e. a phenomenon associated with increases in caspase 3 activation [20]. Similarly, T lymphocytes maintained in a mixed lymphocyte reaction with monocyte-derived dendritic cells show significant apoptosis in the presence of INF. Finally, binding may not predict the cellular effects. For example, a recent study found that ETA, INF, ADA, and certolizumab all bound equally to activated PBT and PBMC, but only ETA, INF, and ADA were able to induce apoptosis in those cells [14].
Genetic makeup of individuals may dictate response to biological treatments. For example, RA patients with a TNFα-308G/G genotype are better infliximab responders than are patients with A/A or A/G genotypes. TNFα-308 genotyping may be a useful tool for predicting response to infliximab treatment [71]. This observation has been extended to other anti-TNF agents independent of the treated rheumatic disease (RA, PsA or AS) [72].
In summary, more than one third of patients (suffering from any approved indication) do not benefit clinically from anti-TNF treatment. Future studies should investigate whether this limitation is due to different pathogenetic mechanisms in different patients or due to involvement of different mediators of disease development and progression in different stages of disease. Other limiting factors could be the genetic make up of individuals or specific properties of the anti-TNF agent used for the treatment.
Mechanism of treatment-limiting adverse responses
Although anti-TNFα therapy is safe and well tolerated, some of the adverse events reported in patients treated with TNFα blockading agents include immunogenicity, infections, delayed hypersensitivity-type reactions and autoimmune diseases such as drug-induced lupus and demyelination (reviewed in [116]).
One study examined the effect of TNFα neutralization on cell-mediated and humoral immunity in a mouse model of acute graft-vs-host disease (GVHD). The anti-TNFα treatment blocked the lymphopenia, but induced lupus-like chronic lymphoproliferation and autoantibody production [73]. There was a complete inhibition of anti-host CTL activity, with preferential inhibition of IFN-γ production and of IFN-γ-dependent upregulation of Fas, but not the production of Th2 cytokines, IL-4, IL-6 and IL-10. This selective inhibition of CTL response that would normally suppress autoreactive B cells may be the mechanism by which TNFα blockade promotes autoimmunity.
While treatment of RA patients with TNFα blockade is associated with a reduction in the levels of specific autoantibodies, such as rheumatoid factor (RF), and anticyclic citrunillated peptides, it has also been associated with the induction of non-organ specific autoantibodies, such as antinuclear Abs (ANA), anti-dsDNA, and antiphospholipid Abs (aPL) [74,75].
In CD patients, one study reported development of ANA in 56.8% of patients within 1 year of INF treatment and drug-induced lupus erythematosus in 2 out of these 71 patients [76]. The effect of INF treatment on the induction of ANAs, anti-dsDNA, antinucleosome, antihistone, and anti-extractable nuclear antigen (anti-ENA) Abs in RA, was investigated by De Rycke et al. [77]. Following treatment, there was a significant increase in the percent of RA patients that tested positive for ANAs. There was also an increase in the percent of patients with anti-dsDNA Abs of the IgM and IgA type. In contrast, following long-term INF therapy in patients with RA, the presence of both anti-ssDNA and anti-dsDNA of the IgG type in the serum correlated with lupus-like symptoms and/or anaphylactoid reactions in these patients [78].
In animal models, TNFα plays an essential role in host defense against tuberculosis (TB), including granuloma formation and containment of disease [79–81]. It is therefore not surprising that TNFα blockade causes reactivation of TB in animal models [82]. In fact, all three approved anti-TNF agents increase the risk of TB in clinics that do not comply with recommendations to prevent reactivation of latent TB infection [83].
Elevated levels of TNFα are seen in serum and hepatocytes of patients with chronic HBV, and are secreted by HBV-specific cytotoxic T lymphocytes (CTL) [84,85]. Animal studies show that TNFα knockout mice have defects in the proliferative capacity of the HBV-specific CTL, suggesting that TNFα may play a role in clearing or controlling HBV [86]. Therefore immunosuppression in chronic HBV could theoretically reactivate or worsen the disease. TNFα promotes viral clearance in hepatitis B infection (at least in animals), a role different than in hepatitis C where it is postulated to promote chronic liver injury. Published case reports indicate that INF ± MTX reactivates chronic HBV infection, yet concurrent treatment of INF ±MTX with lamivudine can stabilize HBV disease activity.
TNFα plays an important role in host defense against intracellular infections. Studies in animal models demonstrate that TNFα plays a critical role in defense against infections due to histoplasma, cryptococcus, coccidiodes, candida, aspergillus and pneumocystis [87–89]. Studies in three patients with histoplasmosis associated with INF use showed that INF interfered with the host's helper T cell response to histoplasma antigens [90].
Injection site reactions (ISRs) that can occur with anti-TNF agents are believed to be T-lymphocyte-mediated, delayed-type hypersensitivity reactions that decrease in intensity over time, possibly due to induced tolerance [91].
Of the CD patients who respond to anti-TNF, only 25% maintain a clinical response at 1 year follow-up. Human anti-chimeric Abs (HACA) seem to be responsible for the majority of the loss of efficacy. Antibodies to INF also correlate with allergic reactions and serum sickness. Recent studies in the RA, however, suggest that the trough INF levels, but not the titers of HACA or ATI (Abs to INF), decrease its clinical efficacy [92]. New dosing strategies may be designed to address this problem. One strategy of dealing with HACA is by increasing the dose of INF from 5 to 10 mg/kg or increasing the frequency of dosing.
In summary, in certain disease settings, use of anti-TNF biologics increases the risk of adverse effects. Understanding the mechanisms of these complications and of pathophysiology associated with elevated or reduced levels of TNFα may help explain some of these limitations.
Looking to the future: research and innovation priorities to improve and extend therapeutic use and safety
Designing new translational research strategies
As not all patients with RA, Crohn's and other diseases benefit from anti-TNF agents, it is critical at this time to investigate predictors of potential responders and adverse consequences. In this regard, traditional disease parameters, such as levels of acute phase reactants, pace of disease progression, as well as pharmacogenetic approaches, are being used. For example, identifying which genetic polymorphisms associate with increased levels and effects of this cytokine and which negatively affect disease progression would be helpful. For example, CD patients bearing mutations in the Fas ligand gene (843-TT) were more likely to be unresponsive to INF therapy [93]. Results were similar, but not as significant, in patients with a mutation in the caspase-9 gene (93-CC/CT). Treatment with concurrent azathioprine/6-MP was able to overcome these pharmacogenetic predispositions to INF failure. Other genetic polymorphisms, such as polymorphisms in the genes encoding TNF receptors, may predict efficacy to INF [94]. Also helpful would be proteomic and functional genomic studies of peripheral blood, synovial fluid, and synovial tissue obtained from patients undergoing these treatments.
Only certain IMIDs seem to have significant therapeutic benefit to anti-TNF agents. Pilot studies and case reports have shown no benefit in other IMIDs. Mechanistic studies must focus on pathways of disease initiation and progression in these diseases.
Gaining a better understanding of how TNF inhibition affects chronic infections such as hepatitis C and B has the potential to guide therapy in patients with concomitant infection or have IMIDs that are associated with chronic infectious diseases. Of particular importance is to gain a better understanding of how TNFα participates in immune surveillance functions against autoreactive T and B cells and cancer would also be helpful.
In summary, more than one third of patients (suffering from any approved indication) do not benefit clinically from anti-TNF treatment. On the other hand, even in the absence of clinical remission, some patients achieve radiological or pathological improvement. These observations have important lessons for designing future studies to understand the pathogenesis of disease initiation and tissue damage. Moreover, it is now well appreciated that the dose of these drugs should be tailored to achieve maximal response, while much lower than the conventional doses are enough in many other patients. Future studies should explore the concept of the need for individualized regimens based on genetic polymorphisms.
Developing ‘smart’ TNFα antagonists
Newer anti-TNF agents in clinical trials
Certolizumab (CDP, Cimzia)
Certolizumab is a polyethylene glycolated (PEG) Fab' fragment of a humanized anti-TNF mAb made by microbial fermentation with Escherichia coli [95]. PEG increases its circulating half-life to approximately 14 days, which is that of a whole Ab [96], and it is much longer than the half-life of unconjugated Fab' fragments. This Ab has been developed to address the concerns that some toxicity associated with INF and ADA might be due to Fc-associated effects on complement activation and ADCC.
Phase 3 trials have been performed in inflammatory diseases including RA and CD. In a randomized, double-blind, placebo-controlled, dose-escalating trial of i.v. infusion of CDP870, followed by a single open-label infusion, certolizumab significantly reduced inflammation and improved symptoms in RA patients [97]. Clinical improvement (ACR20 response) was comparable to that of ETA [98] and INF [96,99]. Certolizumab was very well tolerated in the study and had an extended duration of action after one or more i.v. doses. At higher doses, certolizumab generated only very low levels of Ab response.
An ongoing phase 3 multi-center, open-label, follow-on study of CDP870-027 will assess the efficacy and safety of lyophilized CDP870 as an additional medication to MTX in the treatment of signs and symptoms and preventing structural damage in patients with active RA.
A randomized, double-blind, placebo-controlled multi-center study evaluated the use of certolizumab in patients with moderate to severe CD [100]. Certolizumab 400 mg s.c. showed a significant benefit in clinical response when compared with placebo at weeks 2 (_p_=0.01), 4 (p ≤ 0.01), 8 (p ≤ 0.01), and 10 (_p_=0.006), but the difference in CDAI scores were not statistically significant at week 12. The percentage of patients achieving remission with certolizumab 400 mg s.c. was significantly higher than those given placebo at weeks 4 (p ≤ 0.05) and 8 (p ≤ 0.01), but not at week 12. Post-hoc analysis stratifying patients with serum CRP ≥ 10 (_n_=119) or < 10 (_n_=171) resulted in statistically significant benefit of certolizumab 400 mg s.c. over placebo in terms of both clinical response and remission at all time points.
Golimumab (CNTO-148)
Golimumab is a fully human anti-TNFα IgG1 mAb that targets and neutralizes both soluble and membrane-bound form of TNFα. In a phase 2, randomized, double-blind dose-ranging trial of a combination therapy of s.c. golimumab and oral MTX versus MTX alone in patients with active RA, golimumab was effective in all dosing groups [101]. After 16 weeks of treatment, more patients in the combined group achieved ACR 20 (62% vs. 37%, _P_=0.008), ACR 50 (31% vs. 6%) and ACR 70 responses (12% vs. 0), and DAS28 remission including serum CRP levels (27% vs. 6%, _P_=0.007). The adverse events between the two groups were similar. In addition, there was no clear dose–response relationship between golimumab 50 mg every 4 weeks and golimumab 100 mg every 2 weeks: both statistically significantly more effective than placebo in achieving an ACR 20 response (63%, _P_=0.031 and 79%, P < 0.001, respectively). The medication is currently being investigated for administration by either s.c. injection or i.v. infusion.
CDP-571
In a short-term, double-blind, placebo-controlled study, CDP-571, a humanized IgG4 anti-TNF Ab, was given as a single 5 mg/kg dose to 31 patients with moderate to severe CD [102]. At 2 weeks after the infusion, the median CDAI fell from 263 to 167 in the CDP-treated group, and the change was insignificant in the placebo-treated group. Of the 30 patients evaluated at the primary endpoint, 6/21 in the CDP group achieved remission (CDAI ≤ 150) and another 3 “near remission” (CDAI ≤ 156). This compared to just one patient in the placebo-treated group with a CDAI ≤ 156 at 2 weeks.
In a subsequent 24-week phase 2 study [103], CD patients were randomized to receive either CDP-571 10 mg/kg or 20 mg/kg IV and then redosed with 10 mg/kg CDP-571 or placebo every 8 or every 12 weeks. Only 32% of treated and 19% of placebo patients completed the study. The most common reason for study withdrawal in both groups was disease progression. The rate of clinical response (decrease in CDAI of ≥ 70) at 2 weeks was significantly greater in the CDP-treated group as compared to those receiving placebo (_p_=0.023).
A phase III trial evaluating the efficacy of CDP-571 in a larger cohort of patients with moderate to severe CD soon followed [104]. This multicenter, randomized, double-blind, placebo-controlled study extended the duration of the trial to 28 weeks. 396 patients in 68 centers were randomized in a 2:1 fashion to receive either CDP-571 10 mg/kg i.v. or placebo every 8 weeks until week 24. The population receiving at least one dose of study medication was then assessed for meeting the primary endpoint 4 weeks later, the percentage of patients with a clinical response (decrease in CDAI of ≥ 100) or those who had achieved remission (CDAI ≤ 150). At the conclusion of the study, 80/263 (30.4%) patients treated with CDP had a clinical response compared with 31/132 patients (23.5%) given placebo (_p_=0.102). In a post-hoc analysis, patients with high serum CRP (≥ 10) were more likely to achieve clinical response (29/101 [28.7%]) with CDP-571 than those who had received placebo (7/58 [12.1%]) (_p_=0.018). Thus, CDP-571 is more likely to work in a select group of patients with high serum CRP concentration.
The safety and tolerability of CDP-571 was also evaluated in a small group (_n_=20) of pediatric patients with active CD (Pediatric CD Activity Index (PCDAI) ≥ 20) [105]. Children were given CDP-571 10 mg/kg as a single dose and followed for 12 weeks. 65% of patients had a clinical response to CDP-571 at the 2-week time point, defined as a decrease of ≥ 10 points in PCDAI. 6 of these patients developed detectable Abs to CDP-571.
Pegsunercept (r-metHu-sTNFRI, PEG sTNFRI)
Pegsunercept is a truncated form of soluble, natural TNF p55 type I receptor molecule derived from E. coli, PEGylated with a 30 kDa PEG molecule at the N-terminus (met-1) position to extend its biological half-life [106]. Preclinical studies of rodent models have suggested efficacy in treating both adjuvant and collagen induced arthritis. Treatment with pegsunercept significantly inhibited the amount of joint swelling. Histologically, it also reduced inflammation, pannus, cartilage and bone damage [107].
In a prospective, double-blind, randomized, placebo-controlled, multicenter, phase II trial in the US, treatment with pegsunercept at 800 μg/kg for 12 weeks demonstrated significant improvement for ACR20 response, including morning stiffness and HAQ [108]. It was also well tolerated: rates of adverse events and infectious episodes were similar to those of placebo, and no clinically significant safety concerns were found with pegsunercept in treating RA [109]. Further studies to determine the optimal dose and frequency are still ongoing.
Onercept
Another strategy of avoiding the immunogenicity associated with traditional anti-TNF Abs is to develop a recombinant TNF binding protein, a soluble form of the membrane-bound TNF receptor p55. One such biologic, onercept was developed to bind with high affinity to soluble TNF in the serum of patients with CD. A small phase I pilot study tested the safety and anti-inflammatory properties of onercept [110]. This TNF binding protein (TBP-1) was given to 12 patients with moderate to severe CD who were naïve to other anti-TNF biologics. Patients were randomly assigned to receive either 11.7 or 50 mg s. c. three times weekly for 2 weeks, and were followed for as long as 6 months after the last treatment. The primary efficacy endpoint was defined as the mean change in CDAI from baseline to measurement at day 15 of the study. Mean CDAI values dropped from 291±44 to 196±65 in the low dose group, and from 296±52 to 181±72 in the high dose group. Clinical improvement was noted for as long as 4 months after stopping the study drug, before patients again had to resume traditional therapy. During the first 6 weeks of the study, 7/12 patients achieved clinical response (drop in CDAI of ≥ 100), while 5 of these 7 patients entered remission (CDAI score of ≤ 150). These responses were very rapid, most occurring during the 2-week study period.
Future potential TNF antagonists
Small-molecule inhibitors to disassemble TNFα trimers. A recent study identified a small-molecule inhibitor of TNFα that promotes subunit disassembly of this trimeric cytokine family member [111]. This compound formed an intermediate complex with the intact trimer, resulting in a 600-fold accelerated subunit dissociation rate that leads to trimer dissociation. The compound inhibits TNFα activity in biochemical- and cell-based assays.
Cell signaling inhibitors that inhibit TNFα production
Hoping to develop more specific and less toxic therapies, several groups are developing compounds that can more specifically block intracellular signaling resulting in reduced TNFα production or its effects. For example, a recent report identified an inhibitor of p38 MAP kinase as a potent anti-TNFα drug [112].
Certain drugs already in the clinic can effectively reduce or block TNF production or its effects. Examples include thalidomide and statins in high doses. A better understanding of these effects and how and when to apply these drugs in the treatment of IMIDs would be desirable.
Looking beyond TNFα antagonists
Although anti-TNF agents confer remarkable therapeutic benefit compared to other available therapeutic modalities, only about 50% or fewer RA patients achieved a 50% response (ACR50) in most clinical trials. Thus, a sizeable portion of patients with RA and other inflammatory diseases has an incomplete response, and a small percentage have no response to TNF inhibitors. Unfortunately, combination biologic therapy may not be the solution: the available data using anakinra plus anti-TNF and abatacept plus other biologics show an increased risk of serious infections. Moreover, the anti-TNF agents have been found to have no effect in certain IMIDs and in other cases such as SLE, there is a potential risk of exacerbating IMID. Other therapies based on the role of other cell types and molecules are being examined for such cases. So, the search has to continue for newer, more effective treatments for IMIDs.
Acknowledgments
Drs. Braun, Reed and Singh are recipients of grants from the National Institutes of Health. Dr. Lin is recipient of a fellowship grant from the Southern California Chapter of the Scleroderma Foundation. Dr. Ziring is the recipient of a research fellowship award from the Crohns and Colitis Foundation of America.
We apologize to authors whose contributions could not be cited due to page limitations.
This article was commissioned by the Federation of Clinical Immunology Societies (FOCIS) and supported through an unrestricted educational grant from Abbott Laboratories. The content of this article was formulated solely by the authors.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author’s institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit:
http://www.elsevier.com/copyright
References
- 1.Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl J Med. 2006;355:704–712. doi: 10.1056/NEJMct055183. [DOI] [PubMed] [Google Scholar]
- 2.Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003;14:185–191. doi: 10.1016/s1359-6101(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 3.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–1725. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 4.Harriman G, Harper LK, Schaible TF. Summary of clinical trials in rheumatoid arthritis using infliximab, an anti-TNF alpha treatment. Ann Rheum Dis. 1999;58:i61–i64. doi: 10.1136/ard.58.2008.i61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klinkhoff A. Biological agents for rheumatoid arthritis: targeting both physical function and structural damage. Drugs. 2004;64:1267–1283. doi: 10.2165/00003495-200464120-00001. [DOI] [PubMed] [Google Scholar]
- 6.Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151:1548–1561. [PubMed] [Google Scholar]
- 7.Spencer-Green G. Etanercept (Enbrel): update on therapeutic use. Ann Rheum Dis. 2000;59:i46–i49. doi: 10.1136/ard.59.suppl_1.i46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med. 1992;175:323–329. doi: 10.1084/jem.175.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen C, Assche GV, Colpaert S, Maerten P, Geboes K, Rutgeerts P, Ceuppens JL. Adalimumab induces apoptosis of human monocytes: a comparative study with infliximab and etanercept. Aliment Pharmacol Ther. 2005;21:251–258. doi: 10.1111/j.1365-2036.2005.02309.x. [DOI] [PubMed] [Google Scholar]
- 10.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–298. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 11.Scallon BJ, Moore MA, Trinh H, Knight DM, Ghrayeb J. Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine. 1995;7:251–259. doi: 10.1006/cyto.1995.0029. [DOI] [PubMed] [Google Scholar]
- 12.Mitoma H, Horiuchi T, Hatta N, Tsukamoto H, Harashima S, Kikuchi Y, Otsuka J, Okamura S, Fujita S, Harada M. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology. 2005;128:376–392. doi: 10.1053/j.gastro.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 13.Gustot T, Lemmers A, Louis E, Nicaise C, Quertinmont E, Belaiche J, Roland S, Van Gossum A, Deviere J, Franchimont D. Profile of soluble cytokine receptors in Crohn's disease. Gut. 2005;54:488–495. doi: 10.1136/gut.2004.043554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gramlick A, Fossati G, Nesbitt AM. Neutralization of soluble and membrane tumor necrosis factor (TNF) by infliximab, adalimumab, or certolizumab pegol using p55 or p75 TNF-receptor-specific bioassays. Gastroenterology. 2006;130:697. [Google Scholar]
- 15.Fossati G, Nesbitt AM. In vitro complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity by the anti-TNF agents adalimumab, etanercept, infliximab, and certolizumab pegol (CDP870) Am J Gastroenterol. 2005;100:S287. [Google Scholar]
- 16.Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 17.Van den Brande JM, Braat H, van den Brink GR, Versteeg HH, Bauer CA, Hoedemaeker I, van Montfrans C, Hommes DW, Peppelenbosch MP, van Deventer SJ. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology. 2003;124:1774–1785. doi: 10.1016/s0016-5085(03)00382-2. [DOI] [PubMed] [Google Scholar]
- 18.Mitoma H, Horiuchi T, Tsukamoto H. Binding activities of infliximab and etanercept to transmembrane tumor necrosis factor-alpha. Gastroenterology. 2004;126:934–935. doi: 10.1053/j.gastro.2004.01.036. author reply 935–6. [DOI] [PubMed] [Google Scholar]
- 19.Di Sabatino A, Ciccocioppo R, Cinque B, Millimaggi D, Morera R, Ricevuti L, Cifone MG, Corazza GR. Defective mucosal T cell death is sustainably reverted by infliximab in a caspase dependent pathway in Crohn's disease. Gut. 2004;53:70–77. doi: 10.1136/gut.53.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lugering A, Schmidt M, Lugering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn's disease by using a caspase-dependent pathway. Gastroenterology. 2001;121:1145–1157. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- 21.Shen C, Van Assche G, Rutgeerts P, Ceuppens JL. Caspase activation and apoptosis induction by adalimumab: demonstration in vitro and in vivo in a chimeric mouse model. Inflamm Bowel Dis. 2006;12:22–28. doi: 10.1097/01.mib.0000194185.69800.07. [DOI] [PubMed] [Google Scholar]
- 22.Di Sabatino A, Pender SL, Jackson CL, Prothero JD, Gordon JN, Picariello L, Rovedatti L, Docena G, Monteleone G, Rampton DS, Tonelli F, Corazza GR, MacDonald TT. Functional modulation of Crohn's disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology. 2007;133:137–149. doi: 10.1053/j.gastro.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 23.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 24.Paleolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN. Deactivation of vascular endothelium by monoclonal anti-tumor necrosis factor alpha antibody in rheumatoid arthritis. Arthritis Rheum. 1996;39:1082–1091. doi: 10.1002/art.1780390703. [DOI] [PubMed] [Google Scholar]
- 25.Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, Meinders AE, Maini RN. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1077–1081. doi: 10.1002/art.1780390702. [DOI] [PubMed] [Google Scholar]
- 26.Goldring SR, Gravallese EM. Pathogenesis of bone lesions in rheumatoid arthritis. Curr Rheumatol Rep. 2002;4:226–231. doi: 10.1007/s11926-002-0069-y. [DOI] [PubMed] [Google Scholar]
- 27.Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med. 1985;162:2163–2168. doi: 10.1084/jem.162.6.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, Breedveld FC, Smolen JS, Eberl G, deWoody K, Feldmann M, Maini RN. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163:1521–1528. [PubMed] [Google Scholar]
- 29.Ulfgren AK, Andersson U, Engstrom M, Klareskog L, Maini RN, Taylor PC. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391–2396. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 30.Catrina AI, Lampa J, Ernestam S, af Klint E, Bratt J, Klareskog L, Ulfgren AK. Anti-tumour necrosis factor (TNF)-alpha therapy (etanercept) down-regulates serum matrix metalloproteinase (MMP)-3 and MMP-1 in rheumatoid arthritis. Rheumatology (Oxford) 2002;41:484–489. doi: 10.1093/rheumatology/41.5.484. [DOI] [PubMed] [Google Scholar]
- 31.Brennan FM, Browne KA, Green PA, Jaspar JM, Maini RN, Feldmann M. Reduction of serum matrix metalloproteinase 1 and matrix metalloproteinase 3 in rheumatoid arthritis patients following anti-tumour necrosis factor-alpha (cA2) therapy. Br J Rheumatol. 1997;36:643–650. doi: 10.1093/rheumatology/36.6.643. [DOI] [PubMed] [Google Scholar]
- 32.Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 33.Capsoni F, Sarzi-Puttini P, Atzeni F, Minonzio F, Bonara P, Doria A, Carrabba M. Effect of adalimumab on neutrophil function in patients with rheumatoid arthritis. Arthritis Res Ther. 2005;7:R250–R255. doi: 10.1186/ar1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.den Broeder AA, Wanten GJ, Oyen WJ, Naber T, van Riel PL, Barrera P. Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti-tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. J Rheumatol. 2003;30:232–237. [PubMed] [Google Scholar]
- 35.Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, Klareskog L, Ulfgren AK. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52:61–72. doi: 10.1002/art.20764. [DOI] [PubMed] [Google Scholar]
- 36.Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998;41:1258–1265. doi: 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 37.Klimiuk PA, Sierakowski S, Domyslawska I, Fiedorczyk M, Chwiecko J. Reduction of soluble adhesion molecules (sICAM-1, sVCAM-1, and sE-selectin) and vascular endothelial growth factor levels in serum of rheumatoid arthritis patients following multiple intravenous infusions of infliximab. Arch Immunol Ther Exp (Warsz) 2004;52:36–42. [PubMed] [Google Scholar]
- 38.Hurlimann D, Forster A, Noll G, Enseleit F, Chenevard R, Distler O, Bechir M, Spieker LE, Neidhart M, Michel BA, Gay RE, Luscher TF, Gay S, Ruschitzka F. Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation. 2002;106:2184–2187. doi: 10.1161/01.cir.0000037521.71373.44. [DOI] [PubMed] [Google Scholar]
- 39.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein I, Ben-Horin S, Koltakov A, Chermoshnuk H, Polevoy V, Berkun Y, Amariglio N, Bank I. alpha1beta1 Integrin+ and regulatory Foxp3+ T cells constitute two functionally distinct human CD4+ T cell subsets oppositely modulated by TNFalpha blockade. J Immunol. 2007;178:201–210. doi: 10.4049/jimmunol.178.1.201. [DOI] [PubMed] [Google Scholar]
- 41.Balanescu A, Radu E, Nat R, Regalia T, Bojinca V, Ionescu R, Balanescu S, Savu C, Predeteanu D. Early and late effect of infliximab on circulating dendritic cells phenotype in rheumatoid arthritis patients. Int J Clin Pharmacol Res. 2005;25:9–18. [PubMed] [Google Scholar]
- 42.Catrina AI, af Klint E, Ernestam S, Catrina SB, Makrygiannakis D, Botusan IR, Klareskog L, Ulfgren AK. Anti-tumor necrosis factor therapy increases synovial osteo-protegerin expression in rheumatoid arthritis. Arthritis Rheum. 2006;54:76–81. doi: 10.1002/art.21528. [DOI] [PubMed] [Google Scholar]
- 43.Voulgari PV, Kolios G, Papadopoulos GK, Katsaraki A, Seferiadis K, Drosos AA. Role of cytokines in the pathogenesis of anemia of chronic disease in rheumatoid arthritis. Clin Immunol. 1999;92:153–160. doi: 10.1006/clim.1999.4736. [DOI] [PubMed] [Google Scholar]
- 44.Davis D, Charles PJ, Potter A, Feldmann M, Maini RN, Elliott MJ. Anaemia of chronic disease in rheumatoid arthritis: in vivo effects of tumour necrosis factor alpha blockade. Br J Rheumatol. 1997;36:950–956. doi: 10.1093/rheumatology/36.9.950. [DOI] [PubMed] [Google Scholar]
- 45.Boirivant M, Marini M, Di Felice G, Pronio AM, Montesani C, Tersigni R, Strober W. Lamina propria T cells in Crohn's disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology. 1999;116:557–565. doi: 10.1016/s0016-5085(99)70177-0. [DOI] [PubMed] [Google Scholar]
- 46.Ina K, Itoh J, Fukushima K, Kusugami K, Yamaguchi T, Kyokane K, Imada A, Binion DG, Musso A, West GA, Dobrea GM, McCormick TS, Lapetina EG, Levine AD, Ottaway CA, Fiocchi C. Resistance of Crohn's disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol. 1999;163:1081–1090. [PubMed] [Google Scholar]
- 47.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ringheanu M, Daum F, Markowitz J, Levine J, Katz S, Lin X, Silver J. Effects of infliximab on apoptosis and reverse signaling of monocytes from healthy individuals and patients with Crohn's disease. Inflamm Bowel Dis. 2004;10:801–810. doi: 10.1097/00054725-200411000-00015. [DOI] [PubMed] [Google Scholar]
- 49.Till A, Rosenstiel P, Krippner-Heidenreich A, Mascheretti-Croucher S, Croucher PJ, Schafer H, Scheurich P, Seegert D, Schreiber S. The Met-196→Arg variation of human tumor necrosis factor receptor 2 (TNFR2) affects TNF-alpha-induced apoptosis by impaired NF-kappaB signaling and target gene expression. J Biol Chem. 2005;280:5994–6004. doi: 10.1074/jbc.M411541200. [DOI] [PubMed] [Google Scholar]
- 50.Agnholt J, Kaltoft K. Infliximab downregulates interferon-gamma production in activated gut T-lymphocytes from patients with Crohn's disease. Cytokine. 2001;15:212–222. doi: 10.1006/cyto.2001.0919. [DOI] [PubMed] [Google Scholar]
- 51.Danese S, Sans M, Scaldaferri F, Sgambato A, Rutella S, Cittadini A, Pique JM, Panes J, Katz JA, Gasbarrini A, Fiocchi C. TNF-alpha blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: a novel anti-inflammatory mechanism of infliximab in Crohn's disease. J Immunol. 2006;176:2617–2624. doi: 10.4049/jimmunol.176.4.2617. [DOI] [PubMed] [Google Scholar]
- 52.Gitter AH, Wullstein F, Fromm M, Schulzke JD. Epithelial barrier defects in ulcerative colitis: characterization and quantification by electrophysiological imaging. Gastroenterology. 2001;121:1320–1328. doi: 10.1053/gast.2001.29694. [DOI] [PubMed] [Google Scholar]
- 53.Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302. doi: 10.1136/gut.2003.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shealy DJ, Wooley PH, Emmell E, Volk A, Rosenberg A, Treacy G, Wagner CL, Mayton L, Griswold DE, Song XY. Anti-TNF-alpha antibody allows healing of joint damage in polyarthritic transgenic mice. Arthritis Res. 2002;4:R7. doi: 10.1186/ar430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmdahl R, Andersson ME, Goldschmidt TJ, Jansson L, Karlsson M, Malmstrom V, Mo J. Collagen induced arthritis as an experimental model for rheumatoid arthritis. Immunogenetics, pathogenesis and autoimmunity. APMIS. 1989;97:575–584. doi: 10.1111/j.1699-0463.1989.tb00446.x. [DOI] [PubMed] [Google Scholar]
- 57.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams RO, Mason LJ, Feldmann M, Maini RN. Synergy between anti-CD4 and anti-tumor necrosis factor in the amelioration of established collagen-induced arthritis. Proc Natl Acad Sci U S A. 1994;91:2762–2766. doi: 10.1073/pnas.91.7.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams RO, Ghrayeb J, Feldmann M, Maini RN. Successful therapy of collagen-induced arthritis with TNF receptor–IgG fusion protein and combination with anti-CD4. Immunology. 1995;84:433–439. [PMC free article] [PubMed] [Google Scholar]
- 60.Williams RO, Marinova-Mutafchieva L, Feldmann M, Maini RN. Evaluation of TNF-alpha and IL-1 blockade in collagen-induced arthritis and comparison with combined anti-TNF-alpha/anti-CD4 therapy. J Immunol. 2000;165:7240–7245. doi: 10.4049/jimmunol.165.12.7240. [DOI] [PubMed] [Google Scholar]
- 61.Marinova-Mutafchieva L, Williams RO, Mauri C, Mason LJ, Walmsley MJ, Taylor PC, Feldmann M, Maini RN. A comparative study into the mechanisms of action of anti-tumor necrosis factor alpha, anti-CD4, and combined anti-tumor necrosis factor alpha/anti-CD4 treatment in early collagen-induced arthritis. Arthritis Rheum. 2000;43:638–644. doi: 10.1002/1529-0131(200003)43:3<638::AID-ANR21>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 62.Su X, Zhou T, Yang P, Edwards CK, III, Mountz JD. Reduction of arthritis and pneumonitis in motheaten mice by soluble tumor necrosis factor receptor. Arthritis Rheum. 1998;41:139–149. doi: 10.1002/1529-0131(199801)41:1<139::AID-ART17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 63.Pizarro TT, Arseneau KO, Bamias G, Cominelli F. Mouse models for the study of Crohn's disease. Trends Mol Med. 2003;9:218–222. doi: 10.1016/s1471-4914(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 64.Kosiewicz MM, Nast CC, Krishnan A, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Kozaiwa K, Cominelli F. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J Clin Invest. 2001;107:695–702. doi: 10.1172/JCI10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marini M, Bamias G, Rivera-Nieves J, Moskaluk CA, Hoang SB, Ross WG, Pizarro TT, Cominelli F. TNF-alpha neutralization ameliorates the severity of murine Crohn's-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci U S A. 2003;100:8366–8371. doi: 10.1073/pnas.1432897100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen C, Maerten P, Geboes K, Van Assche G, Rutgeerts P, Ceuppens JL. Infliximab induces apoptosis of monocytes and T lymphocytes in a human-mouse chimeric model. Clin Immunol. 2005;115:250–259. doi: 10.1016/j.clim.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 67.Itai T, Tanaka M, Nagata S. Processing of tumor necrosis factor by the membrane-bound TNF-alpha-converting enzyme, but not its truncated soluble form. Eur J Biochem. 2001;268:2074–2082. doi: 10.1046/j.1432-1327.2001.02085.x. [DOI] [PubMed] [Google Scholar]
- 68.Harashima S, Horiuchi T, Hatta N, Morita C, Higuchi M, Sawabe T, Tsukamoto H, Tahira T, Hayashi K, Fujita S, Niho Y. Outside-to-inside signal through the membrane TNF-alpha induces E expression on activated human CD4+ T cells. J Immunol. 2001;166:130–136. doi: 10.4049/jimmunol.166.1.130. [DOI] [PubMed] [Google Scholar]
- 69.Kaymakcalan SP, Bose S, Sasso EH. Etanercept, infliximab, and adalimumab bind to soluble and transmembrane TNF with similar affinities. Clin Immunol. 2006;119(supplement):S69. [Google Scholar]
- 70.Sieper J, Baraliakos X, Listing J, Brandt J, Haibel H, Rudwaleit M, Braun J. Persistent reduction of spinal inflammation as assessed by magnetic resonance imaging in patients with ankylosing spondylitis after 2 yrs of treatment with the anti-tumour necrosis factor agent infliximab. Rheumatology (Oxford) 2005;44:1525–1530. doi: 10.1093/rheumatology/kei046. [DOI] [PubMed] [Google Scholar]
- 71.Mugnier B, Balandraud N, Darque A, Roudier C, Roudier J, Reviron D. Polymorphism at position-308 of the tumor necrosis factor alpha gene influences outcome of infliximab therapy in rheumatoid arthritis. Arthritis Rheum. 2003;48:1849–1852. doi: 10.1002/art.11168. [DOI] [PubMed] [Google Scholar]
- 72.Seitz M, Wirthmuller U, Moller B, Villiger PM. The-308 tumour necrosis factor-alpha gene polymorphism predicts therapeutic response to TNFalpha-blockers in rheumatoid arthritis and spondyloarthritis patients. Rheumatology (Oxford) 2007;46:93–96. doi: 10.1093/rheumatology/kel175. [DOI] [PubMed] [Google Scholar]
- 73.Via CS, Shustov A, Rus V, Lang T, Nguyen P, Finkelman FD. In vivo neutralization of TNF-alpha promotes humoral auto-immunity by preventing the induction of CTL. J Immunol. 2001;167:6821–6826. doi: 10.4049/jimmunol.167.12.6821. [DOI] [PubMed] [Google Scholar]
- 74.Atzeni F, Turiel M, Capsoni F, Doria A, Meroni P, Sarzi-Puttini P. Autoimmunity and anti-TNF-alpha agents. Ann N Y Acad Sci. 2005;1051:559–569. doi: 10.1196/annals.1361.100. [DOI] [PubMed] [Google Scholar]
- 75.Charles PJ, Smeenk RJ, De Jong J, Feldmann M, Maini RN. Assessment of antibodies to double-stranded DNA induced in rheumatoid arthritis patients following treatment with infliximab, a monoclonal antibody to tumor necrosis factor alpha: findings in open-label and randomized placebo-controlled trials. Arthritis Rheum. 2000;43:2383–2390. doi: 10.1002/1529-0131(200011)43:11<2383::AID-ANR2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 76.Vermeire S, Noman M, Van Assche G, Baert F, Van Steen K, Esters N, Joossens S, Bossuyt X, Rutgeerts P. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn's disease: a prospective cohort study. Gastroenterology. 2003;125:32–39. doi: 10.1016/s0016-5085(03)00701-7. [DOI] [PubMed] [Google Scholar]
- 77.De Rycke L, Kruithof E, Van Damme N, Hoffman IE, Van den Bossche N, Van den Bosch F, Veys EM, De Keyser F. Antinuclear antibodies following infliximab treatment in patients with rheumatoid arthritis or spondylarthropathy. Arthritis Rheum. 2003;48:1015–1023. doi: 10.1002/art.10876. [DOI] [PubMed] [Google Scholar]
- 78.Comby E, Tanaff P, Mariotte D, Costentin-Pignol V, Marcelli C, Ballet JJ. Evolution of antinuclear antibodies and clinical patterns in patients with active rheumatoid arthritis with long term infliximab therapy. J Rheumatol. 2006;33:24–30. [PubMed] [Google Scholar]
- 79.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 80.Senaldi G, Yin S, Shaklee CL, Piguet PF, Mak TW, Ulich TR. Corynebacterium parvum- and Mycobacterium bovis bacillus Calmette–Guerin-induced granuloma formation is inhibited in TNF receptor I (TNF-RI) knockout mice and by treatment with soluble TNF-RI. J Immunol. 1996;157:5022–5026. [PubMed] [Google Scholar]
- 81.Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- 82.Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gomez-Reino JJ, Carmona L, Angel Descalzo M. Risk of tuberculosis in patients treated with tumor necrosis factor antagonists due to incomplete prevention of reactivation of latent infection. Arthritis Rheum. 2007;57:756–761. doi: 10.1002/art.22768. [DOI] [PubMed] [Google Scholar]
- 84.Torre D, Zeroli C, Giola M, Ferrario G, Fiori GP, Bonetta G, Tambini R. Serum levels of interleukin-1 alpha, interleukin-1 beta, interleukin-6, and tumor necrosis factor in patients with acute viral hepatitis. Clin Infect Dis. 1994;18:194–198. doi: 10.1093/clinids/18.2.194. [DOI] [PubMed] [Google Scholar]
- 85.Fang JW, Shen WW, Meager A, Lau JY. Activation of the tumor necrosis factor-alpha system in the liver in chronic hepatitis B virus infection. Am J Gastroenterol. 1996;91:748–753. [PubMed] [Google Scholar]
- 86.Kasahara S, Ando K, Saito K, Sekikawa K, Ito H, Ishikawa T, Ohnishi H, Seishima M, Kakumu S, Moriwaki H. Lack of tumor necrosis factor alpha induces impaired proliferation of hepatitis B virus-specific cytotoxic T lymphocytes. J Virol. 2003;77:2469–2476. doi: 10.1128/JVI.77.4.2469-2476.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Allendoerfer R, Deepe GS., Jr Blockade of endogenous TNF-alpha exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J Immunol. 1998;160:6072–6082. [PubMed] [Google Scholar]
- 88.Bauman SK, Huffnagle GB, Murphy JW. Effects of tumor necrosis factor alpha on dendritic cell accumulation in lymph nodes draining the immunization site and the impact on the anticryptococcal cell-mediated immune response. Infect Immun. 2003;71:68–74. doi: 10.1128/IAI.71.1.68-74.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beaman L. Effects of recombinant gamma interferon and tumor necrosis factor on in vitro interactions of human mononuclear phagocytes with Coccidioides immitis. Infect Immun. 1991;59:4227–4229. doi: 10.1128/iai.59.11.4227-4229.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wood KL, Hage CA, Knox KS, Kleiman MB, Sannuti A, Day RB, Wheat LJ, Twigg HL., III Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med. 2003;167:1279–1282. doi: 10.1164/rccm.200206-563OC. [DOI] [PubMed] [Google Scholar]
- 91.Zeltser R, Valle L, Tanck C, Holyst MM, Ritchlin C, Gaspari AA. Clinical, histological, and immunophenotypic characteristics of injection site reactions associated with etanercept: a recombinant tumor necrosis factor alpha receptor: Fc fusion protein. Arch Dermatol. 2001;137:893–899. [PubMed] [Google Scholar]
- 92.Haraoui B, Cameron L, Ouellet M, White B. Anti-infliximab antibodies in patients with rheumatoid arthritis who require higher doses of infliximab to achieve or maintain a clinical response. J Rheumatol. 2006;33:31–36. [PubMed] [Google Scholar]
- 93.Hlavaty T, Pierik M, Henckaerts L, Ferrante M, Joossens S, van Schuerbeek N, Noman M, Rutgeerts P, Vermeire S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn's disease. Aliment Pharmacol Ther. 2005;22:613–626. doi: 10.1111/j.1365-2036.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 94.Pierik M, Vermeire S, Steen KV, Joossens S, Claessens G, Vlietinck R, Rutgeerts P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20:303–310. doi: 10.1111/j.1365-2036.2004.01946.x. [DOI] [PubMed] [Google Scholar]
- 95.Kaushik V, Moots R. CDP-870 (certolizumab) in rheumatoid arthritis. Expert Opin Biol Ther. 2005;5:601–606. doi: 10.1517/14712598.5.4.601. [DOI] [PubMed] [Google Scholar]
- 96.Rankin E, Choy E, Kassimos D, Kingsley G, Sopwith A, Isenberg D, Panayi G. The therapeutic effects of an engineered human anti-tumour necrosis factor alpha antibody (CDP571) in rheumatoid arthritis. Br J Rheumatol. 1995;34:334–342. doi: 10.1093/rheumatology/34.4.334. [DOI] [PubMed] [Google Scholar]
- 97.Choy E, Hazleman B, Smith M, Moss K, Lisi L, Scott D, Patel J, Sopwith M, Isenberg D. Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized, dose-escalating trial. Rheumatology. 2002;41:1133–1137. doi: 10.1093/rheumatology/41.10.1133. [DOI] [PubMed] [Google Scholar]
- 98.Moreland L, Schiff M, Baumgartner S, Tindall E, Fleischmann R, Bulpitt K, Weaver A, Keystone E, Furst D, Mease P, Ruderman E, Horwitz D, Arkfeld D, Garrison L, Burge D, Blosch C, Lange M, McDonnell N, Weinblatt M. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130:478–486. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- 99.Maini R, St Clair E, Breedveld F, Furst D, Kalden J, Weisman M, Smolen J, Harriman EPG, Feldmann M, Lipsky P. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354:1932–1939. doi: 10.1016/s0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- 100.Schreiber S, Khaliq-Kareemi M, Lawrence I, et al. Certolizumab pegol, a humanised anti-TNF PEGylated Fab' fragment, is safe and effective in the maintenance of response and remission following induction in active Crohn's Disease: a Phase III study (PRECISE) (Abstract) Gut. 2005;54:A82. [Google Scholar]
- 101.Kay J, Matteson E, Dasgupta B, Nash P, Durez P, Hall P, Han J, Rahman M. Subcutaneous Injection of CNTO 148 Compared with Placebo in Patients with Active Rheumatoid Arthritis Despite Treatment with Methotrexate: A Randomized, Double-blind, Dose-ranging Trial. American College of Rheumatology; San Diego: 2005. [DOI] [PubMed] [Google Scholar]
- 102.Stack WA, Mann SD, Roy AJ, Heath P, Sopwith M, Freeman J, Holmes G, Long R, Forbes A, Kamm MA. Randomised controlled trial of CDP571 antibody to tumour necrosis factor-alpha in Crohn's disease. Lancet. 1997;349:521–524. doi: 10.1016/s0140-6736(97)80083-9. [DOI] [PubMed] [Google Scholar]
- 103.Sandborn WJ, Feagan BG, Hanauer SB, Present DH, Sutherland LR, Kamm MA, Wolf DC, Baker JP, Hawkey C, Archambault A, Bernstein CN, Novak C, Heath PK, Targan SR. An engineered human antibody to TNF (CDP571) for active Crohn's disease: a randomized double-blind placebo-controlled trial. Gastroenterology. 2001;120:1330–1338. doi: 10.1053/gast.2001.24042. [DOI] [PubMed] [Google Scholar]
- 104.Colombel JF, Loftus WJ EV, Jr, Tremaine LJ, Egan WS, Harmsen CD, Schleck AR, Zinsmeister WJ. The safety profile of infliximab in patients with Crohn's disease: the Mayo clinic experience in 500 patients. Gastroenterology. 2004;126:19–31. doi: 10.1053/j.gastro.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 105.Mamula P, Markowitz JE, Cohen LJ, von Allmen D, Baldassano RN. Infliximab in pediatric ulcerative colitis: two-year follow-up. J Pediatr Gastroenterol Nutr. 2004;38:298–301. doi: 10.1097/00005176-200403000-00013. [DOI] [PubMed] [Google Scholar]
- 106.Edwards CK, III, Martin SW, Seely J, Kinstler O, Buckel S, Bendele AM, Ellen Cosenza M, Feige U, Kohno T. Design of PEGylated soluble tumor necrosis factor receptor type I (PEG sTNF-RI) for chronic inflammatory diseases. Adv Drug Deliv Rev. 2003;55:1315–1336. doi: 10.1016/s0169-409x(03)00112-1. [DOI] [PubMed] [Google Scholar]
- 107.McComb J, Gould T, Chlipala E, Sennelo G, Frazier J, Kieft G, Seely J, Edwards CK, III, Bendele A. Antiarthritic activity of soluble tumor necrosis factor receptor type I forms in adjuvant arthritis: correlation of plasma levels with efficacy. J Rheumatol. 1999;26:1347–1351. [PubMed] [Google Scholar]
- 108.Sole K. Pegsunercept is a safe and efficacious treatment for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:121. doi: 10.1038/ncprheum0171. [DOI] [PubMed] [Google Scholar]
- 109.Furst D, Fleischmann R, Kopp E, Schiff M, Edwards CR, Solinger A, Macri M, Group S. A phase 2 dose-finding study of PEGylated recombinant methionyl human soluble tumor necrosis factor type I in patients with rheumatoid arthritis. J Rheumatol. 2005;32:2303–2310. [PubMed] [Google Scholar]
- 110.Rutgeerts P, Lemmens L, Van Assche G, Noman M, Borghini-Fuhrer I, Goedkoop R. Treatment of active Crohn's disease with onercept (recombinant human soluble p55 tumour necrosis factor receptor): results of a randomized, open-label, pilot study. Aliment Pharmacol Ther. 2003;17:185–192. doi: 10.1046/j.1365-2036.2003.01414.x. [DOI] [PubMed] [Google Scholar]
- 111.He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, Fung AD, Farrington G, Eldredge JK, Day ES, Cruz LA, Cachero TG, Miller SK, Friedman JE, Choong IC, Cunningham BC. Small-molecule inhibition of TNF-alpha. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- 112.Miwatashi S, Arikawa Y, Kotani E, Miyamoto M, Naruo K, Kimura H, Tanaka T, Asahi S, Ohkawa S. Novel inhibitor of p38 MAP kinase as an anti-TNF-alpha drug: discovery of N-[4-[2-ethyl-4-(3-methylphenyl)-1,3-thiazol-5-yl]-2-pyridyl]benza-mide (TAK-715) as a potent and orally active anti-rheumatoid arthritis agent. J Med Chem. 2005;48:5966–5979. doi: 10.1021/jm050165o. [DOI] [PubMed] [Google Scholar]
- 113.Maini RN, Feldmann M. How does infliximab work in rheumatoid arthritis? Arthritis Res. 2002;4(Suppl 2):S22–S28. doi: 10.1186/ar549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brennan FM, Chantry D, Jackson AM, Maini RN, Feldmann M. Cytokine production in culture by cells isolated from the synovial membrane. J Autoimmun. 1989;2(Suppl):177–186. doi: 10.1016/0896-8411(89)90129-7. [DOI] [PubMed] [Google Scholar]
- 115.Taylor PC, Peters AM, Glass DM, Maini RN. Effects of treatment of rheumatoid arthritis patients with an antibody against tumour necrosis factor alpha on reticuloendothelial and intrapulmonary granulocyte traffic. Clin Sci (Lond) 1999;97:85–89. [PubMed] [Google Scholar]
- 116.Lin J, Ziring D, Desai S, Kim S, Wong M, Korin Y, Braun J, Reed E, Gjertson D, Singh RR. TNFα blockade in human diseases: an overview of efficacy and safety. Clin Immunol. doi: 10.1016/j.clim.2007.08.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]