Nitric oxide-mediated immunosuppression following murine Echinococcus multilocularis infection (original) (raw)
Abstract
In some parasitic infections immunosuppression is a prominent characteristic of the host–parasite interplay. We have used a murine alveolar echinococcosis (AE) model in susceptible C57BL/6 mice to document a suppressed splenocyte proliferative response to concanavalin A (Con A) at the early (1-month) stage and to _Echinococcus multilocularis_-crude antigen (Emc-antigen) at the late (4–6-month) stage of chronic infection. Despite proliferative suppression, splenic cytokine production [interleukin-2 (IL-2), IL-4 and interferon-γ (IFN-γ)] in response to Con A or Emc-antigen stimulation was not suppressed at 1 month postinfection (p.i.). Infection resulted in a strong Mac-1+ cell infiltration of the peritoneal cavity and spleen. Peritoneal cells (PEC) from mice infected at the 1-month stage were rich in macrophages and expressed significantly higher levels of transcripts for the inflammatory cytokine IL-1β and for tumour necrosis factor-α and inducible nitric oxide synthase (iNOS), when compared with PEC from non-infected control mice. Conversely, the IL-10 transcript level remained low and did not change during infection. Spleen cells supplemented with PEC from infected mice induced a marked increase in the levels of nitrite in response to Con A and Emc-antigen stimulation, and also a complete suppression of splenic proliferation. The spleen cells from late-stage infected mice expressed only background levels of IL-10 but greatly increased levels of iNOS, when compared with normal spleen cells. This observation correlated with the immunosuppression demonstrated at the late stage of murine AE. Furthermore, the suppressed splenic proliferative responses observed at the early and late stage were reversed to a large extent by the addition of NG-monomethyl-l-arginine and partially by anti-IFN-γ. Thus, our results demonstrated that the immunosuppression observed in chronic AE was not primarily dependent on IL-10 but rather on nitric oxide production by macrophages from infected animals.
INTRODUCTION
Alveolar echinococcosis (AE), a chronic disease caused by infection with the metacestode (larval) stage of the fox tapeworm Echinococcus multilocularis, is, if it remains untreated, one of the most lethal helminthic infections of humans.1 The natural life cycle involves mainly rodents and accidentally humans as intermediate hosts, where the metacestode stage of the parasite develops predominantly in the liver as a solid tumour-like tissue. Subsequently metastasis formation can occur in adjacent sites such as in the peritoneum, lungs, kidney and others. Extensive experimental studies to elucidate the host–parasite interplay have been carried out in laboratory rodents secondarily infected by intraperitoneal inoculation of metacestode material.2 Primary parasite tissue proliferation and subsequent metastasis of the metacestode in susceptible hosts is associated with the induction of a characteristic host immune response.1 Various studies in susceptible and relatively resistant mice have demonstrated that predominantly cell-mediated immunity plays a crucial role in the control of E. multilocularis infection.1,3–5 One of the interesting features observed during chronic E. multilocularis infection in mice as well as in human patients, is a marked depression of cell-mediated immunity.6–8 Previous studies on immunoregulative processes in experimental murine infections with larval E. multilocularis have pointed towards a relationship between the growth of the cyst mass and the depression in the level of cell-mediated immunity.7 The mechanism(s) of immunosuppression acting during chronic AE, however, are poorly understood so far. Potential contributions to immunosuppression during AE may involve increasing concentrations of immune complexes,9 the presence of high numbers of CD8dull suppressor T cells in the spleen8 and accessory macrophages in the peritoneal cavity of mice.10
Macrophages seem to play a key role in modulating different pathways of cellular immune responses.11 One of these concerns macrophage-mediated immunosuppression which has been found during the course of infection with a variety of, predominantly intracellular, pathogens, including Trypanosoma brucei and T. cruzi,12,13 Listeria monocytogenes,2 Salmonella typhimurium,14 Toxoplasma gondii15 and Plasmodium chabaudi.16 In addition, microbial extracts, for instance peptidoglycans,17 and thermal injury18 may also activate macrophages to mediate immunosuppression. In the present study, we addressed the questions first, whether peritoneal and splenic macrophages from mice infected with the uniquely extracellular metazoan parasite E. multilocularis mediate the suppression of lymphocyte proliferation, and second, whether nitric oxide (NO) produced by macrophages from infected mice is involved in the immunosuppressive status observed in the chronic course of secondary infection with E. multilocularis.
MATERIALS AND METHODS
Mice
Female 6–10-week-old C57BL/6 mice were purchased from Charles River GmbH (Kisslegg, Germany) and used for infection with E. multilocularis or as non-infected control litter mates. Each experimental step/group consisted of five animals (unless otherwise indicated in the Results or the figure legends). The number of groups and the repetition of experiments are given in the Results section and in the legends to the figures. All mice were raised, housed and handled under standard animal laboratory conditions according to the rules of the Swiss regulations for animal experimentation.
Parasites, experimental infections and parasite antigen preparation
The parasite used in this study was a cloned E. multilocularis (KF5) isolate19 maintained by serial passages (vegetative transfer) into Meriones unguiculatus or in C57BL/6 mice.
C57BL/6 mice were injected intraperitoneally (using a syringe and 1·2×40 mm needles) with 50–100 freshly prepared acephalic vesicular cysts suspended in RPMI-1640 (Gibco, Basel, Switzerland) to a final volume of 100 μl per mice. Control mice received a corresponding volume of RPMI-1640.
A crude E. multilocularis metacestode extract antigen (Emc-antigen) was prepared from a homogenate obtained by mincing and repeated cycles of freeze–thawing of cysts as described earlier.20 The 40 000 g supernatant fraction was dialysed against distilled water for 48 hr at 4°. A solution of 1 mg protein per ml was prepared in phosphate-buffered saline (PBS) and stored in aliquots at −80° until use. The antigen batch used for proliferation assay was the same as employed previously,21 which had been shown to be free of any bacterial endotoxins using a standard turbidimetric kinetic-Limulus assay (Amoebocyte lysate assay, Haemachem Inc., St. Louis, MO).
Cell preparations
Spleen cell suspensions were prepared from infected or non-infected control C57BL/6 mice. The cell suspensions were depleted of erythrocytes by treatment with 0·83% NH4Cl in 0·01 m Tris–HCl (pH 7·2) and subsequently resuspended in RPMI-1640 complete medium containing heat-inactivated 10% fetal calf serum (FCS, Gibco), 2 mm l-glutamine, 0·05 mm 2-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco).
Peritoneal cells (PEC) were obtained by performing peritoneal washings with 5 ml RPMI-1640, the sediments of the washings were resuspended in RPMI-1640 complete medium.
Flow cytometry
Cells (2×105–10×105) were labelled with either phycoerythrin-, biotin-, or fluorescein isothiocyanate (FITC)-coupled anti-Mac-1 (M1/70), anti-Iab (AF6-120.1), anti-CD45R/B220 (RA3-6B2), anti-CD4 (RM4-5) and anti-CD8 (53-6.7); biotin-coupled anti-Iab antibody was counterstained with streptavidin/phycoerythrin. Antibody-reagents were purchased from PharMingen, Heidelberg, Germany. Propidium iodide (Sigma Chemical, Basel, Switzerland) staining was used to exclude dead cells. To avoid unspecific binding, peritoneal cell suspensions were preincubated for 30 min in a mixture containing normal mouse serum (at a dilution of 1:40) and unlabelled anti-CD32 (2.4G2) antibody. Between each staining step, cells were washed twice with PBS containing 0·01% NaN3. Cells were analysed on a fluorescence-activated cell sorter (FACScan; Becton Dickinson, Heidelberg, Germany).
Cell cultures and lymphocyte proliferation assays
Spleen cells were cultured in 96-well round-bottom plates at 2×105/well. For some experiments, splenic cell cultures were supplemented with 5×104 of PEC from infected or non-infected control mice. The cell cultures were stimulated with concanavalin A (Con A) (2 μg/ml; Sigma) or crude parasite Emc-antigen (10 μg/ml), or were left unstimulated as negative controls. All tests were performed in quadruplicates.
The acetate salt of NG-monomethyl-l-arginine (NMMA, Calbiochem Novabiochem Corp., San Diego, CA) was used in cultures of spleen cells at a final concentration of 500 μm. Anti-interferon-γ (IFN-γ) neutralizing monoclonal antibody (mAb)22 (clone R4-6A2, a kind gift from Dr F. Brombacher, Max-Planck-Institute for Immunobiology, Freiburg, Germany) were added to the culture at 20 μg/ml.
Cells were stimulated with Emc-antigen for 96 hr and Con A for 72 hr. Subsequently, the cells were pulsed with 1 μCi/well of [3H]thymidine (New England Nuclear, Boston, MA) and harvested 16–18 hr later.20 The results were calculated as the mean of c.p.m./min of quadruplicate wells and expressed as geometric mean minus background (▵c.p.m.), where background is the c.p.m. of wells containing pulsed but unstimulated cells. The background was accepted when being <10% of the values of the stimulated positive wells. All experiments were done with duplicate plates in parallel, one for the proliferation assay and one for collecting supernatants for quantification of cytokine production.
Cytokines and nitrite production in the supernatants
Supernatants were harvested from quadruplicate cultures after 24, 48, or 96 hr and stored at −80°. Cytokines were measured by sandwich enzyme-linked immunosorbent assays (ELISA). The capture and biotinylated detection antibodies for interleukin-2 (IL-2), IL-10, IL-4 and IFN-γ were purchased from PharMingen GmbH, Heidelberg, Germany. Alkaline phosphatase covalently linked to streptavidin (PharMingen GmbH) was used to induce a colour reaction for the quantitative detection of secondary antibodies. The absorbance was subsequently measured at 405 nm.
Nitrite (NO2−) accumulation in the cell culture supernatants was measured by the Griess assay. Briefly, 50 μl of sample and standard reagent sodium nitrite (NaNO2) were added to individual wells of a 96-well plate, followed by 50 μl of Griess reagent (1% sulphanilamide in 2·5% H3PO4, 0·1% naphthylethylenediamine dihydrochloride in 2·5% H3PO4). After a 10-min incubation, the absorbance was read at 515 nm.
Quantification of cytokines and inducible nitric oxide synthase (iNOS) transcripts by competitive reverse transcription–polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated by the single-step guanidinium thiocyanate procedure using TRIZOL® Reagent (Gibco). The cDNA was synthesized for 90 min at 37° in 50 μl containing 16 U/μl Moloney murine leukaemia virus reverse transcriptase (Promega, Heidelberg, Germany) and 1·2 ng random hexamers (Biolabs, Beverley, MA).
Competitive PCR was performed as described previously.23,24 Briefly, constant amounts of cDNA (40 ng) were coamplified in the presence of serial dilutions of an internal competitor plasmid: pNIL for IL-10 and iNOS and pMUS for IL-1, tumour necrosis factor-α (TNF-α) and IFN-γ (a kind gift of Dr M. Kopf, Basel Institute for Immunology, Switzerland). Competitor plasmids were diluted fourfold in nine dilution steps (point 1 to point 9) from a stock concentration of 3·73 ng/ml (1×106 molecules/μl). Under these conditions of PCR, 5 μl cDNA and 5 μl competitor plasmid compete for the PCR primers, leading to the amplification of two fragments that differ 50–100 base pairs (bp) in size. Relative quantification of cDNA was done by calculating how much of the competitor was required to achieve equal amounts of two products. First, cDNAs were standardized to equal concentrations of the house-keeping gene β2-microglobulin.
PCR were performed in 50 μl containing: 0·25 mm of each dNTP, 0·25 μm 5′ and 3′ primers, 10 mm Tris–HCl (pH 8·3), 50 mm KCl, 1·5 mm MgCl2, 0·01% w/v gelatin, and 1·25 U of AmpliTaq DNA polymerase (Perkin Elmer Cetus, Rotkreuz, Switzerland) for 35 cycles (20 seconds at 94°, 20 seconds at 56° and 30 seconds at 72°). Thereafter, 20 μl of the reaction product was analysed on a 1·6% agarose gel in Tris borate–ethylenediaminetetraacetic acid (EDTA) buffer containing 0·2 μg/ml ethidium bromide.
Statistical methods
Comparisons were analysed by Student’s _t_-test using the Microsoft Excel (MacOffice98) program. Differences were considered significant when _P_-values were <0·05.
RESULTS
Suppressive splenic proliferative responses to mitogen and Emc-antigen stimulation during chronic AE
To analyse lymphocyte functions after experimental chronic infection of susceptible C57BL/6 mice with E. multilocularis, the proliferative responses to the T-cell mitogen Con A and to crude parasite Emc-antigen were investigated. Spleen cells were isolated from control mice and from E. multilocularis infected mice 1, 3 and 6 months p.i. and stimulated in vitro with Con A and Emc-antigen. As shown here in Fig. 1, the splenic proliferative responses to Con A stimulation in infected mice significantly decreased at a relatively early stage (1 month) of chronic infection to ≈40% of the values obtained from non-infected control mice. The degree of suppression was determined in four independent experiments and ranged between 40 and 70%. By 3 months p.i., spleen cells from infected mice regained the normal level of responsiveness to Con A, which, however, declined again at the late stage (months 4–6 p.i.).
Figure 1.
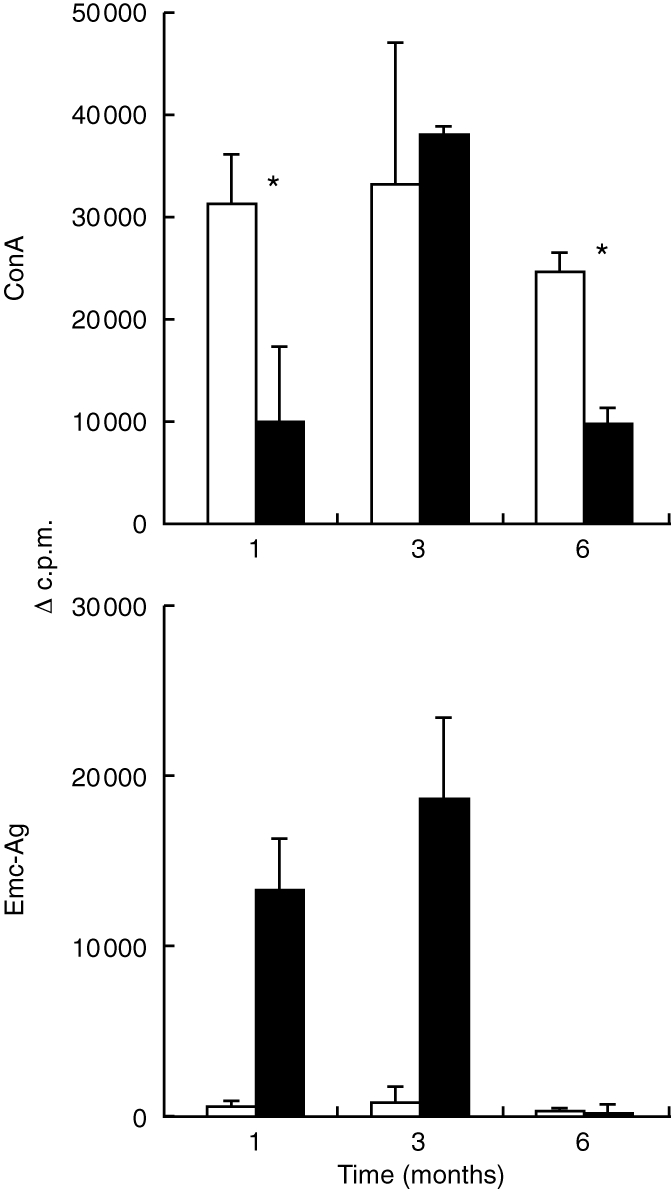
C57BL/6 mice (five mice per group) were infected intraperitoneally with 50–100 freshly isolated vesicular cysts of E. multilocularis. The proliferative responses to Con A and Emc-antigen stimulation of pooled spleen cells from infected (solid bars) and non-infected control (open bars) mice were determined at different time-points p.i. (1 month, 3 months and 6 months). *Indicates the statistically significant differences (P < 0·05). This is one representative out of two independent experiments exhibiting identical results. The suppression of Con A-derived proliferation at 1 month was repeated in four independent experiments with similar results.
A parasite-specific proliferative response of spleen cells to crude Emc-antigen stimulation was only found in mice infected with E. multilocularis, but not in control mice. In contrast to a marked Emc-antigen-specific response in the first phase of chronic infection (1–3 months p.i.), at the late stage (4–6 months p.i.) the spleen cells showed a striking loss of proliferative responsiveness to Emc-antigen (Fig. 1). This immunosuppression was coincident with a high parasite weight. Thus, at 6 months p.i., the larval metacestode had reached an average mass of 16·9±3·6 g per infected mouse (inoculum size at infection: 50 μg).
No suppression of splenic cytokine production after 1-month infection with E. multilocularis
To determine whether immunosuppression also affected splenic cytokine production, cytokine levels in the supernatants collected from spleen cell cultures were measured by ELISA. Table 1 shows the results obtained for supernatants collected after 48 hr. Spleen cells from 1-month infected AE mice exhibited a significantly increased production of IFN-γ (P < 0·01) and slightly, but not statistically significantly, increased IL-4 production (P = 0·18) after Con A stimulation when compared with spleen cells from non-infected control mice, whereas no IFN-γ production by the AE splenocytes was detectable after Emc-antigen stimulation at any of the supernatant collection time-points (24, 48 and 96 hr, see the Materials and Methods).
Table 1.
Cytokine levels in supernatants of spleen cell cultures from control and from 1-month infected _E. multilocularis_-infected mice
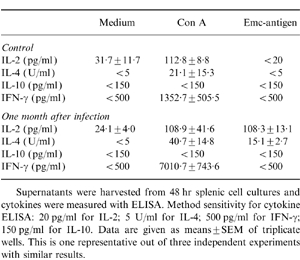
Spleen cells from both AE-infected and control mice exhibited a marked IL-2 production after Con A stimulation, while Emc-antigen-driven IL-2 production was only detectable in spleen cells from the infected mice. Thus, while the splenic proliferative response to Con A was strongly suppressed 1 month p.i. (Fig. 1), splenic IL-2 production in response to Con A stimulation was not altered. Throughout the experiment described above IL-10 production remained undetectable. To determine whether this cytokine was degraded during cell cultivation, supernatants were collected at various time-points (24, 48 and 96 hr) after stimulation and assayed. No IL-10 production on Con A- and Emc-antigen-stimulation was detected at any of the time-points (data not shown).
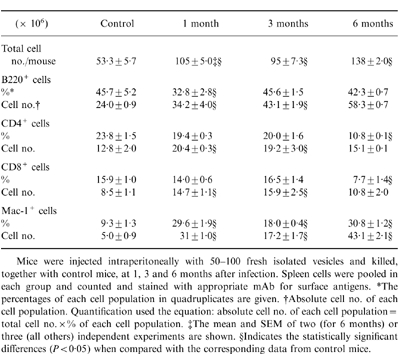
Increased peritoneal and splenic Mac-1+ cell population after infection
To identify the cells involved in the proliferative suppression during chronic AE, the cellular composition of peritoneal and splenic cells was assessed in infected mice by flow cytometry. One month after infection with E. multilocularis, the total PEC numbers were 5- to 12-fold increased when compared with control mice (31·4±3·9×106 cells for AE mice versus 3·5±0·7×106 cells for control mice assessed in four independent experiments). With regard to total PEC phenotypes, 43·7%±0·9% were Mac-1+Iab− (=resident macrophages) in control mice. In infected AE mice, the Mac-1+ cells (further identified by microscopic analysis as macrophages) were significantly increased (66·8%±3·5%), from which about half (33·3%±0·7% of total peritoneal cells) were Mac-1+Iab+ (activated macrophages). These data indicated that intraperitoneal infection with E. multilocularis induced a macrophage infiltration into the peritoneal cavity coupled to an activation of macrophages (increased major histocompatibility complex type II molecular expression).
Chronic AE resulted in splenomegaly observed at 1 and 3 months p.i. which markedly increased at 6 months p.i. The phenotypic compositions of spleen cells are shown in Table 2. Associated with splenomegaly, the total splenic cell number was increased about twofold p.i. Mac-1+ cell percentages were markedly higher in spleens of infected mice compared to the control group values. Infiltration of Mac-1+ cells into the spleen was associated with a decrease of the T-lymphocyte (especially at 6 month p.i.) and B-lymphocyte populations (especially at 1 month p.i.). With regard to the absolute cell numbers, the Mac-1+ cells exhibited a sixfold increase in 1-month infected mice and a 8·6-fold increase in 6-month infected mice, when compared with control mice. The B lymphocytes also exhibited a twofold increase after infection, while T lymphocytes (CD4+ and CD8+) were increased only during the early stage (1–3 months p.i.) but were in the same range as control mice at the late stage.
Table 2.
Phenotype composition of spleen cell suspensions
Suppression of Con A- and Emc-antigen-derived lymphocyte proliferation by peritoneal cells from AE mice
To further investigate the cellular mechanisms involved in the suppression of lymphocyte proliferation in chronic AE, the interaction between accessory cells and responder cells was analysed. Cocultivation of splenocytes (rich in lymphocytes) and PEC (rich in macrophages) was established. The spleen cells of 1-month-infected mice were supplemented with 5×104 PEC derived either from non-infected control mice or from infected AE mice. Cells were subsequently stimulated with Con A or Emc-antigen in vitro. As shown in Fig. 2(a), the PEC from infected mice (AE-PEC) suppressed completely (>90%) both Con A and Emc-derived lymphocyte proliferation (assessed in five independent experiments), while PEC from control mice exhibited no or only significantly lower suppressive effects (dependent on the supplemented PEC number, data not shown). Similar results concerning suppression of Con A stimulation were obtained when spleen cells from normal (non-infected) mice were investigated. Suppression of splenic proliferation by PEC from infected mice was also observed when assessed at later time-points of infection (3 months p.i. and 4 months p.i) (data not shown).
Figure 2.

Effect on Con A- and Emc-antigen-stimulated proliferation and NO production of supplementing spleen cells with PEC from control or 1-month infected mice. Spleen cells from 1-month infected mice were supplemented either with medium alone (open bars) or with 5×104 peritoneal cells from either normal (stippled bars) or 1-month infected mice (solid bars) and stimulated with Con A for 72 hr or Emc-antigen for 96 hr. (a) Incorporation of [3H]thymidine was determined in quadruplicate wells. Data represent the mean c.p.m.±SEM of quadruplicate wells. Similar results were obtained in five independent experiments. (b) Simultaneously, 50 μl supernantants from wells kept in parallel were collected at 96 hr for assessing NO production. One representative out of two independent experiments with similar results is shown.
Simultaneously, a marked increase of nitrite levels in the AE-splenocytes/AE -PEC cocultures was noted in response to Con A as early as 24 hr after stimulation (12 μm), when compared to AE-splenocytes alone (<5 μm) or AE-splenocyte/PEC from control mice cocultures (5·4 μm). Furthermore, an increasingly elevated nitrite production by AE-splenocytes/AE-PEC cocultures was found at 48 and 96 hr after Con A stimulation, respectively. The Emc-antigen-induced nitrite production was only detectable after 96 hr of stimulation in two experiments (Fig. 2b), but the differences of nitrite levels after Con A stimulation at 96 hr between AE-PEC-supplement and control mice PEC-supplement were not as high as at 24 hr.
PEC from AE-infected mice expressed high levels of IL-1β, TNF-α and iNOS but only constitutively low levels of IL-10
In order to assess peritoneal macrophage activation following infection with E. multilocularis and to identify putative peritoneal cell-derived factors responsible for immunosuppression, we performed quantitative RT-PCR with RNA derived from pooled peritoneal cells from 1-month infected mice and from corresponding control mice to determine cytokine- and iNOS-specific transcript levels. We found a marked increase in IL-1β, TNF-α and iNOS mRNA transcript levels in AE-PEC from infected mice (Fig. 3). PEC from control mice expressed very low levels of the inflammatory cytokines IL-1β and TNF-α. The expression of TNF-α and IL-1β by AE-PEC was increased >64-fold as compared to PEC from control mice. The PEC from control mice expressed no detectable iNOS, whereas iNOS mRNA-transcript was clearly expressed at significant levels in AE-PEC (Fig. 3). The expression of IL-10 was low and comparable between AE-PEC and control PEC. As the expression of IFN-γ was lower than 7·5 molecules (point 9 in Fig. 3), we can conclude that both AE-PEC and PEC from control mice were not expressing detectable amounts of IFN-γ.
Figure 3.

Determination of cytokine-and iNOS-mRNA expression in pooled PEC from control (PEC−) and infected (AE-PEC) mice. At 1 month p.i. levels of the cytokine-and iNOS-mRNA transcripts were determined by competitive quantitative RT-PCR after standardization for the expression of the constitutively transcribed β2-microglobulin gene. Appropriate amounts of cDNA were amplified in the presence of serial fourfold dilutions of a multispecific internal plasmid control ranging from point 1 (5×106 molecules) to point 9 (7·5×101 molecules). Numbers reflect to the dilution step. The arrows indicate equal amounts of PCR products of cDNA compared with mimic plasmid controls; c, cDNA amplification alone; −, negative control; m, DNA size marker. * Indicates the cDNA band sites. This is one representative out of two independent experiments with similar results.
Restoration of the proliferative response of spleen cells from 1-month infected mice with NMMA and anti-IFN-γ
The high level of iNOS expression by AE-PEC and the high level of nitric oxide in coculture supernatants after Con A stimulation suggested that these could play a functional role in the immunosuppression during chronic murine AE. Thus, we tested the ability of macrophage activation inhibitors to reverse the suppression of proliferative responses by the addition of either NMMA, an inhibitor of reactive nitrogen intermediate production, or of anti-IFN-γ.
Con A- and Emc-antigen-derived lymphocyte proliferation was completely suppressed after supplementation with AE-PEC (Fig. 2a and Fig. 4) which resulted in an elevated nitrite production measurable in the culture supernatants (see Fig. 2b). When NMMA was added to the same splenocyte cocultures, the nitrite production was suppressed in that nitrite was undetectable (detection limit of method was 2 μm) in the culture supernatants collected after 24 and 48 hr of Con A or Emc-antigen stimulation (data not shown). With regard to the AE-splenocyte proliferative response after Con A stimulation, NMMA was able to increase the proliferation rate by 40% (from 21 100±2900 c.p.m. to 33 000±2800 c.p.m.), thus normalizing the response level to that of Con A-stimulated spleen cells from non-infected control mice (32 000±9000 c.p.m., data not shown in Fig. 4). The background reactivities (cells without stimulation) were for spleen cells alone 2477±157 c.p.m., for spleen cells with addition of NMMA 2828±212 c.p.m. and with addition of anti-IFN-γ 2258±251 c.p.m. These data indicated that splenic macrophages which were already present in AE spleen cell suspensions, were able to synthesize sufficient nitric oxide to suppress to some extent the Con A-induced proliferation. The partial immunosuppression by splenic macrophages was reverted by NMMA treatment to a normal level of response. Furthermore, NMMA treatment also recovered, to a large extent, the AE-PEC-mediated suppression of Con A-and Emc-antigen-derived proliferative response (Fig. 4). Thus, anti-IFN-γ treatment partially restored the suppressed Con A-derived proliferation at the early stage of infection. Conversely, anti-IFN-γ treatment had no influence on the suppression of the Emc-antigen-derived splenocyte proliferation by AE-PEC (Fig. 4).
Figure 4.

Recovery by NMMA and anti-IFN-γ of the suppressed proliferative response of spleen cells from 1-month AE-infected mice. Spleen cells were isolated from four mice and subsequently pooled, then cultivated alone (solid bars) or cocultivated with AE-PEC (cross-hatched bars) and stimulated in vitro with Con A and Emc-antigen, or left unstimulated (background). The cell cultures were supplemented with 500 μm NMMA or 20 μg/ml anti-IFN-γ, or with an appropriate amount of medium (control). The [3H]thymidine uptake results were expressed as geometric mean minus background (▵c.p.m.). Background was determined by unstimulated cells kept in medium alone. A representative out of three independent experiments is shown.
Nitric oxide-mediated immunosuppression at the late stage of AE
We further addressed the phenomenon of immunosuppression observed at the late stage of AE by analysing IL-10 and iNOS expression in spleens from 4-month infected mice. In this experiment, two out of six mice died from AE infection at 121 days p.i. The parasite weights ranged from 11·4 g to 20·3 g. The spleen cells showed a marked proliferative suppression in response to Emc-antigen stimulation (data not shown). As shown in Fig. 5(a), spleen cells from 1-month and 4-month infected mice expressed only background levels of IL-10, comparable to those of spleen cells from control mice. Nevertheless, spleen cells from infected mice expressed increased iNOS already at 1 month p.i., and iNOS expression subs equently increased by >32-fold at 4 months p.i. Conversely, iNOS expression by spleen cells from control mice remained negative. The marked increase in iNOS expression by spleen cells from infected mice correlated with the occurrence of splenic immunosuppression observed at the late stage of AE infection. Furthermore, NMMA treatment was able to restore the suppressed splenic proliferative response to Con A and Emc-antigen stimulation at the late stage (6 months p.i.) (Fig. 5b). To rule out potential errors related to variations in the method of the proliferation assays, we included spleen cells from uninfected mice as a negative control and spleen cells from 3-month infected mice as a positive control for Emc-antigen-stimulation in one experiment. As shown in Fig. 5(b), at 6 months p.i. the splenic proliferative response to Con A was significantly decreased (P < 0·01) when compared with splenocytes from uninfected mice. Furthermore, the splenic proliferative response to exogenous Emc-antigen stimulation in vitro was completely abolished when compared with splenocytes from 3-month infected mice. A NMMA treatment partially restored the Con A-derived proliferation and completely restored the lost proliferative responsiveness to Emc-antigen up to normal levels (3 months p.i.). Anti-IFN-γ treatment also restored to a large extent the suppressed proliferative response to Emc-antigen at the late stage of AE. Our data demonstrated that nitric oxide is responsible for the unresponsiveness to Emc-antigen stimulation of splenocytes obtained from 6-month infected mice.
Figure 5.

(a) Expression of iNOS and IL-10 in spleen cells of four 4-month infected mice. In this experiment, two out of six mice died from AE infection at 121 days p.i. The iNOS and IL-10 mRNA transcripts in spleens from four control mice (Sp−) and four 1-month infected (Sp+ 1M) and 4-month infected mice (Sp+ 4M) were determined by competitive quantitative RT-PCR as described in Fig. 3. (b) Recovery by NMMA and anti-IFN-γ of the suppressed proliferative response of spleen cells from 6-month infected mice. Spleen cells were isolated from four uninfected mice (Sp−), four 3-month infected mice (Sp+3M) and three 6-month infected mice (Sp+6M). The spleen cells from 6-month infected mice were treated with NMMA (Sp+6M NMMA) and anti-IFN-γ (Sp+6M anti-IFN-γ) in the same manner as shown in Fig. 4 and stimulated with Con A or Emc-antigen. The [3H]thymidine-uptake results were expressed as geometric mean minus background (▵c.p.m.).
DISCUSSION
One of the most interesting features observed during experimental murine _E. multilocularis_-infection concerns a marked depression of cell-mediated immunity.6–8,21 We designed the present study to tackle the possible immunosuppression mechanisms characterizing chronic AE. In agreement with previous reports, we found a markedly suppressed splenocyte proliferation first, in response to Con A stimulation at a relatively early phase of infection (1 month p.i.)8,20 and second, in response to crude parasite antigen (Emc-antigen) as well as Con A stimulation at a late stage of AE (4–6 months p.i.)20,25 in susceptible C57BL/6 mice. Rakha et al.10 have shown that peritoneal macrophages from mice infected intraperitoneally with E. multilocularis protoscoleces could suppress Con A- and crude antigen-stimulated lymphocyte transformation. However, it still remained unsolved which macrophage-derived factor(s) were involved in the suppression phenomenon in AE. More generally, macrophages have been shown to be implicated in the suppression of lymphocyte functions following infection with a variety of intracellular pathogens including bacteria, viruses and protozoa2,12–15,18 In these infection models, the host immune response was predominantly associated with T helper type 1 (Th1)-mediated macrophage activation, in contrast to most infections with metazoan (helminthic) parasites, which are predominantly associated with Th2 responses. The present study now demonstrates that an extracellular metazoan parasite (E. multicularis) is also able to induce suppressive mechanisms similar to those of intracellular pathogens with respect to macrophage activation. We found that peritoneal macrophages from _E. multilocularis_-infected mice completely suppressed Con A- and Emc-antigen-induced splenic lymphocyte proliferation, and that the peritoneal and splenic macrophage-mediated immunosuppression was due to nitric oxide production in response to the E. multilocularis infection.
Upon experimental infection by inoculation of infectious E. multilocularis vesicles into the peritoneal cavity of susceptible mice, the subsequently proliferating metacestode is characterized by its principally extracellular localization. In our experiments, intraperitoneal injection of vesicle parasites induced a cellular infiltration into the peritoneum and activation of peritoneal macrophages (increased Iab expression). The Mac-1+ cell infiltration into the peritoneal cavity represented putatively the first line of parasite–host interaction. Spleen cells supplemented with 5×104 AE-PEC suppressed completely the Con A- and Emc-antigen-derived lymphocyte proliferation, while PEC from control mice showed no, or in some experiments lower, suppressive effect when supplemented with high PEC number. This perhaps reflected the degree of enviromental unspecific stimulation for activation of resident PEC.26 The strong suppressive effect of AE-PEC on proliferative response indicates that the activated peritoneal macrophages may be crucial for the induction of immunosuppression at the early stage of infection. It may be noted that, conversely to our experimental approach and subsequent findings, in mice infected by intrahepatic implantation of the parasite, no suppression of the Con A-induced proliferative response was observed during the early stage of infection.25 Furthermore, infection with E. multilocularis also caused an increase of Mac-1+ cells in spleen, which associated with the observation of suppressed splenic proliferation after infection.
AE-PEC expressed significantly increased levels of the inflammatory cytokines IL-1β, TNF-α and of iNOS and spleen cells also expressed increased iNOS which indicated the induction of a strong inflammatory reaction in vivo during chronic AE. Reactive nitrogen intermediates (RNI), predominantly produced by activated macrophages, play an important role in many biological and pathological events.26–28 They are crucial toxic molecules involved in the killing and elimination of intracellular pathogens by macrophages.28 On the other hand, RNI are also known to be implicated in macrophage-mediated suppression of lymphocyte proliferation in mice infected with intracellular bacteria or parasites.2,12–15,18,27 For example in murine infections with Leishmania major, spleen cells from infected iNOS-deficient mice exhibited a significantly higher T-cell proliferation than those from wild-type mice when cultured with leishmanial antigens or Con A in vitro. These data suggested that a high concentration of NO produced by iNOS was necessary for the prevention of the potential overexpansion of T cells, especially Th1 cells.29 In the present study, NO appeared as a relevant factor responsible for the suppression by macrophages in chronic AE. This was based on the observation that (1) AE-PEC and splenocytes from 4-month infected mice both expressed high levels of iNOS ex vivo; (2) that supplementation of spleen cells with AE-PEC significantly increased the NO2− production in vitro; and that (3), most importantly, the nitric oxide synthase inhibitor NMMA blocked (reverted) to a large extent the peritoneal and splenic macrophage-mediated suppression.
The mechanisms of RNI-mediated suppression of lymphocyte proliferation are still unknown. Kwon et al.30 have shown that RNI inhibited T-cell proliferation via the ability to block ribonucleotide reductase activity. The inhibition of ribonucleotide reductase activity would only affect T-lymphocyte proliferation, whereas induction of apoptosis would affect all lymphocyte activities, including T-lymphocytokine production (IL-2, IL-4 and IFN-γ). In some infection models, such as murine toxoplasmosis31 and trypanosomiasis,12 RNI mediated the proliferative suppression and the decrease of IL-2 production, while other investigators showed that T-cell proliferation was suppressed by RNI in contrast to IL-2 production.18,32 Infection with E. multilocularis for 1 month had no effect on the Con A-stimulated IL-2 production, whereas the production of IFN-γ and IL-4 was elevated. These results suggest that the RNI-mediated suppression was restricted to the lymphoproliferative response, but did not significantly affect the cytokine production by T lymphocytes.
On the other hand, cytokines exhibit also complex regulatory effects on the T-cell proliferation and RNI production by macrophages. Comparable IL-2 production by splenic cells of 1-month infected mice and of control mice allowed us to exclude the possibility that the suppressed splenic proliferation during early AE was due to a potentially reduced IL-2 production. In contrast to human AE,21 the important suppressive cytokine IL-10 seemed not to be involved in the macrophage-mediated immunosuppression during chronic murine AE, since IL-10 expression in AE-PEC and in spleen cells remained constantly low during the entire course of infection. IFN-γ is an important cytokine for stimulating NO production by macrophages.33 IFN-γ-deficient mice exhibited uncontrolled splenocyte proliferation in response to Con A which illustrated the importance of IFN-γ in the regulation of T-cell proliferative responses.33 In many infection models using intracellular pathogens, RNI production by IFN-γ-activated macrophages appeared as an important event in the control of proliferating pathogens, but also within immunosuppressive events.2,12,14,31 Anti-IFN-γ treatment resulted in a partial reversion of the suppressed Con A- and Emc-antigen-derived lymphocyte proliferation. This suggested that IFN-γ may play a significant role in the macrophage-mediated suppression. AE-PEC expressed no IFN-γ but high iNOS, and suppressed completely the lymphoproliferative responsiveness in vitro. Anti-IFN-γ treatment had no influence on the AE-PEC suppressed Emc-antigen-derived lymphocyte proliferation, while NMMA treatment restored the suppressed Emc-antigen-derived lymphocyte proliferation to a large extent. These data indicated that NO but not IFN-γ was the actual effector factor for the AE-PEC-mediated suppression. The suppressive effect of IFN-γ was probably due to an up-regulation of NO synthesized by macrophages.
The relative importance of macrophage-mediated immunosuppression for the pathogenesis of chronic AE deserves further studies. The suppressive role of RNI during murine toxoplasmosis contrasted remarkably with the capacity of RNI to inhibit replication of T. gondii within macrophages.15,31 In murine AE, earlier experiments have shown that treatment with bacillus Calmette–Guérin (BCG) prior to infection induced a non-specific activation of macrophages which seemed to protect mice against secondary infection with protoscolices.34 RNI was also shown to be instrumental in the in vitro killing of protoscolices of E. multilocularis by activated macrophages.35 In human infections with E. multilocularis, however, protoscolices are not synthesized.1 The effect of RNI in the killing of biologically more relevant metacestode tissue (including the germinal and laminated layer) in vivo has, however, still not been well documented. In contrast to this hypothetical benefit to the host, however, the release of RNI by macrophages clearly has a potentially detrimental effect on the lymphocyte function, as shown in our present study. Future experiments will address the effect of early RNI release within chronic murine AE more precisely by using iNOS-deficient mice.
In summary, the present study revealed that peritoneal as well as splenic macrophages derived from AE-infected mice could account for a suppression of the splenocyte proliferative responses during murine AE, but without altering IL-2 production. Nitric oxide, rather than IL-10, appeared as the effector element responsible for the macrophage-mediated immunosuppression, and IFN-γ also played a significant role in NO-mediated suppression. Further studies are planned to elucidate the biochemical mechanisms involved and the potential adverse consequences to the host of nitric oxide-mediated immunosuppression.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant no. 31-45575.95) and the Roche-Research Foundation. We thank Dr F. Brombacher (Max-Planck Institute for Immunbiology, Freiburg, Germany) and Dr M. Kopf (Basel Institute for Immunology, Basel, Switzerland) for supplying pMUS and pNIL plasmids for cytokine RT-PCR and anti-IFN-γ antibodies; we are grateful to Professor T. Jungi (Institute of Veterinary Virology, University of Berne, Switzerland), PD Dr A. Hemphill, PD Dr N. Müller, Drs R. Felleisen, B. Connolly and S. Eperon (Institute of Parasitology, University of Berne, Switzerland) for critically reviewing the manuscript. We are indebted to Dr T. Cavaliero (Institute of Parasitology, University of Zürich, Switzerland) for the maintenance of our parasite isolates in vivo.
Abbreviations
AE
alveolar echinococcosis
INOS
inducible nitric oxide synthase
NMMA
NG-monomethyl-l-arginine
NO
nitric oxide
PEC
peritoneal cells
RNI
reactive nitrogen intermediates
RT-PCR
reverse transcription–polymerase chain reaction
REFERENCES
- 1.Gottstein B, Hemphill A. Immunopathology of echinococcosis. Chem Immunol. 1997;66:177. doi: 10.1159/000058670. [DOI] [PubMed] [Google Scholar]
- 2.Gregory SH, Wing EJ, Hoffman RA, Simmons RL. Reactive nitrogen intermediates suppress the primary immunologic response to Listeria. J Immunol. 1993;150:2901. [PubMed] [Google Scholar]
- 3.Bresson-Hadni S, Liance M, Meyer JP, Houin R, Bresson JL, Vuitton DA. Cellular immunity in experimental Echinococcus multilocularis infection. II. Sequential and comparative phenotypic study of the periparasitic mononuclear cells in resistant and sensitive mice. Clin Exp Immunol. 1990;82:378. doi: 10.1111/j.1365-2249.1990.tb05457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liance M, Vuitton DA, Guerret-Stocker S, Carbillet JP, Grimaud JA, Houin R. Experimental alveolar echinococcosis. Suitability of a murine model of intrahepatic infection by Echinococcus multilocularis for immunological studies. Experientia. 1984;40:1436. doi: 10.1007/BF01951932. [DOI] [PubMed] [Google Scholar]
- 5.Playford MC, Ooi HK, Oku Y, Kamiya M. Secondary Echinococcus multilocularis infection in severe combined immunodeficient (scid) mice: biphasic growth of the larval cyst mass. Int J Parasitol. 1992;22:975. doi: 10.1016/0020-7519(92)90056-q. [DOI] [PubMed] [Google Scholar]
- 6.Baron RW, Tanner CE. The effect of immunosuppression on secondary Echinococcus multilocularis infections in mice. Int J Parasitol. 1976;6:37. doi: 10.1016/0020-7519(76)90008-4. [DOI] [PubMed] [Google Scholar]
- 7.Devouge M, ALI-Khan Z. Intraperitoneal murine alveolar hydatidosis: relationship between the size of the larval cyst mass, immigrant inflammatory cells, splenomegaly and thymus involution. Tropenmed Parasitol. 1983;34:15. [PubMed] [Google Scholar]
- 8.Kizaki T, Kobayashi S, Ogasawara K, Day NK, Good RA, Onoe K. Immune suppression induced by protoscoleces of Echinococcus multilocularis in mice. Evidence for the presence of CD8dull suppressor cells in spleens of mice intraperitoneally infected with E multilocularis. J Immunol. 1991;147:1659. [PubMed] [Google Scholar]
- 9.Treves S, ALI-Khan Z. Characterization of the inflammatory cells in progressing tumor-like alveolar hydatid cyst. 2. Cell surface receptors, endocytosed immune complexes and lysosomal enzyme content. Tropenmed Parasitol. 1984;35:231. [PubMed] [Google Scholar]
- 10.Rakha NK, Dixon JB, Carter SD, Craig PS, Jenkins P, Folkard S. Echinococcus multilocularis antigens modify accessory cell function of macrophages. Immunology. 1991;74:652. [PMC free article] [PubMed] [Google Scholar]
- 11.Unanue ER, Allen PM. The basis for the immunoregulatory role of macrophages and other accessory cells. Science. 1987;236:551. doi: 10.1126/science.2437650. [DOI] [PubMed] [Google Scholar]
- 12.Abrahamsohn IA, Coffman RL. Cytokine and nitric oxide regulation of the immunosuppression in Trypanosoma cruzi infection. J Immunol. 1995;155:3955. [PubMed] [Google Scholar]
- 13.Sternberg J, McGuigan F. Nitric oxide mediates suppression of T cell responses in murine Trypanosoma brucei infection. Eur J Immunol. 1992;22:2741. doi: 10.1002/eji.1830221041. [DOI] [PubMed] [Google Scholar]
- 14.Schwacha MG, Eisenstein TK. Interleukin-12 is critical for induction of nitric oxide-mediated immunosuppression following vaccination of mice with attenuated Salmonella typhimurium. Infect Immun. 1997;65:4897. doi: 10.1128/iai.65.12.4897-4903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1261. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behrouz CA, Jacobs P, Stevenson MM. Role of macrophages-derived nitric oxide in suppression of lymphocyte proliferation during blood-stage malaria. J Leukoc Biol. 1995;58:23. doi: 10.1002/jlb.58.1.23. [DOI] [PubMed] [Google Scholar]
- 17.Dziarski R. Splenic macrophages: mediators of immunosuppressive activity of staphylococcal peptidoglycan. J Reticuloendothel Soc. 1979;26:239. [PubMed] [Google Scholar]
- 18.Schwacha GM, Somers SD. Thermal injury-induced immunosuppression in mice: the role of macrophage-derived reactive nitrogen intermadiates. J Leuko Bio. 1998;63:51. doi: 10.1002/jlb.63.1.51. [DOI] [PubMed] [Google Scholar]
- 19.Gottstein B. Echinococcus multilocularis infection: immunology and immunodiagnosis. Advances in Parasitology. 1992;31:321. doi: 10.1016/s0065-308x(08)60024-x. [DOI] [PubMed] [Google Scholar]
- 20.Gottstein B, Wunderlin E, Tanner I. Echinococcus multilocularis: parasite-specific humoral and cellular immune response subsets in mouse strains susceptible (AKR, C57B1/6J) or ‘resistant’ (C57B1/10) to secondary alveolar echinococcosis. Clin Exp Immunol. 1994;96:245. doi: 10.1111/j.1365-2249.1994.tb06549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godot V, Harraga S, Deschseaaux M, et al. Increased basel production of interleukin-10 by peripheral blood monoclear cells in human alveolar echinococcosis. Eur Cytokine Netw. 1997;8:401. [PubMed] [Google Scholar]
- 22.Sarawar S, Sangste M, Coffman R, Doherty P. Administration of anti-IFN-γ antibody to β2-microglobulin-deficient mice delays influenza virus clearance but does not switch the response to help cell 2 phenotype. J Immunol. 1994;153:1246. [PubMed] [Google Scholar]
- 23.Dai WJ, Bartens W, Kohler G, Hufnagel M, Kopf M, Brombacher F. Impaired macrophage listericidal and cytokine activities are responsible for the rapid death of Listeria monocytogenes-infected IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5297. [PubMed] [Google Scholar]
- 24.Kopf M, Brombacher F, Kohler G, et al. IL-4-deficient Balb/c mice resist infection with Leishmania major. J Exp Med. 1996;184:1127. doi: 10.1084/jem.184.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emery IM, Liance M, Deriaud E, Vuitton D, Houin R, Leclerc C. Characterization of T-cell immune response to Echinococcus multilocularis-infected C57BL/6J mice. Parasite Immunol. 1996;18:463. doi: 10.1111/j.1365-3024.1996.tb01030.x. [DOI] [PubMed] [Google Scholar]
- 26.Unanue ER. Macrophages, antigen-presenting cells, and the phenomena of antigen handling and presentation. In: Paul WE, editor. Fundamental Immunology. 3. New York: Raven Press; 1994. p. 111. [Google Scholar]
- 27.Eisenstein TK, Huang D, Meissler JJJR, AL-Ramadi B. Macrophage nitric oxide mediates immunosuppression in infectious inflammation. Immunobiology. 1994;191:493. doi: 10.1016/S0171-2985(11)80455-9. [DOI] [PubMed] [Google Scholar]
- 28.Liew FY, Cox FE. Nonspecific defence mechanism: the role of nitric oxide. Immunol Today. 1991;12:A17. doi: 10.1016/S0167-5699(05)80006-4. [DOI] [PubMed] [Google Scholar]
- 29.Wei XQ, Charles IG, Smith A, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 30.Kwon NS, Stuehr DJ, Nathan CF. Inhibition of tumor cell ribonucleotide reductase by macrophage-derived nitric oxide. J Exp Med. 1991;174:761. doi: 10.1084/jem.174.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Candolfi E, Hunter CA, Remington JS. Roles of gamma interferon and other cytokines in suppression of the spleen cell proliferative response to concanavalin A and Toxoplasma antigen during acute toxoplasmosis. Infect Immun. 1995;63:751. doi: 10.1128/iai.63.3.751-756.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huot AE, Moore AL, Roberts JD, Hacker MP. Nitric oxide modulates lymphocyte proliferation but not secretion of IL-2. Immunol Invest. 1993;22:319. doi: 10.3109/08820139309063411. [DOI] [PubMed] [Google Scholar]
- 33.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 34.Rau ME, Tanner CE. BCG suppresses growth and metastasis of hydatid infections. Nature. 1975;256:318. doi: 10.1038/256318a0. [DOI] [PubMed] [Google Scholar]
- 35.Kanazawa T, Asahi H, Hata H, Mochida K, Kagei N, Stadecker MJ. Arginine-dependent generation of reactive nitrogen intermediates is instrumental in the in vitro killing of protoscoleces of Echinococcus multilocularis by activated macrophages. Parasite Immunol. 1993;15:619. doi: 10.1111/j.1365-3024.1993.tb00575.x. [DOI] [PubMed] [Google Scholar]