An essential role for p300/CBP in the cellular response to hypoxia (original) (raw)
Abstract
p300 and CBP are homologous transcription adapters targeted by the E1A oncoprotein. They participate in numerous biological processes, including cell cycle arrest, differentiation, and transcription activation. p300 and/or CBP (p300/CBP) also coactivate CREB. How they participate in these processes is not yet known. In a search for specific p300 binding proteins, we have cloned the intact cDNA for HIF-1α. This transcription factor mediates hypoxic induction of genes encoding certain glycolytic enzymes, erythropoietin (Epo), and vascular endothelial growth factor. Hypoxic conditions lead to the formation of a DNA binding complex containing both HIF-1α and p300/CBP. Hypoxia-induced transcription from the Epo promoter was specifically enhanced by ectopic p300 and inhibited by E1A binding to p300/CBP. Hypoxia-induced VEGF and Epo mRNA synthesis were similarly inhibited by E1A. Hence, p300/CBP–HIF complexes participate in the induction of hypoxia-responsive genes, including one (vascular endothelial growth factor) that plays a major role in tumor angiogenesis. Paradoxically, these data, to our knowledge for the first time, suggest that p300/CBP are active in both transformation suppression and tumor development.
Tumor expansion beyond a certain size requires neovascularization. This process is mediated, at least in part, by local hypoxia-induced production of angiogenic factors, such as vascular endothelial growth factor (VEGF) (1, 2). Hypoxia activates the heterodimeric transcription factor hypoxia-inducible factor (HIF)-1 (3). HIF-1 is composed of α and β subunits, both of which belong to the basic helix–loop–helix (bHLH)-per-arnt-sim (PAS) protein family (3). HIF-1 binds DNA at conserved promoter/enhancer-linked HIF sites and stimulates transcription of hypoxia-responsive genes such as VEGF, erythropoietin (Epo), and various glycolytic enzymes (3). How HIF-1 activates transcription, however, is not yet understood.
Adenovirus E1A-binding p300 and CREB-binding protein (CBP) are homologous transcriptional adaptor proteins active in multiple transcriptional events (4–7). They are thought to act, at least in part, by acting as signaling conduits between specific DNA-bound transcription factors and the basal transcriptional machinery. For example, when a cell is exposed to cAMP, the heretofore inactive transcription factor, CREB, bound to a cAMP-responsive element, recruits CBP (and perhaps p300) to act as a transcriptional adaptor, and thereby stimulates transcription of cAMP-responsive genes (6,8–10). Other factors that potentially use p300 and/or CBP (p300/CBP) as adaptors include jun, fos, c-myb, MyoD, RAR-α, SRC-1, and YY1 (11–16). In addition, p300 and CBP are both physically and functionally targeted by the adenovirus E1A and simian virus 40 large tumor (T) oncoproteins (6, 7, 17). Significantly, the ability of E1A to transform cells into a malignant phenotype requires the integrity of its p300/CBP-binding domain, implying that targeting these proteins is integral to E1A action (18). This suggests a prominent role for p300/CBP in the suppression of neoplastic transformation.
In an effort to begin to understand the p300 mechanism of action, a search for specific p300-associated proteins was initiated by an interactive protein expression cloning method (19). We used as probe a region of p300 that differs from those which serve as CREB and E1A binding sites. Using this approach, we have identified HIF-1α and subsequently found that p300/CBP and HIF-1α exist in a hypoxia-induced DNA-bound complex that appears to signal at multiple hypoxia-activated genes. These data suggest a major role for p300/CBP in the response to oxygen deprivation.
MATERIALS AND METHODS
Plasmid Constructions.
Plasmids encoding glutathione_S_-transferase (GST) fusion proteins were constructed by PCR amplification of the indicated region of p300, followed by subcloning into pGEX-2TK. C/H1 (aa 300–528) encompasses the first cysteine/histidine-rich region of p300 plus approximately 100 residues on either side. C/H1Δ (contains aa 300–345 and 411–528) is equivalent to C/H1 but lacks the actual cysteine/histidine-rich region (present in residues 346–410). Recombinant baculoviruses were constructed using the BaculoGold system (PharMingen), following manufacturer’s instructions; the ΔC/H1 deletion is the same as that in the GST fusion protein noted above. pCMVβ-HA-HIF-1α was created by inserting a fragment of HIF-1α cDNA into pCMVβ containing an in-frame 3′ hemagglutinin (HA) tag (4). The fragment used contains the complete ORF and the 5′ untranslated region.
Expression Cloning.
GST fusion protein was synthesized in bacteria, as described elsewhere (19), using pGEX-2TK-p300C/H-1 (described above). The protein was radiolabeled in vitro and used to screen a 293 λZAP cDNA library (courtesy of William G. Kaelin, Dana–Farber Cancer Institute), as described (19). Excision of inserts was performed by the manufacturer’s instructions (Stratagene).
Cell Culture.
U-2 OS human osteosarcoma cells and Hep3B human hepatocellular carcinoma cells were maintained in DMEM/10% fetal calf serum at 37°C in 10% CO2/90% air and 5% CO2/95% air, respectively. sf9 insect cells were maintained in suspension at 28°C in Grace’s insect medium supplemented with lactalbumin, yeastolate (GIBCO/BRL), and 10% fetal calf serum.
Electrophoretic Mobility-Shift Assay (EMSA).
Nuclear extracts were prepared and EMSA assays performed as described (20), except that the final dialysis step in the nuclear extract preparation was omitted and gel conditions were as in ref. 21. The wild-type (WT) and mutant (mut) probes were synthesized to match W18 and M18, respectively, in ref. 20. The XRE (xenobiotic response element) and CME (central midline element) probes were synthesized and correspond to sequences GGAGTTGCGTGAGAAGAGCCTGGAGG and AAATTTGTACGTGCCACAGA, respectively.
Monoclonal Antibodies.
Monoclonal antibody, OZ15, was raised against a GST fusion protein containing HIF-1α amino acids 530–826 (Z.A. and D.M.L., unpublished results), and monoclonal antibody AC 240 was raised against a GST fusion protein containing CBP amino acids 720-1676 (R.E. and D.M.L., unpublished results).
Transient Transfections.
Transfections were performed using the calcium phosphate method. Total cytomegalovirus (CMV)-bearing plasmid per transfection was kept constant at 3 μg by using pRC/CMV backbone vector (Invitrogen) as carrier. Luciferase assays were promptly performed using extract quantities normalized to the observed β-galactosidase activity (22). For the experiment in Fig. 3, cells were split into aliquots and incubated for 44 hr prior to lysis and analysis of luciferase activity. Twenty-four hours prior to lysis, one aliquot was exposed to 1% O2, while the other remained at 21% O2. For the experiment in Fig. 4, cells were transferred, where indicated, into 1% O2/100 μM CoCl2 36 hr after transfection for 12–18 hr.
Figure 3.
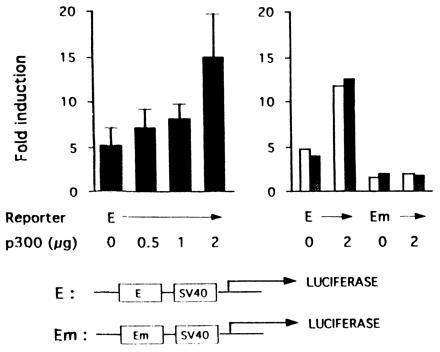
Cotransfected p300 enhances hypoxic induction of transcription from the Epo enhancer (30). Transient transfection of Hep3B cells was performed with 1 μg of a luciferase reporter gene (E) containing the Epo enhancer upstream of the simian virus 40 promoter or one in which the HIF-1 binding site has been mutated (Em). Cells were cotransfected with 0–2 μg (Left) and 0 or 2 μg (Right) of pCMV-p300 (4) with 1 μg of pCMV-lacZ (to correct for variations in transfection efficiency). The_y_ axis shows the ratio of luciferase expression at 1% O2 to that at 21% O2. Results on the_Left_ are the mean of triplicate experiments ± 1 SD and on the Right are duplicate experimental points.
Figure 4.
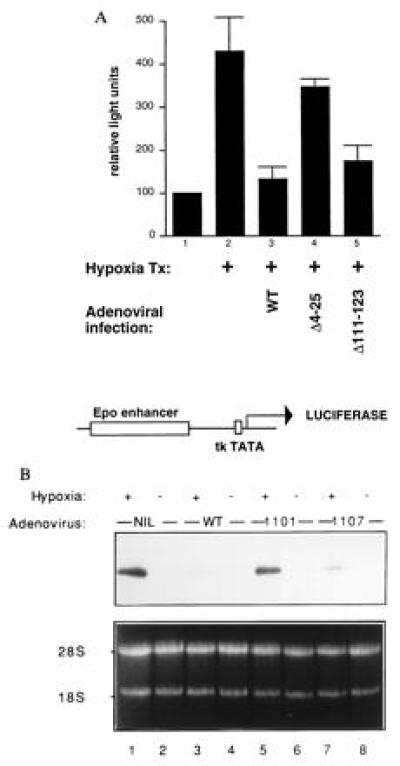
E1A inhibits hypoxia-driven transcription (31). (A) Hep3B cells were cotransfected with 1 μg of pCMV-lacZ and 0.5 μg of Epo49-Luc (32) (diagramed below the bar graph) and brought to 16 μg of total with Bluescript DNA (Stratagene). Twenty-four hours later, cells were infected with adenovirus type 5 bearing the indicated E1A deletion mutations. The 1101 mutant deletes E1A residues 4–25, while the 1107 mutant deletes residues 111–123. E1AΔ111-123 interacts with p300/CBP but not with pRB (33), while E1AΔ4-25 does not recognize p300/CBP but does bind to pocket proteins (33). The adenoviruses also bore a mutation (dl520) rendering them unable to synthesize 13S E1A (34). (B) Hep3B cells were infected with either wild-type (wt) or with mutant adenoviruses as in_A_. After infection, cells were placed in either 1% or 21% O2 for 6 hr; total cellular RNA was extracted, and Northern blots analyses using a VEGF probe were performed.
Northern Blot Analysis.
Northern blot analysis with a VEGF probe was performed as described (23).
RESULTS
The C/H1 Region of p300 Interacts with HIF-1α.
An expression library was probed with a labeled GST fusion protein encompassing the first cysteine/histidine-rich region of p300 (GST–C/H1). The longest clone (which we called CHIP-1) isolated among a set of overlapping cDNAs contained an 826-amino acid ORF predicting a protein bearing basic helix–loop–helix (bHLH) (24) and PAS domains (25). CHIP-1 cDNA was transcribed and translated in vitro, yielding a ≈120-kDa product (Fig.1A, lane 1). This translation product was capable of binding specifically, in vitro, to GST–C/H1 produced in bacteria (Fig. 1A, lane 3) and full-length p300 produced in insect cells (Fig. 1B, lane 3). Binding depended on the integrity, in p300, of the first cysteine/histidine-rich region (Fig. 1 A and B, lanes 4). Similarly, CHIP-1 expressed in U-2 OS human osteosarcoma cells also bound specifically to full-length p300 produced in insect cells (Fig. 1C). Subsequently, the cloning of the two hypoxia-inducible factor-1 subunits (HIF-1α and HIF-1β) (3) led to the realization that CHIP-1 and HIF-1α are identical.
Figure 1.
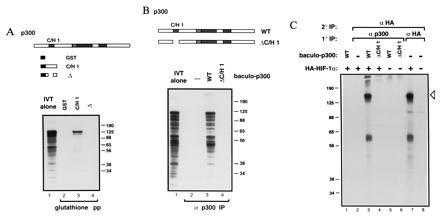
p300 binds HIF-1α (28). (A) GST fusion proteins containing the indicated portions of p300 C/H1 were constructed. 35S-Labeled HIF-1α was transcribed and translated in vitro and mixed with the indicated GST fusion proteins immobilized on glutathione beads. Bead-bound proteins were visualized by SDS/PAGE and autoradiography. (B) Full-length p300 (WT) and p300 deleted within the first cysteine/histidine-rich region (C/H1) were synthesized in insect cells using the baculovirus system. 35S-labeled HIF-1α was synthesized in vitro, and translation products were mixed with the indicated baculo-p300 species and immunoprecipitated with a p300 antibody [RW128 (4)] using protein-A Sepharose beads. Radiolabeled bead-bound proteins were visualized by SDS/PAGE and autoradiography. Lanes 1 in A and B each contain 20% of the input translation products analyzed in the other lanes. (C) U-2 OS cells were transfected with 10 μg of pCMVβ-HA-HIF-1α (+) or vector alone (−). Cells were labeled with [35S]methionine, and cellular extracts were prepared, mixed (where indicated) with the relevant baculovirus-p300 species, and then immunoprecipitated with either anti-p300 or anti-HA antibody, as in B. Bead-bound proteins were released by boiling, and reimmunoprecipitated with antibody to the hemagglutinin (HA) epitope (12CA5). Proteins bound to beads in this second round were visualized by SDS/PAGE and autoradiography. The open arrowhead indicates HA–HIF-1α. The identity of the faster migrating band is not clear but may represent a degradation product. Standard molecular weights (kDa; Sigma) are indicated. IVT, in vitro translate; pp, precipitation; IP, immunoprecipitation.
HIF-1α Interacts with p300/CBP During the Response to Hypoxia.
Using an EMSA, we asked whether p300/CBP exists in a stable complex with hypoxia-activated HIF-1α. Nuclear extracts from both hypoxic and normoxic HeLa cells were prepared, mixed with labeled HIF probe, and electrophoresed in nondenaturing polyacrylamide gels. As shown in Fig. 2A, at least three specific complexes were present in normoxic extracts (lane 6, bands C, i.e., constitutive), and at least two more complexes appeared after oxygen deprivation (lane 7, bands I, i.e., inducible), as reported (28). These complexes were competed by excess unlabeled probe (lane 5) as well as probes containing cross-reacting XRE (xenobiotic response element) (29) and CME (central midline element) (26) sequences (lanes 2 and 3). The XRE sequence is bound by heterodimers of bHLH-PAS proteins (AHR and ARNT/HIF-1β) (27, 29), and the CME sequence is bound by the PAS protein product of the single minded gene (26). The complexes were not competed, however, by excess unlabeled probe containing a mutated HIF site (lane 4) or nonspecific sequences (lane 1). The fastest migrating band appears to be nonspecific, as it is competed by unlabeled excess unrelated oligonucleotide (lane 1).
Figure 2.
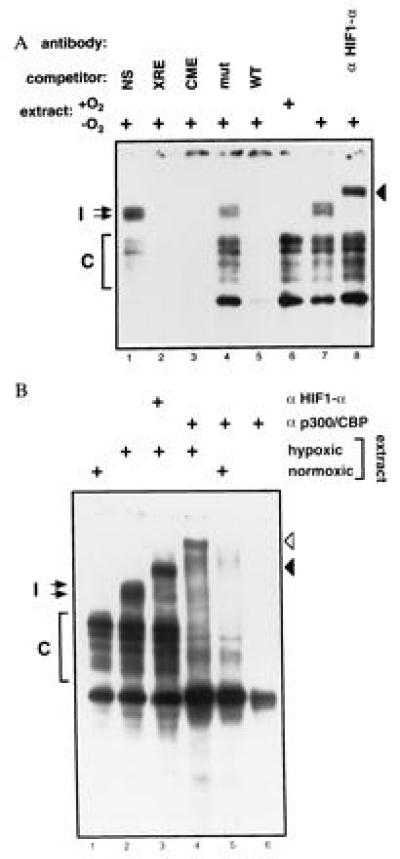
One or more members of the p300/CBP family interact(s) with HIF-1α in a DNA-bound complex (27). (A) Nuclear extracts from Hep-3B cells were analyzed by EMSA using a HIF-site-containing probe. Extracts in lanes 1–5 and 7–8 were prepared from cells treated for 5 hr with 1% O2, and the extract in lane 6 was prepared from normoxic cells. An 100-fold excess of unlabeled wild-type Epo 3′ enhancer probe (WT, lane 5), mutant probe (mut, lane 4), probe containing a central midline element (CME, lane 3), probe containing a xenobiotic response element (XRE, lane 2), or nonspecific probe (NS: myc E2F site, lane 1) were used as competitors. HIF-1α antibody (OZ15) was tested as a potential supershifting reagent in lane 8. In both experiments, free probe was in excess and is not shown. (B) Nuclear extracts from Hep-3B cells were analyzed as in A. Extracts in lanes 2–4 were prepared from cells treated for 5 hr with 1% O2. Extracts in lanes 1 and 5 were prepared from normoxic cells, and lane 6 contains no cellular protein. HIF-1α antibody (OZ15) and p300/CBP monoclonal antibody (AC240) were analyzed as potential supershifting reagents in lanes 3 and 4–6, respectively. Induced (I) and constitutive (C) complexes are indicated.
To investigate for the presence of HIF-1α and p300/CBP in these complexes, we tested antibodies to these proteins as potential supershifting reagents. As shown in Fig. 2, the hypoxia-induced complexes were supershifted by either the presence of a monoclonal antibody to HIF-1α (solid arrowheads in Fig. 2 A, lane 8, and B, lane 3) or a monoclonal antibody that reacts with both p300 and CBP (open arrowhead in Fig. 2B, lane 4). Hence, after hypoxia, complexes exist in vivo containing both HIF-1α and at least one member of the p300/CBP family.
We noted that the mobility of a fraction of the faster-migrating constitutive complexes also decreased in the presence of antibody to p300/CBP (Fig. 2B, lanes 4 and 5). These complexes were not supershifted by antibody to HIF-1α and, therefore, may not contain it. A similar effect was noted with another p300/CBP monoclonal antibody (data not shown). Hence, in both normoxic or hypoxic cells, p300 and/or CBP exist in complexes capable of binding to HIF sites. The detailed nature of the constitutive complexes is unclear, but one might speculate that they contain ATF-1, CREB-1, or AP-1, all of which can bind constitutively to HIF-1 sites (30, 31) and interact with CBP (and possibly p300) (6, 7).
p300 Increases the Inducibility of the Erythropoietin (Epo) Enhancer in Response to Hypoxia.
Cotransfection of a reporter gene containing the 3′ Epo enhancer with pCMV-p300 resulted in a dose-dependent increase in the ratio of luciferase activity in hypoxic versus normoxic cells (Fig. 3 Left). Specifically, it rose from 5.3 ± 1.8 to 14.9 ± 4.8. This 3-fold increase in hypoxia induction was dependent on HIF-1, since cotransfection of p300 had no effect on luciferase production from a reporter gene in which the HIF-1 binding site was mutated (Fig. 3 Right). Hence, p300 increases the hypoxia inducibility of the Epo enhancer in a HIF-1-dependent fashion. The effect is most likely limited to 3-fold due to the large quantities of p300 already present in the cell, prior to transfection of pCMV-p300.
E1A Inhibits the Hypoxic Response of Both the Epo Enhancer and the VEGF Gene by Specifically Targeting p300/CBP.
E1A can suppress the transcriptional activity of systems that use p300/CBP (e.g., the cAMP-mediated transcription response), as well as directly inhibit p300 transactivation function (4, 6, 7). If p300/CBP also participates in the HIF-1-mediated hypoxia response, then E1A should inhibit the hypoxia effect as well. To test this hypothesis, we transfected reporter plasmids bearing the luciferase gene under the control of the Epo enhancer linked to a thymidine kinase TATA box. As mentioned above, this enhancer contains a hypoxia-responsive HIF site. As shown in Fig.4A, the activity of this reporter was stimulated 4-fold by hypoxia (bars 1 versus 2). This stimulation was completely abrogated by E1A, introduced either by coinfection (bar 3), or by cotransfection (data not shown). This inhibition by E1A was dependent on its ability to interact with p300 and CBP: adenovirus encoding a mutant E1A species selectively unable to bind p300 and CBP did not inhibit the hypoxic response (bar 4), while one encoding E1A unable to bind pRB-related proteins, but able to bind p300/CBP, did inhibit (bar 5). E1A had no effect on the reporter in normoxic cells (data not shown). We conclude that E1A abrogates the activation of the Epo enhancer in response to hypoxia and that it does so by targeting p300/CBP. In keeping with these findings, we also found that E1A was a powerful suppressor of hypoxia-stimulated endogenous Epo mRNA synthesis (L.E.H. and H.F.B., unpublished data).
Finally, we asked whether E1A also inhibits the induction of VEGF mRNA in response to hypoxia. As shown in Fig. 4B, infection of Hep3B cells with wild-type adenovirus markedly inhibited the rise in VEGF mRNA after hypoxia (lane 3). By contrast, a mutant virus encoding an E1A species unable to bind p300 and CBP failed to inhibit the hypoxic response (lane 5), demonstrating that the response is, at least in part, mediated by p300/CBP binding. On the other hand, an E1A species capable of interacting with p300/CBP, but defective in binding to pRB-related proteins, inhibited VEGF mRNA synthesis almost as efficiently as wild-type E1A (lane 7). Hence, as with repression of the Epo enhancer, binding of p300/CBP by E1A correlates well with its ability to repress hypoxia-induced VEGF mRNA levels.
DISCUSSION
The data presented herein suggest a critical role for p300/CBP in transcriptional regulation of hypoxia-responsive genes. p300/CBP is present in vivo in hypoxia-induced complexes with DNA-bound HIF-1 (Fig. 2), and it can increase the response of the Epo enhancer to hypoxia, in a HIF-1-dependent fashion (Fig. 3). Furthermore, functional p300/CBP is necessary for hypoxic induction of VEGF and Epo (Fig. 4). We conclude that p300/CBP plays a prominent role in the transcriptional response of VEGF and Epo to hypoxia and that, most likely, p300/CBP is also involved in the transcriptional induction of other hypoxia-responsive genes. The simplest model consistent with these data proposes that p300/CBP act as adaptor proteins, recruited to the HIF site by binding to HIF-1α, after which they stimulate the transcriptional machinery. In this scenario, E1A inhibits hypoxia-responsive transcription by binding to and inactivating p300/CBP. E1A most likely carries this out by directly inhibiting p300/CBP transactivation potential, rather than disturbing the p300/CBP-HIF-1α complex, since E1A can also inactivate p300 fusion proteins that probably bind to DNA constitutively (6).
As discussed earlier, p300/CBP participate in a growing variety of transcriptional regulatory systems (serum response, myogenesis, hormonal responses, etc.). Given such a wide spectrum of interactive transcription regulation function, one might speculate that p300/CBP act as scaffolds, binding simultaneously to various DNA binding (and other) transcriptional factors, and thereby integrating information from various sources. For example, p300/CBP may mediate, by binding both CREB and HIF-1α, the observed synergism during hypoxia between a cAMP-responsive element and a HIF site in the LDHA gene (35). In such a manner, p300/CBP may contribute to the normal integration of multiple incoming signals destined to modulate the behavior of a given promoter.
Of what advantage to the virus is the E1A blockade of hypoxia-responsive gene activation? One possibility is that the response to hypoxia is deleterious to viral replication (36, 37). Neutralization of hypoxia-induced genes might, thus, favor viral production. Alternatively, the effect of E1A may simply be an epiphenomenon of its concomitant effect on a different regulatory system which also involves p300/CBP. For example, p300/CBP are also adaptors in the interferon α (IFN-α) pathway (38), which has potent antiviral proliferative effects. Inhibition by E1A of the IFN-α pathway is well documented (39–41). Thus, inhibition by E1A of hypoxia-responsive genes may simply be secondary to its more purposeful inhibition of IFN-α-responsive genes.
The data presented herein imply strongly that p300/CBP contribute to the hypoxia-induced activation of the VEGF promoter and, hence, to VEGF synthesis. Hypoxia-induced elaboration of VEGF is believed to play a pivotal role in hypoxia-induced angiogenesis and, hence, tumor expansion (1, 2). Thus, p300/CBP, a known transformation-suppressing element, may have the paradoxical feature of supporting tumor progression. In such a setting, the p300/CBP–HIF complex, a hypoxia-specific structure, might become a possible target for rational therapy of tumors and other disorders characterized by aberrant hypoxia-induced neovascularization.
Acknowledgments
We are indebted to Dr. James DeCaprio for his invaluable help in generating the HIF-1α monoclonal antibodies. We also appreciate Dr. Stan Bayley’s generous gift of adenoviruses encoding mutant E1A proteins. This work was supported by National Institutes of Health grants to D.M.L., H.F.B., and M.A.G. and by a grant from the Dana–Farber Cancer Institute/Sandoz Drug Development Program (to D.M.L.). L.E.H., R.E., and S.B. were supported by a National Research Service Award, the Swiss National Science Foundation, and the British Heart Foundation, respectively.
Footnotes
Abbreviations: Epo, erythropoietin; VEGF, vascular endothelial growth factor; GST, glutathione _S_-transferase; EMSA, electrophoretic mobility-shift assay.
References
- 1.Shweiki D, Itin A, Soffer D, Keshet E. Nature (London) 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 2.Plate K H G, Breier G, Weich H A, Risau W. Nature (London) 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 3.Wang G L, Jiang B-H, Rue E A, Semenza G L. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckner R, Ewen M E, Newsome D, Gerdes M, DeCaprio J A, Bentley Lawrence J, Livingston D M. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 5.Arany Z, Sellers W R, Livingston D M, Eckner R. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 6.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. Nature (London) 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 7.Lundblad J R, Kwok R P S, Laurance M E, Harter M L, Goodman R H. Nature (London) 1995;374:85–87. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 8.Chrivia J C, Kwok R P S, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Nature (London) 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 9.Kwok R P S, Lundblad J R, Chrivia J C, Richards J P, Bächinger H P, Brennan R G, Roberts S G E, Green M R, Goodman R H. Nature (London) 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 10.Arias J, Alberts A S, Brindle P, Claret F X, Smeal T, Karin M, Feramisco J, Montminy M. Nature (London) 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 11.Bannister A J, Kouzarides T. EMBO J. 1995;14:4758–4762. doi: 10.1002/j.1460-2075.1995.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bannister A J, Oehler T, Wilhelm D, Angel P, Kouzarides T. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- 13.Yuan W, Condorelli G, Caruso M, Felsani A, Giordano A. J Biol Chem. 1996;271:9009–9013. doi: 10.1074/jbc.271.15.9009. [DOI] [PubMed] [Google Scholar]
- 14.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S C, Heyman R A, Rose D W, Glass C K, Rosenfeld M G. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 15.Dai P, Akimaru H, Tanaka Y, Hou D X, Yasukawa T, Kanei I C, Takahashi T, Ishii S. Genes Dev. 1996;10:528–540. doi: 10.1101/gad.10.5.528. [DOI] [PubMed] [Google Scholar]
- 16.Lee J-S, Galvin K M, See R H, Eckner R, Livingston D, Moran E, Shi Y. Genes Dev. 1995;9:1188–1198. doi: 10.1101/gad.9.10.1188. [DOI] [PubMed] [Google Scholar]
- 17.Eckner R, Ludlow J W, Lill N L, Oldread E, Arany Z, Modjtahedi N, DeCaprio J A, Livingston D M, Morgan J A. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran E. Curr Opin Genet Dev. 1993;3:63–70. doi: 10.1016/s0959-437x(05)80342-9. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin W G J, Krek W, Sellers W R, DeCaprio J A, Ajchenbaum F, Fuchs C S, Chittenden T, Li Y, Farnham P J, Blanar M A, Livingston D M, Flemington E K. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- 20.Wang G L, Semenza G L. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 21.Barberis A, Superti-Furga G, Busslinger M. Cell. 1987;50:347–359. doi: 10.1016/0092-8674(87)90489-2. [DOI] [PubMed] [Google Scholar]
- 22.Ausubel F M, Brent R, Kingston R E, Moore D D, Smith J A, Seidman J G, Struhl K. Current Protocols in Molecular Biology. Vol. 2. New York: Greene & Wiley; 1989. [Google Scholar]
- 23.Goldberg M A, Schneider T J. J Biol Chem. 1994;269:4355–4359. [PubMed] [Google Scholar]
- 24.Murre C, Bain G, van Dijk M A, Engel I, Furnari B A, Massari M E, Matthews J R, Quong M W, Rivera R R, Stuiver M H. Biochim Biophys Acta. 1994;1218:129–135. doi: 10.1016/0167-4781(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 25.Huang Z J, Edery I, Rosbash M. Nature (London) 1993;364:259–262. doi: 10.1038/364259a0. [DOI] [PubMed] [Google Scholar]
- 26.Wharton K A J, Franks R G, Kasai Y, Crews S T. Development (Cambridge, UK) 1994;120:3563–3569. doi: 10.1242/dev.120.12.3563. [DOI] [PubMed] [Google Scholar]
- 27.Hankinson O. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 28.Semenza G L, Wang G L. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsushita N, Sogawa K, Ema M, Yoshida A, Fujii-Kuriyama Y. J Biol Chem. 1993;268:21002–21006. [PubMed] [Google Scholar]
- 30.Kvietikova I, Wenger R H, Marti H H, Gassmann M. Nucleic Acids Res. 1995;23:4542–4550. doi: 10.1093/nar/23.22.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy A P, Levy N S, Wegner S, Goldberg M A. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- 32.Blanchard K L, Acquaviva A M, Galson D L, Bunn H F. Mol Cell Biol. 1992;12:5373–5385. doi: 10.1128/mcb.12.12.5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barbeau D, Charbonneau R, Whalen S G, Bayley S T, Branton P E. Oncogene. 1994;9:359–373. [PubMed] [Google Scholar]
- 34.Jelsma T N, Howe J A, Mymryk J S, Evelegh C M, Cunniff N F A, Bayler S T. Virology. 1989;171:120–130. doi: 10.1016/0042-6822(89)90518-7. [DOI] [PubMed] [Google Scholar]
- 35.Firth J D, Ebert B L, Ratcliffe P J. J Biol Chem. 1995;270:21021–21027. doi: 10.1074/jbc.270.36.21021. [DOI] [PubMed] [Google Scholar]
- 36.Naldini A, Carraro F, Fleischmann W R J, Bocci V. J Interferon Res. 1993;13:127–132. doi: 10.1089/jir.1993.13.127. [DOI] [PubMed] [Google Scholar]
- 37.Baron S, Porterfield J S, Isaacs A. Virology. 1961;14:444–449. doi: 10.1016/0042-6822(61)90336-1. [DOI] [PubMed] [Google Scholar]
- 38.Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, Livingston D M. Nature (London) 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 39.Ackrill A M, Foster G R, Laxton C D, Flavell D M, Stark G R, Kerr I M. Nucleic Acids Res. 1991;19:4387–4393. doi: 10.1093/nar/19.16.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gutch M J, Reich N C. Proc Natl Acad Sci USA. 1991;88:7913–7917. doi: 10.1073/pnas.88.18.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalvakolanu D V, Bandyopadhyay S K, Harter M L, Sen G C. Proc Natl Acad Sci USA. 1991;88:7459–7463. doi: 10.1073/pnas.88.17.7459. [DOI] [PMC free article] [PubMed] [Google Scholar]