Renal Aplasia in Humans Is Associated with RET Mutations (original) (raw)
Abstract
In animal models, kidney formation is known to be controlled by the proteins RET, GDNF, and GFRA1; however, no human studies to date have shown an association between abnormal kidney development and mutation of these genes. We hypothesized that stillborn fetuses with congenital renal agenesis or severe dysplasia would possess mutations in RET, GDNF, or GFRA1. We assayed for mutations in these genes in 33 stillborn fetuses that had bilateral or unilateral renal agenesis (29 subjects) or severe congenital renal dysplasia (4 subjects). Mutations in RET were found in 7 of 19 fetuses with bilateral renal agenesis (37%) and 2 of 10 fetuses (20%) with unilateral agenesis. In two fetuses, there were two different RET mutations found, and a total of ten different sequence variations were identified. We also investigated whether these mutations affected RET activation; in each case, RET phosphorylation was either absent or constitutively activated. A GNDF mutation was identified in only one fetus with unilateral agenesis; this subject also had two RET mutations. No GFRA1 mutations were seen in any fetuses. These data suggest that in humans, mutations in RET and GDNF may contribute significantly to abnormal kidney development.
Introduction
Congenital renal agenesis or dysgenesis is rare in humans, occurring at a frequency of 1/3000–1/5000 births.1 Several lines of evidence suggest a genetic contribution to the development of these anomalies. For example, the incidence of renal agenesis is increased in fetuses who have first degree relatives with congenital renal anomalies.2 Moreover, renal anomalies are associated with some autosomal recessive and dominantly inherited disorders including Meckel-Gruber (MIM #249000), Townes-Brocks (MIM #107480), and Ivemark (MIM 208530) syndromes. Finally, animal genetic models have implicated many genes important for renal development.3
More than 40 genes have been shown to be involved in murine kidney development.4 Major developmentally important proteins include Ret (MIM #164761), Gdnf (MIM ∗600837), Gfra1 (MIM ∗601496), Pax2 (MIM ∗167409), and Wt1 (MIM ∗607102). In mice, renal agenesis is commonly found in animals lacking the genes for the tyrosine kinase receptor Ret, its coreceptor Gfra1, or the Gfra1 ligand Gdnf.5–7 However, in humans, the genes responsible for kidney development have remained elusive.3 Anecdotal reports in humans noting the finding of renal dysplasia in multiple endocrine neoplasia (MEN) 2A (MIM #171400) kindreds with RET mutations have suggested that genes from the RET axis may be important in human kidney development.8,9 However, the RET, GDNF, and GFRA1 genes have not been carefully studied in humans with severe renal anomalies, perhaps owing to the frequent stillbirth of such infants. We hypothesize that mutations in RET, GDNF, and GFRA1 might occur with high frequency in stillborn fetuses having renal agenesis or dysplasia.
Material and Methods
Identification of RET and GDNF Mutations
Paraffin-embedded tissue samples were collected from 33 autopsied stillborn fetuses with bilateral renal agenesis (n = 19), unilateral renal agenesis (n = 10), and severe renal dysgenesis including two patients with Meckel-Gruber syndrome (n = 4). The study was approved by the Institutional Review Board, and in accordance with our IRB, consent was waived because the paraffin-embedded samples were archival and were studied in a completely anonymous fashion. Based on their autopsy findings including biopsy of the colon, none of the patients had Hirschsprung disease (MIM #142623). Moreover, there was no evidence of multiple endocrine neoplasia (MEN) 2A or 2B (MIM #162300) or familial medullary thyroid carcinoma (FMTC) (MIM #155240), but these conditions generally present with clinical findings later in childhood; they may have been present in our subjects and were not detected at standard autopsy in these infants.
We extracted DNA from the paraffin-embedded liver and spleen tissue through xylene deparaffinization of multiple 10-micron tissue sections followed by protein digestion and ethanol precipitation of DNA. This DNA was then used as the template to amplify by PCR each exon in the RET, GDNF, and GFRA1 genes, and these PCR products were screened for mutations by conformation sensitive gel electrophoresis (CSGE).10,11 To generate exonic DNA segments for CSGE, oligonucleotides were designed to flank exonic regions 200–400 basepairs in length by means of published sequence from the Human Genome Database or other published studies.10,11 PCR was performed with a touchdown protocol (94.0°C/2:00, 20 × [94°C/:30, 63.5°C/:45∗, 72.0°C/1:00], 25 × [94.0°C/:30, 53.5°C/:45, 72.0°C/1:00] ∗annealing temperature decreases 0.5°C each cycle) and Platinum Pfx Polymerase with 10 μl reactions (Invitrogen, Carlsbad, CA) according to the manufacturer's specifications. Abnormally migrating or ambiguous exons found by CSGE were then amplified twice independently by PCR and sequenced in both directions. Normal human DNA served as the control for both PCR and CSGE.
Normal and Mutant RET Expression
Plasmid Constructs
Expression plasmids containing the identified RET mutations were created by site-directed PCR mutagenesis with wild-type RET51 (1114 amino acids, RET51) as the template. The upstream RET primer (gatctctagagccgccaccatggcgaaggcgacgtccggt) contained an XbaI restriction site followed by a consensus Kozak sequence. The downstream primer (gatgcggccgcttaactatcaaacgtgtcc) contained a NotI site for directional cloning. The resultant products were then cut and directionally ligated (XbaI-NotI) into the mammalian expression vector, pCI-Neo (Clontech, Mountain View, CA). The parental human neuroblastoma cell line, SK-N-MC, was utilized to establish cell lines expressing normal RET51 and each mutant (cDNA)RET construct, as well as an empty vector control. Cells were transfected with the DNA constructs with a cationic lipid and protocol from Bio-Rad (siLentFect) (Bio-Rad Laboratories, Hercules, CA) and then selected in 800 μg/ml G418 (Invitrogen) until the mock-transfected cells were all dead. Then, entire transfected cell populations were used in subsequent experiments. Transfectant cells were maintained in 400 μg/ml G418.
Chemicals
GDNF (R&D Systems, Inc., Minneapolis, MN) was reconstituted to 10 μg/ml in sterile PBS with 0.1% bovine serum albumin (BSA) and stored at −70°C under light-free conditions.
Cell Culture
All cultures were maintained under a fully humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were grown in minimal essential media (MEM) supplemented with 10% FBS, 1× nonessential amino acids, and penicillin/streptomycin antibiotic mix (Sigma Chemical Co., St. Louis, MO). For experimental incubations, cells in log-phase growth were plated at a density of 1 × 105 cells/ml and allowed to attach overnight. Each cell line was then treated with 100 ng/ml GDNF for 3 hr in serum-free MEM media. Experimental incubations were terminated by collecting cells, pelleting at 500 × g for 5 min, and washing with 1× phosphate-buffered saline (PBS) and directly lysing cells in 1× Laemmli (1% SDS, 10% glycerol, 100 mM dithiothreitol, 50 mM Tris [pH 6.8]) for western blots.
Assays for RET51 Expression and Phosphorylation
To perform western blots, cell lysates were briefly sonicated and centrifuged at 12,800 × g for 5 min. All samples were boiled for 5 min and equal amounts of protein (40 μg) were separated by SDS-PAGE (4%–20% gradient) and electroblotted to 0.45 μM nitrocellulose membranes. Blots were stained with amido black (0.5% [w/v] in 5% acetic acid) and destained with water to confirm transfer and equal loading and then blocked in 1× Tris-buffered saline (0.8% NaCl, 2.7 mM KCl, 25 mM Tris [pH 7.4]) with 0.05% Tween-20 (TBS-T) and 1% milk/1% BSA for 1 hr at 22°C. The blots were incubated in fresh blocking solution and probed for 1 hr with a 1:1000 dilution of either anti-RET51 (C-20), anti-phospho-RET (Tyr 1062-R), or anti-GFRA1 (H70) (Santa Cruz Biotechnology, Santa Cruz, CA) primary antibody. The blots were washed 3 × 5 min in TBS-T and then incubated with a 1:10,000 dilution of peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) in TBS-T for 1 hr at 22°C. Blots were again washed 3 × 5 min in TBS-T and then developed by enhanced chemiluminesence (Pierce, Rockford, IL). Individual blots were stripped for 30 min with Restore (Pierce) and reprobed with primary and secondary antibodies as described above.
Results
Mutation Analysis
We studied the DNA from 33 stillborn human fetuses with renal aplasia or severe dysplasia to determine the incidence of mutation in RET, GDNF, and GFRA1. To screen for genetic mutations, amplified DNA from transcribed regions of RET (22 exons), GDNF (3 exons), and GFRA1 (18 exons) were subjected to conformation sensitive gel electrophoresis (CSGE).10,11 Abnormally migrating or ambiguous exons identified by CSGE were subsequently sequenced. In addition, to control for the possibility of PCR-induced errors during amplification, each suspect exon was amplified twice independently and sequenced in both directions to confirm and identify the presence of mutations. Normal human DNA served as the control for PCR and CSGE. Human RET mutations were found in 37% of fetuses with bilateral renal agenesis and in 20% of fetuses with unilateral renal agenesis (Figure 1). We did not identify any RET mutations in fetuses with renal dysplasia or in normal control DNA. To assess the statistical significance of our findings, we have performed a Bayesian analysis (Appendix A); this analysis demonstrates the approximate likelihood of finding such misfunctioning RET mutations in the general population to be very low (0.00028 = P(θ)). In fetuses with bilateral agenesis, mutations occurred throughout the RET gene and were about evenly divided between the extracellular and intracellular regions of the molecule (Figure 2). Four of the mutations occurred in exon 6. Likewise, in the fetuses with unilateral renal agenesis, mutations occurred with equal frequency in both in the extracellular domain and in the intracellular carboxy-terminal tail of RET (Figure 2). Lastly, one mutation created a severe truncation (KP26). It should be noted that in this study, we assayed for mutations only in the RET coding region, and mutations in noncoding regions of the gene may have been present but undetected.
Figure 1.
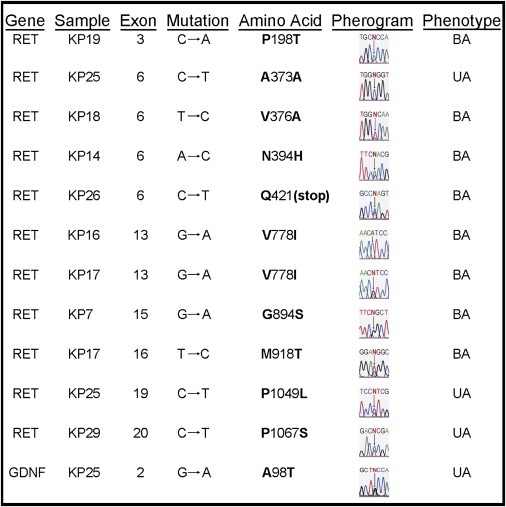
RET and GDNF Mutations in Fetuses with Bilateral or Unilateral Renal Agenesis
Eight RET mutations were seen in 7/19 fetuses (37%) with bilateral renal agenesis (BA), and three RET mutations were discovered in 2/10 fetuses (20%) with unilateral renal agenesis (UA). One GDNF mutation occurred in 1/10 fetuses (10%) with unilateral renal agenesis.
Figure 2.
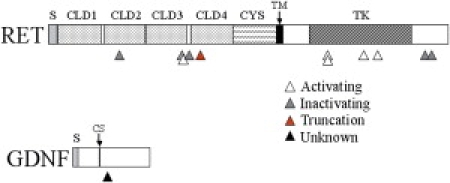
Schematic Illustration of the Identified Mutations in RET54 and GDNF in Stillborn Fetuses with Either Bilateral Renal Agenesis or Unilateral Agenesis
Triangles signify the location and type of mutations. RET: S, signaling domain; CLD, cadherin-like domain; CYS, cysteine-rich region; TM, transmembrane region; TK, tyrosine kinase domain. GDNF: S, signaling domain; CS, cleavage site.
Mutations in GDNF were found in only one fetus with unilateral renal agenesis (Figure 1). Fetuses with renal dysplasia had no identified mutations in GDNF. Of note, the only sample with a GDNF mutation (KP25) also had two mutations in RET. No mutations in GFRA1 were found in any of our samples.
Biochemical Analysis
We performed additional biochemical studies to determine whether the identified RET mutations significantly altered RET activation in a cell-culture system. We created (cDNA)RET constructs possessing the identified mutations as well as normal (cDNA)RET51. We chose to use RET51 as the cDNA template because the KP29 mutation is located with the alternatively spliced carboxy-tail of RET51. We evaluated RET51 phosphorylation in a cell-based assay by transfecting the cDNA constructs into SK-N-MC cells. These cells endogenously express GFRA1, and when soluble GDNF is added to the media, a RET signaling complex can be created, leading to RET phosphorylation. RET51-expressing SK-N-MC cells (normal and mutants) were incubated for 3 hr in either serum-free media (−GDNF) or media supplemented with 100 ng/ml soluble GDNF (+GDNF). Western blots of the resulting cell lysates (Figure 3) showed appropriate levels of expression of normal and each mutant RET. Interestingly, we found that RET mutants KP7, KP16, KP17, and KP18 are constitutively phosphorylated at tyrosine 1062, while KP14, KP19, KP25, and KP29 are inactivated (not activated by GDNF as is normal RET51). Additionally, the constitutively activated mutants seem to be further activated by the addition of soluble GDNF. Lastly, RET normally presents as a doublet of 150 kDa and 170 kDa on western blots, with the larger form representing the RET on the cell surface and the smaller form representing a preprocessed intracellular form.12,13 Although constitutively activated, both KP7 and KP18 seem to express only the 150 kDa form of RET51 instead of the doublet usually observed.
Figure 3.
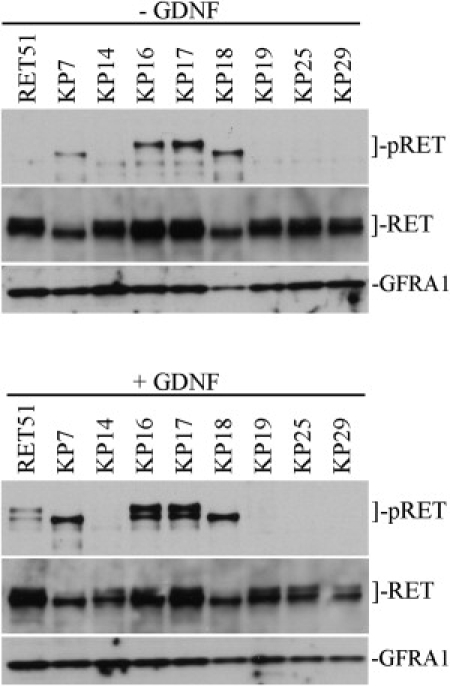
Western Blots of Normal and Mutant RET51 Expressed in SK-N-MC Neuroblastoma Cells ± GDNF
Blots were sequentially probed for phospho-RET51 (Y1062) (pRET), total RET51 (RET), and GFRA1.
Top gels: cell lysates with no GDNF added to the media. KP7, KP16, KP17, and KP18 are constitutively phosphorylated.
Bottom gels: cell lysates treated with GDNF. Normal RET51 is phosphorylated and KP7, KP16, KP17, and KP18 seem further phosphorylated. The RET mutations in KP14, KP19, KP25, and KP29 appear to inactivate RET51 (prevent autophosphorylation in response to GDNF).
Discussion
Mutations in the RET proto-oncogene are responsible for several diseases in humans. Initially, translocations in the gene were found to be associated with papillary thyroid cancer (MIM #188550);14 the frequency of this genetic finding is especially high in thyroid cancer resulting from radiation exposure.15,16 Subsequently, it was discovered that certain inherited mutations in RET are responsible for the multiple endocrine neoplasia (MEN) 2A and 2B syndromes, as well as the familial medullary thyroid carcinoma (FMTC).17–21 In these _RET_-related cancers and neoplastic syndromes, there is constitutive activation of the tyrosine kinase domain, with increased downstream signaling ultimately resulting in cellular transformation and malignancy.22,23 Conversely, Hirschprung disease is a nonneoplastic disease associated with RET mutations that reduce RET tyrosine kinase activity, probably resulting in an embryonic microenvironment that does not allow the normal development of enteric ganglion cells in the affected bowel.24–28
We have shown that RET is frequently mutated in humans with renal agenesis. This finding is consistent with other experimental evidence supporting the importance of RET in renal development. For example, RET is normally expressed in the Wolffian duct of the developing kidney in humans and mice,29,30 and mice lacking Ret exhibit severe renal dysplasia or agenesis as a result of defective ureteric bud outgrowth.29,30 Moreover, there are reports describing the occurrence of familial unilateral renal aplasia and FMTC8,9 and the rare coincidence of renal aplasia and Hirschsprung disease.31,32 Taken together, these findings suggest that congenital renal aplasia joins Hirschsprung disease as another congenital human condition resulting from RET mutations that prevent or impede the embryonic development of RET-dependant structures.
In this study, we defined RET activation as the phosphorylation of RET at residue Y1062, because phosphorylation of this tyrosine, in particular, has been shown to be critical in kidney development.33 We detected four inactivating RET mutations (KP14, KP19, KP25, KP29), four activating RET mutations (KP7, KP16, KP17, KP18), and one RET truncation (KP26). Four of these RET mutations have been previously noted in humans. The mutations found in samples KP14 and KP7 are associated with Hirschsprung disease,34,35 and the mutations from samples KP16 and KP17 are associated with sporadic medullary thyroid carcinoma (MTC).36 Moreover, the M918T mutation in sample KP17 causes the MEN2B syndrome, in addition to occurring in sporadic MTC.
As depicted in Figure 2, inactivating RET mutations occurred in both the extracellular and the intracellular regions of RET. There are several mechanisms through which these particular mutations may abrogate RET activity.28 For example, mutations in the intracellular carboxy-terminal tail of RET, such as the ones we found in KP25 and KP29, have been shown to impair SHC binding to RET resulting in a RET-mediated signal defect.37,38 Alternatively, mutations in the extracellular region have been shown to impair protein maturation as well as protein-protein interactions between RET and its coreceptors.39–41 In any case, the association of inactivating RET mutations with renal aplasia in humans is certainly consistent with the presence of severe renal anomalies in mice lacking Ret.5
What is perhaps surprising is that we also found renal aplasia in individuals possessing activating RET mutations. One of the four such mutations was located in the extracellular region (KP18) and three were in the intracellular region (KP7, KP16, KP17). Other investigators have clearly shown that activating mutations in RET can be associated with both gain-of-function and loss-of-function phenotypes.42 This phenomenon was recently reported in a transgenic murine model in which animals were created possessing Ret with the C620R mutation.43 This mutation is generally transforming, and in humans has been associated with FMTC, MEN 2A, and in a minority of cases, Hirschsprung disease. Mice with the homozygous C620R Ret mutation demonstrated the absence of enteric ganglia and kidneys, whereas the heterozygote transgenic animals exhibited no such developmental abnormalities, but developed premalignant tumors of the thyroid and adrenal glands.44 We postulate that despite the presence of a constitutively activated RET, there is a RET signaling defect to cause a loss-of-function phenotype. Supporting this hypothesis are reports in humans describing the occurrence of familial unilateral renal aplasia and FMTC8,9 and the rare coincidence of renal aplasia and Hirschsprung disease.31,32
There are several possible mechanisms by which an activating RET mutation results in a loss-of-function phenotype. First, as noted above, two of the mutations we detected (KP16 and KP17) are known to be associated with MTC, and the cosegregation of Hirschsprung disease with MEN 2 has been previously noted; it is likely that in our samples, renal aplasia occurs through mechanism analogous to the aganglionosis in MEN 2 patients.42,45 In one recent report, it was noted that in tissue culture, the Ret C620R mutation has the ability to promote cellular proliferation, but impairs the GDNF effects of Ret on cell migration, differentiation, and survival.46 This duality in function may help explain the ability of mutant RET to induce both the gain-of-function and loss-of-function phenotypes.
In our in vitro expression experiments, neither KP7 nor KP18 exhibited the 170 kDa form of the protein on a western blot that represents the mature cell surface receptor. Indeed, others have reported that certain RET mutations associated with Hirschsprung disease result in the expression of only the immature, 150 kDa RET protein,39,40 and that this RET protein is localized to the rough endoplasmic reticulum and Golgi.40 Through a mechanism that prevents maturation and migration of the RET molecule to the cell surface,40,47 such mutations could disrupt ureteric branching of the kidney that is dependent on RET signaling.5 Thus, each of the RET mutations detected in this study either inactivates RET or has the potential to reduce overall RET signaling to account for our phenotypic findings.
Hirschprung disease is a nonneoplastic disease associated with RET mutations that reduce RET tyrosine kinase activity, probably resulting in an embryonic microenvironment that does not allow the normal development of enteric ganglion cells in the affected bowel.24–28 Thus, it might be expected that the RET mutations leading to renal aplasia in our subjects should also cause intestinal aganglionosis or Hirschsprung disease. However, it is likely that RET is only one of a number of different genes cooperating to support the embryonic development of kidneys and enteric ganglia. Indeed, segregation analysis has suggested that three genetic loci are involved with familial Hirschprung disease.48 Moreover, a polygenic process is further supported by the incomplete penetrance of familial Hirschsprung disease related to RET mutations.24,25 The finding in the current study that RET mutations can induce either unilateral or bilateral renal agenesis also supports the notion that other genes may be important. Finally, that RET-related renal agenesis and aganglionosis are not strictly linked is further supported by a recent study demonstrating that the transgenic introduction of a mutated human RET gene into the Ret null murine background induced the development of nearly normal kidneys, but not enteric ganglia.49
Our study suggests that abnormal renal development is rarely related to mutations in GDNF. This is consistent with the extreme rarity of reported GDNF mutations, which have been found in only a handful of patients with Hirschsprung disease.50–52 It may be that such mutations are not usually pathologic, or that in many cases GDNF mutations cause early fetal demise. Indeed, in one animal model, mutations in GDNF were found to be insufficient to cause Hirschsprung disease.51 Interestingly, the fetus in our study with a GDNF mutation also had two RET mutations. The association of mutations in both RET and GDNF has also been reported in Hirschsprung disease, suggesting that RET and GDNF mutations may act in concert to cause the intestinal aganglionosis, as well as the renal anomalies seen in our study.50 This synergistic heterozygosity phenomenon has recently been described, in which there is cooperation between two mutated genes in a common pathway to induce a disease phenotype,53 and we speculate that the interaction between GDNF and RET behaves similarly in renal development, requiring in some cases both molecular events to occur before there is renal aplasia.
In conclusion, this is the first report to our knowledge describing the association of RET mutations with renal agenesis in humans. It is possible that the lack of previous reports is due to the devastating nature of renal agenesis, causing most fetuses to die in utero. The high rate of RET mutation in fetuses with renal agenesis suggests that heterozygous mutations in RET may be responsible for a significant fraction of humans with abnormal kidney development. Furthermore, the low mutation rate for GDNF suggests a minor role for this gene in renal agenesis or dysplasia. These data have significant relevance to the genetic communities in that it provides evidence for a possible mechanism of abnormal kidney development in humans. Our findings suggest that congenital renal aplasia joins Hirschsprung disease as another congenital human condition resulting from RET mutations that prevent or impede the embryonic development of RET-dependant structures. Moreover, these data may support genetic screening for RET mutations in individuals who have had children born with abnormal kidneys or have miscarried a child with abnormal kidneys.
Appendix A
Statistical Analysis
We used a Bayesian analysis to approximate the expected likelihood of misfunctioning RET mutations in the general population, to determine the statistical significance of our genetic findings.
Let:
P(θ)=probability of a person in the general population possessing a RET mutation causing protein misfunction
P(Y)=probability of renal aplasia in the general population(=1/3000)
and restating Bayes theorem:
| P(θ|Y)=P(θandY)/P(Y). | (A1) |
|---|
More commonly written:
| P(θ|Y)=P(Y|θ)×P(θ)/P(Y). | (A2) | | -------------------------- | ---- |
The left side of this equation, which is the probability of finding a misfunctioning RET gene in patients with renal aplasia, was found in our experiements to be approximately 0.31. In particular, we found such mutations in 9 of 29 subjects. By evaluating the binomial probability distribution, the 95% confidence interval for this experimental finding in our population sample is consistent with an actual population probability as low as 21%. That is, the actual probability of RET misfunction in the population of newborns with renal aplasia could be this low and still be consistent with our finding of 9 cases in 29 subjects.
It remains to be shown that the probability of such mutations in the general population, P(θ), is significantly less than our (most conservative) experimental finding of 21% in subjects having renal aplasia.
Rearranging equation (A2) above,
| P(θ|Y)×P(Y)/P(Y|θ)=P(θ). | (A3) | | -------------------------- | ---- |
The term in the denominator of this equation, the probability that a subject with a RET misfunction mutation will exhibit renal aplasia, is simply the penetrance of such mutations, and the value must be assumed. The penetrance of RET mutations in Hirschsprung disease has been determined to be 51% in females and 72% in males; for the sake of conservatism in the current analysis, we will assume the penetrance, P(Y|θ), is only 25%.
Substituting the most conservative values into equation (A3),
| (0.21)(1/3000)/(0.25)=0.00028=P(θ). | (A4) |
|---|
In conclusion, under our assumptions, we estimate that the incidence of RET mutations causing abnormal protein activation in the general population is approximately 1:3800, which varies significantly from our finding of such mutations in 9/29 subjects with renal aplasia.
Web Resources
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
This work was sponsored, in part, by donations to The Children's Miracle Network.
References
- 1.Wilson R.D., Baird P.A. Renal agenesis in British Columbia. Am. J. Med. Genet. 1985;21:153–169. doi: 10.1002/ajmg.1320210123. [DOI] [PubMed] [Google Scholar]
- 2.Roodhooft A.M., Birnholz J.C., Holmes L.B. Familial nature of congenital absence and severe dysgenesis of both kidneys. N. Engl. J. Med. 1984;319:1341–1344. doi: 10.1056/NEJM198405243102101. [DOI] [PubMed] [Google Scholar]
- 3.Bates C.M. Kidney development: regulatory molecules crucial to both mice and men. Mol. Genet. Metab. 2000;71:391–396. doi: 10.1006/mgme.2000.3072. [DOI] [PubMed] [Google Scholar]
- 4.Yu J., McMahon A.P., Valerius M.T. Recent genetic studies of mouse kidney development. Curr. Opin. Genet. Dev. 2004;14:550–557. doi: 10.1016/j.gde.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Schuchardt A., D'Agati V., Larsson-Blomberg L., Constantini F., Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 6.Pichel J.G., Shen L., Hui S.Z., Granholm A.-C., Drago J., Grinberg A., Lee E.J., Huang S.P., Saarma M., Hoffer B.J. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 7.Cacalano G., Farinas I., Wang L.-C., Hagler K., Forgie A., Moore M., Armanini M., Phillips H., Ryan A.M., Reichardt L.F. GFRa1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lore F., Di Cairano G., Talidis F. Unilateral renal agenesis in a family with medullary thyroid carcinoma. N. Engl. J. Med. 2000;342:1218–1219. doi: 10.1056/NEJM200004203421615. [DOI] [PubMed] [Google Scholar]
- 9.Lore F., Talidis F., Di Cairano G., Renieri A. Multiple endocrine neoplasia type 2 syndromes may be associated with renal malformations. J. Intern. Med. 2001;250:37–42. doi: 10.1046/j.1365-2796.2001.00846.x. [DOI] [PubMed] [Google Scholar]
- 10.Korkko J., Annunen S., Pihlajamaa T., Prockop D.J., Ala-Kokko L. Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc. Natl. Acad. Sci. USA. 1998;95:1681–1685. doi: 10.1073/pnas.95.4.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen L.A., Christiansen M., Vuust J., Andersen P.S. Recent developments in high-throughput mutation screening. Pharmacogenomics. 2001;2:387–399. doi: 10.1517/14622416.2.4.387. [DOI] [PubMed] [Google Scholar]
- 12.Iwamoto T., Taniguchi M., Asai N., Ohkusu K., Nakashima I., Takahashi M. cDNA cloning of mouse ret proto-oncogene and its sequence similarity to the cadherin superfamily. Oncogene. 1993;8:1087–1091. [PubMed] [Google Scholar]
- 13.Tansey M.G., Baloh R.H., Milbrandt J., Johnson E.M. GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/s0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 14.Grieco M., Santoro M., Berlingieri M.T., Melillo R.M., Donghi R., Bongarzone I., Pierotti M.A., Della Porta G., Fusco A., Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 15.Klugbauer S., Lengfelder E., Demidchik E.P., Rabes H.M. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene. 1995;11:2459–2467. [PubMed] [Google Scholar]
- 16.Bounacer A., Wicker R., Caillou B., Cailleux A.F., Sarasin A., Schlumberger M., Suarez H.G. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. 1997;15:1263–1273. doi: 10.1038/sj.onc.1200206. [DOI] [PubMed] [Google Scholar]
- 17.Donis-Keller H., Dou S., Chi D., Carlson K.M., Toshima K., Lairmore T.C., Howe J.R., Moley J.F., Goodfellow P., Wells S.A. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993;2:851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 18.Mulligan L.M., Kwok J.B.J., Healey C.S., Elsdon M.J., Eng C., Gardner E., Love D.R., Mole S.E., Moore J.K., Papi L. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 19.Carlson K.M., Dou S., Chi D., Scavarda N., Toshima T., Jackson C.E., Wells S.A., Goodfellow P.J., Donis-Keller H. Single missense mutation in the tyrosine kinase domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc. Natl. Acad. Sci. USA. 1994;91:1579–1583. doi: 10.1073/pnas.91.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofstra R.M.W., Landsvater R.M., Ceccherini I., Stulp R.P., Stelwagon T., Luo Y., Pasini B., Hoppener J.W.M., van Amstel H.K.P., Romeo G. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367:375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- 21.Eng C., Smith D.P., Mulligan L.M., Nagal M.A., Healey C.S., Ponder M.A., Gardner E., Scheumann G.F.W., Jackson C.E., Tunacliffe A., Ponder B.A.J. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum. Mol. Genet. 1994;3:237–241. doi: 10.1093/hmg/3.2.237. [DOI] [PubMed] [Google Scholar]
- 22.Santoro M., Rosati R., Grieco M., Berlingieri M.T., D'Amato G.L., de Franciscis V., Fusco A. The ret proto-oncogene is consistently expressed in human pheochromocytomas and thyroid medullary carcinomas. Oncogene. 1990;5:1595–1598. [PubMed] [Google Scholar]
- 23.Santoro M., Carlomagno F., Romano A., Bottaro D.P., Dathan N.A., Grieco M., Fusco A., Vecchio G., Matoskova B., Kraus M.H., Di Fiore P.P. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267:381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 24.Romeo R., Ronchetto P., Luo Y., Barone V., Serl M., Ceccherini I., Pasini B., Bocclardl R., Lerone M., Kaalainen H., Marucciello G. Point mutations affecting the tyrosine domain of the RET proto-oncogene in Hirschsprung's disease. Nature. 1994;367:377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 25.Edery P., Lyonnet S., Mulligan L.M., Pelet A., Dow E., Abel L., Holder S., Nihoul-Fekete C., Ponder B.A.J., Munnich A. Mutations of the RET proto-oncogene in Hirschsprung's disease. Nature. 1994;367:378–380. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 26.Pelet A., Attie T., Goulet O., Eng C., Ponder B.A.J., Munnich A., Lyonnet S. De novo mutations of the RET proto-oncogenein Hirschsprung's disease. Lancet. 1994;344:1769–1770. doi: 10.1016/s0140-6736(94)92908-4. [DOI] [PubMed] [Google Scholar]
- 27.Edery P., Pelet A., Mulligan L.M., Abel L., Attie T., Dow E., Bonneau D., David A., Flintoff W., Jan D. Long segment and short segment familial Hirschsprung's disease: variable clinical expression at the RET locus. J. Med. Genet. 1994;31:602–606. doi: 10.1136/jmg.31.8.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelet A., Geneste O., Edery P., Pasini A., Chappuis S., Atti T., Munnich A., Lenoir G., Lyonnet S., Billaud M. Various mechanisms cause RET-mediated signaling defects in Hirschsprung's disease. J. Clin. Invest. 1998;101:1415–1423. doi: 10.1172/JCI375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Attie-Bitach T., Abitbol M., Gerard M., Delezoide A.L., Auge J., Pelet A., Amiel J., Pachnis V., Munnich A., Lyonnet S., Vekemans M. Expression of the RET proto-oncogene in human embryos. Am. J. Med. Genet. 1998;80:481–486. doi: 10.1002/(sici)1096-8628(19981228)80:5<481::aid-ajmg8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Pachnis V., Mankoo B., Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- 31.Sinnassamy P., Yazbeck S., Brochu P., O'Regan S. Renal anomalies and agenesis associated with total intestinal aganglionosis. Int. J. Pediatr. Nephrol. 1986;7:1–2. [PubMed] [Google Scholar]
- 32.Santos H., Mateus J., Leal M.J. Hirschsprung disease associated with polydactyly, unilateral renal agenesis, hypertelorism, and congenital deafness: a new autosomal recessive syndrome. J. Med. Genet. 1988;25:204–205. doi: 10.1136/jmg.25.3.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong A., Bogni S., Kotka P., de Graaff E., D'Agati V., Costantini F., Pachnis V. Phosphotyrosine 1062 is critical for the in vivo activity of the Ret9 receptor tyrosine kinase isoform. Mol. Cell. Biol. 2005;25:9661–9673. doi: 10.1128/MCB.25.21.9661-9673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolk S., Pelet A., Hofstra R.M., Angrist M., Salomon R., Croaker D., Buys C.H., Lyonnet S., Chakravarti A. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc. Natl. Acad. Sci. USA. 2000;97:268–273. doi: 10.1073/pnas.97.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitze G., Cramer J., Ziegler A., Schierz M., Schreiber M., Kuhlisch E., Roesner D., Schackert H.K. Association between c135G/A genotype and RET proto-oncogene germline mutations and phenotype of Hirschsprung's disease. Lancet. 2002;359:1200–1205. doi: 10.1016/S0140-6736(02)08218-1. [DOI] [PubMed] [Google Scholar]
- 36.Miyauchi A., Matsuzuka F., Hirai K., Yokozawa T., Kobayashi K., Ito Y., Nakano K., Kuma K., Futami H., Yamaguchi K. Prospective trial of unilateral surgery for nonhereditary medullary thyroid carcinoma in patients without germline RET mutations. World J. Surg. 2002;26:1023–1028. doi: 10.1007/s00268-002-6665-1. [DOI] [PubMed] [Google Scholar]
- 37.Iwashita T., Kurokawa K., Qiao S., Murakami H., Asai N., Kawai K., Hashimoto M., Watanabe T., Ichihara M., Takahashi M. Functional analysis of RET with Hirschsprung mutations affecting its kinase domain. Gastroenterology. 2001;121:24–33. doi: 10.1053/gast.2001.25515. [DOI] [PubMed] [Google Scholar]
- 38.Ishiguro Y., Iwashita T., Murakami H., Asai N., Iida K., Goto H., Hayakawa T., Takahashi M. The role of amino acids surrounding tyrosine 1062 in ret in specific binding of the shc phosphotyrosine-binding domain. Endocrinology. 1999;140:3992–3998. doi: 10.1210/endo.140.9.7003. [DOI] [PubMed] [Google Scholar]
- 39.Iwashita T., Murakami H., Asai N., Takahashi M. Mechanism of ret dysfunction by Hirschsprung mutations affecting its extracellular domain. Hum. Mol. Genet. 1996;5:1577–1580. doi: 10.1093/hmg/5.10.1577. [DOI] [PubMed] [Google Scholar]
- 40.Cosma M.P., Cardone M., Carlomagno F., Colantuoni V. Mutations in the extracellular domain cause RET loss of function by a dominant negative mechanism. Mol. Cell. Biol. 1998;18:3321–3329. doi: 10.1128/mcb.18.6.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjaer S., Ibanez C.F. Intrinsic susceptibility to misfolding of a hot-spot for Hirschsprung disease mutations in the ectodomain of RET. Hum. Mol. Genet. 2003;12:2133–2144. doi: 10.1093/hmg/ddg227. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi M., Iwashita T., Santoro M., Lyonnet S., Lenoir G.M., Billaud M. Co-segregation of MEN2 and Hirschsprung's disease: the same mutation of RET with both gain and loss-of-function? Hum. Mutat. 1999;13:331–336. doi: 10.1002/(SICI)1098-1004(1999)13:4<331::AID-HUMU11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 43.Carniti C., Belluco S., Riccardi E., Cranston A.N., Mondellini P., Ponder B.A., Scanziani E., Pierotti M.A., Bongarzone I. The Ret(C620R) mutation affects renal and enteric development in a mouse model of Hirschsprung's disease. Am. J. Pathol. 2006;168:1262–1275. doi: 10.2353/ajpath.2006.050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin L., Puliti A., Bonora E., Evangelisti C., Conti V., Tong W.M., Medard J.J., Lavoue M.F., Forey N., Wang L.C. C620R mutation of the murine ret proto-oncogene: loss of function effect in homozygotes and possible gain of function effect in heterozygotes. Int. J. Cancer. 2007;121:292–300. doi: 10.1002/ijc.22378. [DOI] [PubMed] [Google Scholar]
- 45.Mulligan L.M., Eng C., Attie T., Lyonnet S., Marsh D.J., Hyland V.J., Robinson B.G., Frilling A., Verellen-Dumoulin C., Safar A. Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum. Mol. Genet. 1994;3:2163–2167. doi: 10.1093/hmg/3.12.2163. [DOI] [PubMed] [Google Scholar]
- 46.Arighi E., Popsueva A., Degl'Innocenti D., Borrello M.G., Carniti C., Perala N.M., Pierotti M.A., Sariola H. Biological effects of the dual phenotypic Janus mutation of ret cosegregating with both multiple endocrine neoplasia type 2 and Hirschsprung's disease. Mol. Endocrinol. 2004;18:1004–1017. doi: 10.1210/me.2003-0173. [DOI] [PubMed] [Google Scholar]
- 47.Pasini B., Borrello M.G., Greco A., Bongarzone I., Luo Y., Mondellini P., Alberti L., Miranda C., Arighi E., Bocciardi R. Loss of function effect of RET mutations causing Hirschsprung disease. Nat. Genet. 1995;10:35–40. doi: 10.1038/ng0595-35. [DOI] [PubMed] [Google Scholar]
- 48.Gabriel S.B., Salomon R., Pelet A., Angrist M., Amiel J., Fornage M., Attie-Bitach T., Olson J.M., Hofstra R., Buys C. Segregation at three loci explains familial and population risk in Hirschsprung disease. Nat. Genet. 2002;31:89–93. doi: 10.1038/ng868. [DOI] [PubMed] [Google Scholar]
- 49.Skinner M.A., Kalyanaraman S., Safford S.D., Heuckeroth R.O., Tourtellotte W., Goyeau D., Goodfellow P., Milbrandt J.D., Freemerman A. A human yeast artificial chromosome containing the multiple endocrine neoplasia type 2B Ret mutation does not induce medullary thyroid carcinoma but does support the growth of kidneys and partially rescues enteric nervous system development in Ret-deficient mice. Am. J. Pathol. 2005;166:265–274. doi: 10.1016/S0002-9440(10)62250-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angrist M., Bolk S., Halushka M., Lapchak P.A., Chakravarti A. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat. Genet. 1996;14:341–344. doi: 10.1038/ng1196-341. [DOI] [PubMed] [Google Scholar]
- 51.Salomon R., Attie T., Pelet A., Bidaud C., Eng C., Amiel J., Sarnacki S., Goulet O., Ricour C., Nihoul-Fekete C. Germline mutations of the RET ligand GDNF are not sufficient to cause Hirschsprung disease. Nat. Genet. 1996;14:345–347. doi: 10.1038/ng1196-345. [DOI] [PubMed] [Google Scholar]
- 52.Ivanchuk S.M., Myers S.M., Eng C., Mulligan L.M. De novo mutation of GDNF, ligand for the RET/GDNFR-alpha receptor complex, in Hirschsprung disease. Hum. Mol. Genet. 1996;5:2023–2026. doi: 10.1093/hmg/5.12.2023. [DOI] [PubMed] [Google Scholar]
- 53.Vockley J., Rinaldo P., Bennett M.J., Matern D., Vladutiu G.D. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol. Genet. Metab. 2000;71:10–18. doi: 10.1006/mgme.2000.3066. [DOI] [PubMed] [Google Scholar]
- 54.Anders J., Kjar S., Ibanez C.F. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J. Biol. Chem. 2001;276:35808–35817. doi: 10.1074/jbc.M104968200. [DOI] [PubMed] [Google Scholar]