Low reduction potential of Ero1α regulatory disulphides ensures tight control of substrate oxidation (original) (raw)
Abstract
Formation of disulphide bonds within the mammalian endoplasmic reticulum (ER) requires the combined activities of Ero1α and protein disulphide isomerase (PDI). As Ero1α produces hydrogen peroxide during oxidation, regulation of its activity is critical in preventing ER-generated oxidative stress. Here, we have expressed and purified recombinant human Ero1α and shown that it has activity towards thioredoxin and PDI. The activity towards PDI required the inclusion of glutathione to ensure sustained oxidation. By carrying out site-directed mutagenesis of cysteine residues, we show that Ero1α is regulated by non-catalytic disulphides. The midpoint reduction potential (E°′) of the regulatory disulphides was calculated to be approximately −275 mV making them stable in the redox conditions prevalent in the ER. The stable regulatory disulphides were only partially reduced by PDI (E°′∼−180 mV), suggesting either that this is a mechanism for preventing excessive Ero1α activity and oxidation of PDI or that additional factors are required for Ero1α activation within the mammalian ER.
Keywords: disulphide formation, Ero1α, protein disulphide isomerase, reduction potential
Introduction
The endoplasmic reticulum (ER) of mammalian cells contains a number of enzymes, the role of which is to ensure efficient formation of native disulphide bonds in proteins entering the secretory pathway. Central to the formation of disulphides is the initial oxidation of cysteine residues catalysed by protein disulphide isomerase (PDI) (Freedman, 1995). PDI is one of a large family of homologous oxidoreductases present within the ER (Ellgaard and Ruddock, 2005). Oxidation by PDI results in the introduction of a disulphide into the substrate protein with the concomitant reduction of a disulphide within one of the active sites in PDI (Kulp et al, 2006). A second enzyme, Ero1α or Ero1β in mammalian cells (Cabibbo et al, 2000; Pagani et al, 2000), is thought to catalyse the reoxidation of the PDI active site (Sevier and Kaiser, 2002). The Ero1 enzymes are flavoproteins that can couple the introduction of a disulphide into PDI with the reduction of oxygen to liberate hydrogen peroxide (Gross et al, 2006). Hence, for every disulphide introduced into a substrate protein one molecule of hydrogen peroxide theoretically can be produced by Ero1, providing a potential source of reactive oxygen species (ROS) and ER-generated oxidative stress (Chakravarthi et al, 2006).
To prevent the deleterious accumulation of ROS in the ER lumen, the enzymatic activity of Ero1 needs to be tightly regulated. Ero1α and Ero1β levels are regulated by hypoxia (Gess et al, 2003) and the unfolded protein response (UPR) (Pagani et al, 2000), respectively, providing a mechanism for responding to changing conditions within the ER. Yeast Ero1 (Ero1p) is also regulated by the UPR (Frand and Kaiser, 1998; Pollard et al, 1998) but, in addition, is regulated post-translationally by the formation of non-catalytic disulphides that need to be reduced to activate the enzyme (Sevier et al, 2007). Such a mechanism allows the enzyme to be activated during conditions when the ER is more reducing but inactivated when oxidising conditions prevail. It is highly likely that a similar system of regulation exists with the mammalian Ero1 proteins; however, the cysteine residues outside the active site are not conserved between the yeast and human Ero1s (Sevier and Kaiser, 2008) making it difficult to predict which cysteines form the regulatory disulphides.
In addition to enzymes catalysing the oxidation of disulphide bonds, the ER lumen also contains enzymes that catalyse the reduction or isomerisation of non-native disulphides (Jansens et al, 2002). PDI is able to catalyse both the formation and reduction of disulphides (Freedman, 1995), whereas other members of the PDI family of oxidoreductases such as ERp57 may exert an effect primarily to resolve non-native disulphides (Jessop et al, 2007). To catalyse these reactions, the active site disulphides within these enzymes need to be reduced. Indeed, most of the PDI family members, apart from PDI, which is partially oxidised, are reduced at steady state (Jessop and Bulleid, 2004). Glutathione (GSH) is required to maintain such a reduced state, depletion of GSH does not prevent disulphide formation but can lead to a defect in folding due to a lack of resolution of non-native disulphides (Chakravarthi et al, 2006). Hence, the ratio of reduced to oxidised GSH (GSH/GSSG) must be sufficiently reducing to maintain the ER oxidoreductases in a reduced state, while allowing correct disulphide formation in substrate proteins. Ero1p can oxidise PDI even at quite high GSH/GSSG ratios (Tu et al, 2000) potentially explaining how both oxidation and reduction reactions can occur within the same intracellular organelle. Interestingly, the phenotype of the temperature-sensitive ero1-1 mutant can be suppressed by perturbing the biosynthesis of GSH, suggesting that the presence of this low molecular weight thiol increases the load on Ero1p (Cuozzo and Kaiser, 1999). Although GSH is a poor substrate for Ero1p, it can reduce oxidised PDI (Gilbert, 1989; Darby and Creighton, 1995), hence Ero1 could oxidise GSH indirectly through the oxidation of PDI or other PDI family members.
In this study, we set out to identify the regulatory disulphides within human Ero1α and to evaluate the ability of GSH and PDI to activate the enzyme. To be able to characterise Ero1α, we purified full-length recombinant protein expressed in Escherichia coli and characterised its activity towards thioredoxin and PDI. We found that although thioredoxin was an excellent substrate for the enzyme, PDI required the presence of GSH for sustained activity. In addition, the regulatory disulphides were shown to have a midpoint reduction potential of approximately −275 mV, making these disulphides relatively stable within the ER lumen. Such a low reduction potential explains why PDI partially reduces this disulphide and raises the possibility of an additional regulatory mechanism for Ero1α.
Results
Characterisation of recombinant Ero1
To be able to characterise the enzymatic activity, regulatory disulphides and biophysical properties of Ero1α, we first needed to produce sufficient purified protein. To this end, we expressed and purified the full-length protein in the Origami B(DE3)pLysS strain of E. coli. Both the wild-type and several mutant proteins containing single- or double-point mutations changing individual cysteine residues to alanine were soluble and expressed efficiently, unless stated otherwise. All proteins contained similar amounts of bound FAD with a protein/FAD ratio of approximately 1:0.85. An absorbance scan of the purified wild-type protein revealed a peak at 454 nm characteristic of a bound FAD moiety (Supplementary Figure S1A). The purified protein was analysed by multi-angle light scattering coupled to gel filtration. A single major peak of 57 kDa was observed along with a minor peak of 99 kDa, consistent with the majority of the protein being monomeric with some dimerisation (Supplementary Figure S1B). These results are consistent with previous data, which suggest that the Ero1 family member, Ero1β, can form a dimer (Dias-Gunasekara et al, 2005). The folding characteristics of the protein was analysed by CD (Supplementary Figure S2A), confirming that the secondary structure was dominated by α-helices (minima at 208 and 222 nm), as expected from the known structure of the Ero1p (Gross et al, 2004). In addition, we determined the intact mass of the recombinant wild-type protein by ESI-TOF mass spectrometry giving a value of 52 945 Da (mass accuracy ±2 Da). This compares with a predicted mass of 52 960 Da, indicating that up to seven disulphides had formed (the wild-type protein has 15 cysteine residues). We also carried out an Ellman's assay on the denatured wild-type protein to determine the number of free thiol groups (Ellman, 1959). The result indicated that there were approximately three free thiols per molecule, indicating that six disulphides had formed. In addition, recombinant Ero1α migrated as a single species by rpHPLC (Supplementary Figure S2B) with a retention time distinct from the reduced protein, indicating that a single disulphide-bonded species was present.
Previous work with yeast recombinant Ero1p (Sevier et al, 2007) and with mammalian Ero1s (Benham et al, 2000) has demonstrated that the hydrodynamic volume of the denatured protein changes significantly on reduction of long-range disulphide bonds. These disulphides have been shown to be regulatory within Ero1p with their reduction causing an activation of enzymatic activity (Sevier et al, 2007). A large shift in mobility was seen when recombinant Ero1α was separated on non-reducing gels either before or after reduction with 12.5 mM dithiothreitol (DTT) for 5 min (see gel inset Supplementary Figure S2A). This result strongly suggests that the recombinant protein contains long-range disulphides. An obvious change in overall secondary structure of the native protein did not occur on reduction as judged by CD as both the untreated and protein treated with 12.5 mM DTT gave identical spectra (Supplementary Figure S2A). However, an increase in the intrinsic tryptophan/tyrosine fluorescence was observed for the wild-type protein on reduction of the long-range disulphides (Supplementary Figure S2C), demonstrating that a change does occur in the microenvironment around aromatic residues. These results suggest that the breaking of long-range disulphides within Ero1α does not cause gross changes to secondary structure in the native protein but are consistent with a conformational change giving rise to enzyme activation.
Recombinant Ero1 activity assays
To characterise the enzymatic properties of recombinant Ero1α, we assayed the ability of the enzyme to oxidise thioredoxin or PDI. The thioredoxin assay directly measures the redox state of the substrate by using a gel-based assay (Sevier et al, 2007). Thioredoxin is modified with the thiol-alkylating agent 4-acetamido-4′-maleimidylstilbene-2,2′-disulphonic acid (AMS), which increases the mass so that the modified, reduced protein migrates slower through the gel than oxidised protein. Complete oxidation of thioredoxin occurred after 10 min in the presence of Ero1α (Figure 1A, lane 7); however, no thioredoxin was oxidised in the absence of Ero1α (Figure 1A, lanes 2–8). To determine the redox state of Ero1α during the reaction with thioredoxin, we assayed changes in its electrophoretic mobility under non-reducing conditions (Figure 1B). At the beginning of the reaction, Ero1α is fully oxidised (Figure 1B, lane 1); however, after 20 s most of the Ero1α migrates slower through the gel, indicating reduction of long-range disulphides (Figure 1B, lane 2). As the thioredoxin substrate becomes fully oxidised, the long-range disulphides in Ero1α reform so that after 1200 s most of the Ero1α are oxidised (Figure 1B, lane 7). There was a distinct lag between the oxidation of thioredoxin and the change in redox status of Ero1α; at 400 s most of the thioredoxin is oxidized, yet the Ero1α regulatory disulphides are still partially reduced (Figure 1A and B). Hence, the rate of reformation of the Ero1α regulatory disulphide is slower than the oxidation of thioredoxin by Ero1α. These results mirror what happens with Ero1p (Sevier et al, 2007), strongly suggesting that the human protein is regulated in a similar way to the yeast protein.
Figure 1.

The redox state of Ero1α changes transiently during the efficient oxidation of thioredoxin. (A) Ero1α oxidises Trx1 (100 μM) as confirmed by the change of redox status judged by alkylation with AMS when incubated in the presence of Ero1α (0.5 μM) but not in its absence. Proteins were visualised by reducing SDS–PAGE and Coomassie blue staining. (B) Ero1α is transiently reduced following incubation with Trx1. Reactions were quenched with NEM, the redox state of Ero1α was visualised by non-reducing SDS–PAGE and silver staining. The reduced and oxidised forms of thioredoxin and Ero1α are indicated as r and ox, respectively. (A, B) Representative gels; the experiment was repeated at least three times with similar results.
Having shown that recombinant Ero1α is active towards thioredoxin, we then wanted to see whether it had activity towards its physiological substrate, PDI. To assay activity, we first monitored the consumption of oxygen when PDI was incubated in the presence of Ero1α, either in the presence or absence of GSH (Figure 2A and B). We found that in the absence of GSH the rate of oxygen consumption was initially faster than the rate in the absence of Ero1 but quickly ceased (Figure 2A) after consumption of approximately 40 μM oxygen. Given the fact that the assay contains 100 μM PDI with two active sites, this consumption of oxygen indicates that only 20% of the active sites were oxidised. However, in the presence of GSH, oxygen was consumed for a much more prolonged period of time, so even in the presence of 10 mM GSH, Ero1α was active towards PDI. The maximal turnover number was found to be approximately three disulphides per molecule of Ero1α per minute, which although low, compares well with the turnover rate for Ero1p assayed under identical conditions (Sevier et al, 2007). It should be noted that not all the oxygen is consumed during the reaction with PDI even in the presence of GSH (Figure 2B). Here, approximately 160 μM oxygen was consumed, which equates to 80% of the PDI active sites oxidised.
Figure 2.

Ero1α oxidises PDI and shows selectivity towards the a′ active site. (A) Kinetics of oxygen consumption by Ero1α (2 μM) during reaction with reduced PDI (100 μM) was assayed either in the absence (A) or presence (B) of GSH (10 mM). Control reactions following oxygen consumption in the absence of Ero1α are as indicated. (C) The oxygen consumption by Ero1α (2 μM) during reaction with either a ΔS1 or ΔS2 mutant of PDI in the presence of 10 mM GSH. The experiments were carried out three times with similar activity profiles.
It has been shown earlier that Ero1p displays distinct reactivity towards the two active sites of yeast PDI (Kulp et al, 2006). We also found that the mammalian enzyme differentially oxidised the two active sites in PDI (Figure 2C). When mutant PDI containing a CGHC to SGHA mutation in the ‘a' domain active site (ΔS1) was incubated with Ero1α and GSH, wild-type rates of oxygen consumption were observed. However, a similar mutation of the a′ domain (ΔS2) greatly diminished the rate of oxygen consumption. It would appear from these results that Ero1α preferentially oxidises the a′ domain of PDI.
To explain these results further, we determined whether PDI was being oxidised by Ero1α using a similar gel-based assay as used with thioredoxin. Unfortunately, the redox state of human PDI cannot be assayed using AMS as the difference in mobility of the reduced and oxidised forms is minimal (Mezghrani et al, 2001; Chakravarthi and Bulleid, 2004). However, the redox state can be determined using an assay based on the modification of cysteine residues using methoxy polyethyleneglycol 5000 maleimide (mPEG-mal) (Xiao et al, 2004; Appenzeller-Herzog and Ellgaard, 2008). In this assay, the two non-active site thiols and four active site thiols are modified if PDI is fully reduced resulting in a dramatic increase in mass and an increase in Coomassie blue staining (Figure 3A, lanes 1 and 2). If PDI is oxidised, then additional faster migrating species should be apparent. When PDI was incubated alone, no additional faster migrating species were seen after 60 min (Figure 3A, lanes 2 and 3). However, on addition of Ero1α, faster migrating, oxidised forms of PDI were apparent both in the absence and presence of GSH (Figure 3A, lanes 4–7). Several different mPEG-modified forms were apparent, suggesting that intermediate oxidation states of PDI were present even after 60 min of incubation with Ero1α. The most prominent oxidised species co-migrated with fully reduced and mPEG-modified ΔS2 mutant PDI (Figure 3A, lane 9), which migrates with a distinct mobility to the reduced ΔS1 mutant (lane 8) as described earlier (Appenzeller-Herzog and Ellgaard, 2008). Oxidation of the a′ domain would prevent mPEG modification of this active site, so the mobility of the protein would be identical to the reduced ΔS2 mutant. This result further supports the idea that the a′ domain is preferentially oxidised by Ero1α. Unlike the situation with thioredoxin, PDI was not quantitatively oxidised by Ero1α with the fully reduced form present even after 60 min.
Figure 3.

Ero1α oxidises PDI but the long-range disulphides in Ero1α are only partially reduced. (A) The oxidation of PDI by Ero1α was followed by the change in redox state of reduced PDI as judged by the access of free thiols to alkylation by mPEG-mal. Reduced PDI (100 μM) was incubated in the absence (lanes 2 and 3) or presence (lanes 4–7) of Ero1α and in the absence (lanes 2–5) or presence (lanes 6 and 7) of GSH (10 mM). Reactions proceeded for 5 or 60 min as indicated before being terminated by the addition of TCA followed by alkylation with mPEG-mal. Protein was separated by reducing SDS–PAGE and visualised by Coomassie staining. An untreated and mPEG-mal-treated reduced PDI sample (lanes 1 and 2) and an mPEG-mal-treated sample of reduced ΔS1 and ΔS2 mutants of PDI (lanes 8 and 9) are shown to indicate the mobility shift on mPEG-mal modification of reduced PDI. (B) The redox state of Ero1α was assayed during the reaction with PDI either in the absence (lanes 1–4) or presence (lanes 5–7) of GSH (10 mM). Reactions were quenched with NEM and the redox state of Ero1α was visualised by non-reducing SDS–PAGE followed by western blotting with an anti-Ero1α antibody. Thioredoxin-reduced Ero1α was loaded in lane 8 for comparison. (C) The redox state of Ero1α was determined after incubation (15 min) with increasing concentrations of GSH (titrated to pH 7.5) as indicated. Reactions were quenched with NEM and the redox state of Ero1α was visualised by non-reducing SDS–PAGE and silver staining. The experiments in (A–C) were repeated three times with similar results obtained.
To determine whether PDI can reduce the regulatory disulphide within Ero1α, we determined the redox state of Ero1α during its reaction with PDI both in the presence and absence of GSH (Figure 3B). In the absence of GSH, PDI reduces only a fraction of the available Ero1α in the first 10 min, which can then become reoxidised (Figure 3B, lanes 1–4). This result is consistent with the oxygen consumption assay and supports the idea that PDI can partially reduce the long-range disulphides in Ero1α. In the presence of GSH, PDI again reduces only a fraction of Ero1α and this fraction remains reduced over the 60 min (Figure 3B, lanes 5–7). If we assume that the long-range disulphides are regulating Ero1α activity (see later), then PDI is no more able to activate Ero1α in the presence of GSH than in its absence. However, in the presence of GSH, PDI can activate sufficient Ero1α to maintain activity, presumably because GSH can continually reduce PDI. We also determined whether GSH itself can reduce the regulatory disulphide in Ero1α. At concentrations of GSH up to 10 mM, there was minimal reduction of Ero1α (Figure 3C) and the regulatory disulphides were only fully reduced at concentrations above 100 mM. Hence, GSH itself cannot activate Ero1α at physiological concentration possibly explaining why this low molecular weight thiol is a poor substrate for Ero1α.
Identification of regulatory disulphides within Ero1
Human Ero1α contains 15 cysteine residues, of which four are within two active sites (Bertoli et al, 2004). Previous mutagenesis studies have identified the active site disulphides to be between cys394–397 and cys94–99 with cys85, cys391 and cys131 affecting the redox state of Ero1α at steady state when transiently expressed in mammalian cells (Bertoli et al, 2004). The regulatory disulphides within Ero1p form between cys90–349 and cys150–295 (Sevier et al, 2007). In human Ero1α, the first of these disulphides is conserved in the primary amino-acid sequence so it was assumed that cys85–391 will form one regulatory disulphide. However, additional regulatory disulphide(s) cannot be predicted from comparison of the yeast and human sequences. Hence, to identify the cysteine residues involved, we converted individual cysteines to alanine and determined whether these mutations result in a change in the long-range disulphides formed in recombinant Ero1α. The first cluster of cysteine residues (cys35, 37, 46 and 48) were not analysed as the corresponding region of the protein in Ero1p can be deleted without any effect on regulation (Sevier et al, 2007). Of all the remaining non-active site cysteine mutants, only C391A and C85/391A did not form soluble protein when expressed in E. coli. In addition, the C85A mutant gave a poor yield and was intrinsically unstable on storage, restricting the analysis to its redox status on non-reducing gels. When the cysteine mutant proteins were analysed on non-reducing gels and their mobility was compared with that of oxidised and reduced wild-type proteins, C85A, C94A, C131A and to a lesser effect C104A showed additional bands migrating with an intermediate mobility between the reduced and oxidised wild-type proteins (Figure 4A). The fact that the single cysteine mutants gave rise to two bands indicates that alternate cysteines may be able to form disulphides to generate new long-range disulphides. The shift in mobility with C85A was expected as cys85 was predicted to form a long-range disulphide with cys391, so the second regulatory disulphide was hypothesised to be between cys131, 94 or possibly 104. A double mutation C94/99A ran as a single band with a mobility corresponding to the slowest band in the single mutants C94A and C131A (Figure 4A, lane 12). These results suggest that cys94 is the partner for cys131 and that the presence of two bands with the C94A mutant could be due to cys99 behaving as an alternate partner for cys131. In addition, a double mutation C85/131A ran even slower through the gel (Figure 4A, lane 11), indicating that this mutant was now unable to form two long-range disulphides. The slight difference in mobility between C85/131A and the fully reduced wild-type protein is likely to be due to other disulphides still present in the double mutant. The results obtained from the gel shift assay have recently been confirmed independently (Appenzeller-Herzog et al, 2008). They identified disulphide bonds between cys85–391 and cys94–131 by mass spectrometry analysis of Ero1α purified from human cells. The data, however, do not rule out the possibility of further disulphides involved in the modulation of Ero1α activity.
Figure 4.

Identification of the cysteine residues involved in the formation of long-range disulphides in Ero1α. (A) Reduced (lanes 10 and 14) and non-reduced (lanes 1 and 13) wild-type Ero1α were separated by SDS–PAGE along with non-reduced cysteine mutants of Ero1α as indicated. Protein was visualised following SDS–PAGE and Coomassie staining. (B) Schematic illustration comparing the disulphide bonds within human Ero1α and yeast Ero1p that are either active site disulphides ( ) or regulatory disulphides (
) or regulatory disulphides ( ). Known disulphide pairings are as indicated as they exist in the inactive state of the protein. Note that in the human protein cys94 and cys99 are engaged in disulphide pairings with cys131 and cys104, respectively, whereas in the active state these would form a disulphide with each other.
). Known disulphide pairings are as indicated as they exist in the inactive state of the protein. Note that in the human protein cys94 and cys99 are engaged in disulphide pairings with cys131 and cys104, respectively, whereas in the active state these would form a disulphide with each other.
We also assessed whether the cysteine mutants gave the same changes in tryptophan/tyrosine fluorescence when reduced, as the wild-type protein. Both the C131A and the C85/131A showed no increase in fluorescence at 345 nm (Figure 5A, panels 2 and 3) on reduction with 12.5 mM DTT, indicating that there were no major conformational changes to the micromolecular environment of the tryptophan/tyrosine residues on DTT treatment. However, the C104A mutant did show a change in fluorescence on reduction, very similar to the wild-type protein (Figure 5A, panel 1), indicating that cys104 does not form a disulphide bond that affects tryptophan/tyrosine fluorescence. Taken together, these results support the idea that cys94 and cys131 form a disulphide bond that when broken is sufficient to cause the change in conformation giving rise to an increase in tryptophan/tyrosine fluorescence.
Figure 5.

Ero1α long-range disulphides regulate conformation changes and enzymatic activity. (A) The fluorescence emission spectra of reduced (grey line) or non-reduced (black line) Ero1α mutants C104A (1), C131A (2) and C85/131A (3) were analysed following excitation at 280 nm. (B) Oxygen consumption was assayed with PDI (100 μM) in the presence of 10 mM GSH and the Ero1α mutants (2 μM) C131A (1), C85/131A (2) or C104/131 (3). These are representative profiles from three separate experiments. The activity of wild-type Ero1α towards PDI is also included in each graph as indicated.
To determine whether the long-range disulphides regulated enzymatic activity, we carried out enzyme assays using thioredoxin and PDI as substrates. Only the C94/99A double mutant and the C99A mutant showed a total lack of activity towards thioredoxin (Supplementary Figure S3). Interestingly, C94A did have some weak activity towards thioredoxin consistent with previous data, which suggest that this mutant is weakly active in an in vivo folding assay and can complement the yeast ero1-1 mutant (Bertoli et al, 2004). All the cysteine mutants outside the active site were enzymatically active, indicating that they had folded correctly. However, the thioredoxin assay could not distinguish whether the mutants had an enhanced activity. To further evaluate the potential regulatory disulphides, we determined the activity of C131A and C85/131A towards PDI in the presence of GSH using the oxygen consumption assay (Figure 5B). C131A showed a clear difference in kinetics when compared with wild type (Figure 5B, panel 1) lacking both a lag phase and having a greater rate of oxygen turnover (about twice that of wild type). In addition, the activity of the C85/131A towards PDI was significantly quicker than wild-type Ero1α (Figure 5B, panel 2). Our conclusion from these data and the previous work on Ero1p was that the long-range disulphides cys85–391 and cys94–131 are regulatory as their reduction results in a cumulative increase in the activity of Ero1α towards PDI.
The fact that cys94 forms a regulatory disulphide with cys131 means that in its inactive state Ero1α, cys99 and cys104 could potentially form a disulphide. Our gel shift data with the C104A mutant suggest that breaking this potential disulphide could destabilise the cys94–cys131 disulphide. In addition, the activity of the C131A mutant protein could also be modulated by the formation of a cys99–cys104 disulphide. To test this possibility, we made a further Ero1α mutation, C104/131A. When the activity of this mutant was tested in the oxygen electrode assay, we now obtained even faster kinetics than the C85/131A mutant (Figure 5B, panel 3), suggesting that a disulphide does indeed exist between cys99 and cys104, which can also modulate Ero1α activity. In addition, we now saw almost complete consumption of oxygen during the assay, which contrasts with the wild-type protein and the C85/131A mutant (Figure 5B). These results would suggest that the lack of complete consumption of oxygen with the wild-type protein and C85/131A mutant is due to the formation of a disulphide between cys104 and cys99, which is not efficiently reduced even in the presence of GSH. A schematic diagram illustrating the positions of the active site and regulatory disulphides in Ero1α and Ero1p is shown in Figure 4B.
Determining the reduction potential of the regulatory disulphide bonds
To ascertain whether the lack of efficient reduction of the regulatory disulphides in Ero1α by PDI could be attributed to their relative stability, we set out to determine their reduction potential. We used thioredoxin as a redox couple as it efficiently reduces Ero1α. In addition, we used the C99A mutant of Ero1α as it is inactive towards thioredoxin (Supplementary Figure S3) ensuring no changes to thioredoxin redox state due to enzymatic activity. In addition, this mutant will not contain the potential cys99–104 disulphide. Ero1α was incubated with various ratios of reduced/oxidised thioredoxin and the redox state was evaluated by non-reducing gel analysis. A clear transition from reduced to oxidised Ero1α was seen as the ratio of reduced/oxidised thioredoxin decreased (Figure 6A). The equilibrium constant (_K_eq) was calculated to be 2.10±0.64 (average and standard deviation from three separate measurements) (Figure 6B). Only a single transition in the redox state of Ero1α was observed, indicating that the breaking of the regulatory disulphides occurs at a similar reduction potential. Assuming that two molecules of thioredoxin are required to reduce the cys85–391 and cys94–131 disulphides, the calculated midpoint reduction potential would be −275 mV. The stability of the disulphide suggests it would be oxidised within the ER and in part explains its partial reduction by PDI.
Figure 6.

Measuring the midpoint reduction potential of the regulatory disulphides in Ero1α. (A) The enzymatically inactive C99A mutant (1 μM) was equilibrated with various ratios of reduced/oxidised thioredoxin before alkylation with NEM and separation by non-reducing SDS–PAGE. Protein was visualised by Coomassie staining and the fraction of reduced Ero1α was quantified by densitometry. (B) Values for the fraction of reduced Ero1α were plotted against the ratio of reduced/oxidised thioredoxin. Plots were interpreted by equation (1), and _K_eq values were determined as described. The experiment was repeated three times and the average _K_eq was used to calculate the reduction potential.
Discussion
Previous work has demonstrated that the enzymatic activity of Ero1p can be regulated by the breaking and reforming of two disulphide bonds (Sevier et al, 2007). Here, we show that the human enzyme, Ero1α, is regulated in a similar manner but, unlike the yeast enzyme, forms regulatory disulphides between non-catalytic cysteines and cysteines located within the first active site. The formation of such bonds would inevitably inhibit enzyme activity as the first active site is required to accept electrons from the substrate protein (Sevier and Kaiser, 2006). The enzyme could only become active on reduction of these disulphides, either by the substrate or by an additional regulatory protein.
The initial identification of regulatory disulphides within Ero1p demonstrated that they were the same disulphides that dramatically alter the hydrodynamic volume of the denatured protein (Sevier et al, 2007). We also show that the same is true for Ero1α and additionally demonstrate that breaking these disulphides also alters the conformation of the native protein. The reduction of the disulphides does not grossly change the secondary structure but does alter the environment around specific aromatic residues thereby altering their fluorescence. The yeast structure has been solved (Gross et al, 2004) and revealed a 10 helix core that contains the bound FAD and second active site disulphide. The first active site disulphide (or shuttle disulphide), which accepts electrons from substrates, is located on a non-helical loop that forms a ‘cap' over the helical core. Sevier et al (2007) speculate that ‘the regulatory disulphides may tether a non-helical cap, with the helical core of Ero1p'. The identification of cys94–131 as a regulatory disulphide is, therefore, somewhat unexpected as both residues would be expected to be in the non-helical cap as judged by homology modelling (data not shown). However, it is difficult to make direct comparisons between the yeast and human enzyme in the absence of structural data for the latter, as homology modelling between the human and yeast proteins also predicts that cys94 and cys131 are not close enough to form a disulphide.
The presence of an additional cysteine residue (cys104) around the first active site in Ero1α that is not present in the yeast enzyme suggested an additional mechanism both for Ero1α activity and for its regulation. The C94A mutant would be predicted to be inactive if the cys94–99 disulphide is the first active site. However, we and others (Bertoli et al, 2004) have shown that this mutant still has some weak activity, suggesting that, in the absence of cys94, cys99 might form an alternative disulphide with cys104, which is itself able to accept electrons, albeit at a much reduced rate, from substrate proteins. In addition, we noticed a partial reduction of one of the long-range disulphides in the C104A mutant, which could be due to cys99 being able to resolve the cys94–131 disulphide if it is not paired with cys104. In addition, when recombinant Ero1α was treated with _N_-ethylmaleimide (NEM), digested with trypsin and the peptides were analysed by MALDI mass spectrometry, we consistently observed a peptide with an internal disulphide between cys99 and cys104 (results not shown). The near complete oxygen consumption seen with this mutant contrasts with a lack of complete oxygen consumption by the wild-type protein or the C85/131A mutant. This result suggests that the formation of a cys99–104 disulphide may lead to the inhibition of Ero1α in the in vitro assay. The ability of Ero1α to exchange internal disulphide bonds may not be restricted to the two active sites, indeed the ability to reform the regulatory disulphides in the absence of substrate demonstrates that several disulphide exchange reactions are not only possible but are also necessary for the tight regulation of this enzyme.
We have extended the previous studies on Ero1p to demonstrate that the regulatory disulphides within Ero1α are relatively stable, which means that only molecules capable of forming an equally stable disulphide would be able to reduce these bonds efficiently. Our activity data support this finding; thioredoxin, which has a relatively stable active site disulphide, can readily reduce the regulatory disulphides in Ero1α, whereas PDI with less stable active site disulphide can only reduce a small fraction of Ero1α. The reduction potential of PDI active sites has been calculated to be approximately −180 mV (Lundstrom and Holmgren, 1993), which means that in the absence of additional factors PDI would need to be over 99% reduced to be able to reduce the regulatory disulphide in Ero1α. Once the regulatory disulphides are broken, the enzyme is active until the regulatory disulphides reform. The distinct lag between oxidation of thioredoxin and the reformation of the regulatory disulphides in Ero1α suggests that the rate of reformation of the inhibitory disulphide is slower than the rate of oxidation of substrate. Hence, once the enzyme is activated, it would be able to complete several oxidation cycles prior to reformation of the regulatory disulphide. Although PDI may be able to activate Ero1α in our in vitro assay due to the starting material being reduced, it is unlikely to be true in vivo as PDI will be at least partially oxidised. This argues for the presence of additional factors regulating the activity of Ero1α in the ER lumen. Alternatively, PDI binding to Ero1α may lead to conformational changes resulting in an alteration of the reduction potentials of the disulphides within Ero1α and PDI.
The ability of Ero1p to oxidise the individual active sites of yeast PDI has been shown to be non-equivalent (Kulp et al, 2006). Ero1p preferentially oxidises the a′ domain of Pdi1p. We also show that the a′ domain of human PDI is oxidised almost exclusively by Ero1α, with little or no oxidation of the a domain. Such a distinct specificity could result in distinct activities of the active sites of PDI towards its substrates; the a domain acting as an isomerase or reductase with the a′ domain acting as an oxidase as suggested for Pdi1p (Kulp et al, 2006).
Our in vitro data demonstrate that although PDI is not fully oxidised by Ero1α, the presence of GSH can prolong the reaction. The mechanism underlying the ability of GSH to facilitate the activity of Ero1α towards PDI is difficult to explain as GSH itself does not seem to be able to reduce the regulatory disulphides and is a poor substrate for Ero1α even though it has a lower reduction potential (−240 mV) than PDI. It is likely that GSH will reduce the active site disulphides in PDI (Gilbert, 1989), so one possibility is that PDI is reduced in the presence of GSH allowing further oxidation by Ero1α, thereby prolonging oxygen consumption. Indeed, as only one active site in PDI is efficiently oxidised by Ero1α and there is 100 μM PDI in the reaction, the fact that ∼160 μM oxygen is consumed would suggest that the PDI is being reduced by GSH. Such a system may well operate in vivo with the transfer of electrons between GSH and oxygen through PDI and Ero1α ultimately leading to the formation of GSSG and H2O2. In support of such a situation is the fact that the depletion of GSH from yeast cells allows the temperature-sensitive ero1-1 mutant to survive at the non-permissive temperature and, in addition, the steady-state level of GSSG is regenerated more quickly in cells that are overexpressing Ero1p (Cuozzo and Kaiser, 1999). Given the differences in the reduction potentials of the regulatory disulphide in Ero1α and the active site disulphides in PDI, one would predict that, similar to in vitro, only a small fraction of PDI would be fully oxidised in vivo. Recent measurements of the redox state of PDI estimate that only 16% of PDI present in mammalian cells is fully oxidised at both active sites (Appenzeller-Herzog and Ellgaard, 2008). It is interesting to note that yeast PDI is predominantly oxidised in vivo (Xiao et al, 2004), suggesting clear differences between the regulation of Ero1p and Ero1α in terms of their ability to be activated and to oxidise PDI.
The concept that GSH is required to reduce PDI thereby maintaining Ero1α activity does have problems when we consider the situation within the lumen of the ER. If this reaction were to go unchecked, one would establish a ‘futile cycle', which only requires GSH, PDI and Ero1α to continue (Thorpe and Coppock, 2007). However, our in vitro data show that Ero1α activity as measured by oxygen consumption is inhibited at later time points even in the presence of high concentrations of GSH. As mentioned above, this inhibition could be due to the formation of an additional disulphide (cys99–104). Alternatively, an additional protein may be required to reduce the regulatory disulphide in Ero1α; given the low turnover rate of the enzyme in vitro, this may well be the case. Clearly more work needs to be done to address these issues, but the fact that the regulatory disulphide in Ero1α is relatively stable suggests that this enzyme may have several layers of regulation specifically to prevent ‘futile cycling' with GSH.
Materials and methods
Plasmid construction and mutagenesis of Ero1
The Ero1α signal sequence was replaced with a hexahistidine tag and the sequence coding for the full-length mature protein was inserted into a pGEX-4T-3 vector (GE Healthcare) downstream from the GST coding sequence to create a GST–His–Ero1α fusion. Cysteine to alanine mutations for Ero1α were created using Quickchange site-directed mutagenesis with Pfu DNA polymerase (Stratagene). All constructs were verified by sequencing.
Expression and purification of Ero1
Ero1α was expressed in the Origami B (DE3)pLysS strain of E. coli. Cells were grown in LB broth containing 100 μg/ml ampicillin, 30 μg/ml chloramphenicol, 10 μg/ml tetracycline and 10 μg/ml kanamycin at 37°C for 4–6 h until an optical density of 1.0 was reached. An additional 1/5 volume of LB and 20 μg/ml ampicillin was then added along with 10 μM FAD and 0.5 mM IPTG to induce the expression of Ero1α and the culture was grown at 16°C for 24 h.
Pelleted cells were resuspended in 20 ml ice-cold lysis buffer (1 mg/ml hen egg white lysozyme, 20 μg/ml DNase I (Roche), 2.5 μg/ml RNase A, two complete EDTA-free protease inhibitor cocktail tablets (Roche), 1 mM EDTA and 0.1% (v/v) Triton X-100 in phosphate-buffered saline (PBS)) per 1 l bacterial culture. The cells were then lysed by freeze–thawing in liquid nitrogen and the lysates were clarified by centrifugation at 12 760 g for 15 min. The soluble fraction was incubated with 1 ml of equilibrated glutathione Sepharose beads (GE Healthcare) at 4°C for 4 h. The beads were washed three times with 10 bed volumes of 1 × PBS before being mixed with 1 ml of PBS containing 100 U of thrombin (GE Healthcare). Thrombin cleavage was left to proceed overnight at 4°C with gentle agitation. The cleaved protein was buffer exchanged using a PD10 column (GE Healthcare) into Ni-NTA-binding buffer (50 mm NaH2PO4 buffer, pH 8.0, 300 mM NaCl and 2.5 mM imidazole).
Ni-NTA beads (0.5 ml bed volume) (Qiagen) were added to Ero1α in Ni-NTA-binding buffer and binding was carried out for 90 min at 4°C. The beads were washed twice with 10 ml of Ni-NTA wash buffer (50 mM NaH2PO4 buffer, pH 8.0, 300 mM NaCl and 20 mM imidazole) before being eluted in the same buffer containing 250 mM imidazole. The Ero1α was then buffer exchanged into 50 mm Tris–HCl buffer, pH 7.5. Flavoprotein concentrations were determined using the experimentally determined bound FAD absorption coefficient of 12.9 mM−1 cm−1 at 454 nm (see Supplementary data).
Expression and purification of thioredoxin and PDI
A construct expressing His-tagged E. coli thioredoxin 1 (Trx1) in a pET28 vector was obtained from Deborah Fass (Rehovot, Israel). Trx1 was expressed, purified and reduced as described earlier (Gross et al, 2006). The concentration of reduced Trx1 was quantified using the Ellman's assay (Ellman, 1959). Constructs expressing His-tagged PDI and the ΔS1 and ΔS2 active site mutants were obtained from Lloyd Ruddock (Oulu, Finland). PDI was expressed and purified in the same manner as Trx1. PDI was reduced for 1 h with 10 mm DTT and then buffer exchanged with a PD10 column into 50 mM Tris–HCl buffer, pH 7.5. PDI was quantified using the 280 nm PDI absorption extinction coefficient of 47.12 mM−1 cm−1 (Gill and von Hippel, 1989).
Electrophoresis and western blotting
Samples for SDS–PAGE were resuspended in SDS–PAGE sample buffer (25 mM Tris–HCl buffer, pH 6.8, 2% w/v SDS, 20% v/v glycerol and 0.004% w/v bromophenol blue). Reduced samples contained 50 mM DTT. Gels were either transferred to a nitrocellulose membrane for western blotting or stained with Coomassie blue or silver stain. For western blotting, gels were transferred to nitrocellulose and blocked using 3% milk in TBST (10 mM Tris–HCl buffer, 150 mM NaCl, pH 7.5, 0.1% Tween-20). A rabbit polyclonal antibody for Ero1α (from Adam Benham, Durham, UK) was used as a primary antibody at a dilution of 1:500 in 1% milk. Products were visualised using a chemiluminescent substrate (Perbio) and Fuji Super RX film (Fujifilm).
Oxygen electrode assays with PDI
Oxygen consumption was measured using a Clark-type oxygen electrode (Rank Brothers, Cambridge, UK). The chamber was covered with foil to prevent photoreduction of flavins. Assays with PDI were carried out in 500 μl using 100 μM reduced PDI and 2 μM Ero1α in 50 mM Tris–HCl buffer, pH 7.5 with or without 10 mM GSH.
Gel-based thioredoxin and PDI oxidation assays
Reactions with Trx1 were initiated by adding 0.5 μM Ero1α to 100 μM reduced Trx1 in 50 mM Tris–HCl, pH 7.5. At the indicated times, reactions were quenched for 30 min at room temperature with an equal amount of SDS–PAGE sample buffer containing either 10 mM AMS (Invitrogen) or 50 mM NEM. Reactions quenched with AMS were separated on 15% polyacrylamide gels and were stained to visualise the redox state of Trx1. Samples quenched with NEM were separated on 10% polyacrylamide gels and silver stained to visualise the redox status of Ero1α. Reactions with PDI were carried out using 100 μM reduced PDI and 2 μM Ero1α in 50 mM Tris–HCl buffer, pH 7.5 with 10 mM GSH where stated. Reactions were terminated with 10% (v/v) trichloroacetic acid (TCA). Samples were incubated on ice for 20 min and then centrifuged at 12 760 g for 10 min. Supernatants were removed and protein pellets were washed twice with ice-cold acetone before finally resuspending the pellets in SDS–PAGE buffer containing mPEG for 30 min (0.2 M Tris–HCl buffer, pH 6.8, 6% w/v SDS, 20% v/v glycerol, 0.004% w/v bromophenol blue and 5 mM mPEG-mal 5000 (Layson Bio, US)). Reactions were visualised by SDS–PAGE and Coomassie blue staining.
Redox state determination of Ero1 with PDI and GSH
Reactions with PDI were carried out in 50 mM Tris–HCl buffer, pH 7.5 with 100 μM reduced PDI and 2 μM Ero1α with or without 10 mM GSH. At the indicated times, an equal amount of SDS–PAGE sample buffer containing 50 mM NEM was added to quench the reactions. These reactions were diluted 1 in 40 into SDS–PAGE sample buffer containing 10 mM NEM to lower the amount of PDI on the gel that otherwise distorts the running of the Ero1α. The reactions were resolved through non-reducing SDS–PAGE followed by western blotting with the anti-Ero1α antibody. For reactions of Ero1α with GSH only, 2 μM Ero1α was incubated with varying concentrations of GSH for 15 min in a 50 mM KPO4 buffer, pH 7.5. Each GSH solution was neutralised to pH 7.5. Reactions were quenched and precipitated with the addition of 10% v/v TCA before finally resuspending the pellets in 20 μl SDS–PAGE sample buffer containing 25 mM NEM.
Redox potential determination of Ero1 regulatory disulphides
The C99A mutant was used to determine the redox potential of the regulatory disulphides in Ero1-Lα. C99A Ero1α (1 μM) was left to equilibrate with 100 μM Trx1 with varying ratios of reduced/oxidised Trx1 for 1 h 30 min. As a control for air oxidation, we incubated reduced thioredoxin in buffer for 1 h 30 min. No change in the oxidation state of thioredoxin was found after this time as judged by assay with Ellman's reagent (Ellman, 1959) and less than 1% change was seen as judged by rpHPLC (data not shown). Such a low level of air oxidation does not affect the calculation of the equilibrium constant. Oxidised Trx1 was prepared by incubating Trx1 for 1 h with 10 mM GSSG and reduced Trx1 was prepared by incubating with 10 mM DTT before buffer exchange using Micron centrifugal YM10 filters (500 μl concentrated to 10 μl three times to give a final concentration of <80 nM DTT or GSSG). The concentrations of reduced and oxidised Trx1 were determined using the Bradford assay (Bradford, 1976). Reactions were quenched with an equal amount of SDS–PAGE buffer containing 50 mM NEM. The redox state of Ero1α was determined by non-reducing SDS–PAGE. The amount of reduced Ero1α in each reaction was determined by densitometry and expressed as a fraction of total Ero1α. The equilibrium constant _K_eq for the redox reaction was calculated according to equation (1) assuming two molecules of thioredoxin are required to reduce the two long-range disulphides in Ero1α.
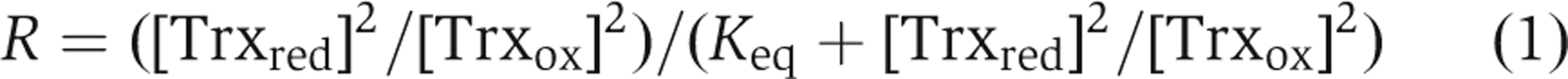
where R is the fraction of reduced Ero1α. The standard reduction potential for the Ero1α regulatory disulphides could then be derived from the Nernst equation [_E_°′(Ero1-Lα)=_E_°′(Trx)–(RT/nF) In(_K_eq)] using the calculated _K_eq from equation (1), _T_=25°C, _n_=4 and the standard reduction potential of Trx1=−270 mV as determined earlier (Krause and Holmgren, 1991).
Supplementary Material
Supplementary Figures S1–S3
Acknowledgments
We thank Adam Benham, Lloyd Ruddock and Deborah Fass who provided reagents for this project. We also thank Lars Ellgaard for sharing unpublished work. The multi-angle light scattering analysis was carried out by Tom Jowitt (University of Manchester) and the mass spectrometry by David Knight (University of Manchester). This study was funded by grants from The Wellcome Trust (ref: 74081 and 75335).
References
- Appenzeller-Herzog C, Ellgaard L (2008) In vivo reduction–oxidation state of protein disulphide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal 10: 55–64 [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Christensen B, Sørensen ES, Ellgaard L (2008) A novel disulphide switch mechanism in Ero1α balances ER oxidation in human cells. EMBO J (advance online publication, 2 October 2008; doi:10.1038/emboj.2008.202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham AM, Cabibbo A, Fassio A, Bulleid N, Sitia R, Braakman I (2000) The CXXCXXC motif determines the folding, structure and stability of human Ero1-Lalpha. EMBO J 19: 4493–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli G, Simmen T, Anelli T, Molteni SN, Fesce R, Sitia R (2004) Two conserved cysteine triads in human Ero1alpha cooperate for efficient disulphide bond formation in the endoplasmic reticulum. J Biol Chem 279: 30047–30052 [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72: 248–254 [DOI] [PubMed] [Google Scholar]
- Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, Sitia R (2000) ERO1-L, a human protein that favors disulphide bond formation in the endoplasmic reticulum. J Biol Chem 275: 4827–4833 [DOI] [PubMed] [Google Scholar]
- Chakravarthi S, Bulleid NJ (2004) Glutathione is required to regulate the formation of native disulphide bonds within proteins entering the secretory pathway. J Biol Chem 279: 39872–39879 [DOI] [PubMed] [Google Scholar]
- Chakravarthi S, Jessop CE, Bulleid NJ (2006) The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep 7: 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuozzo JW, Kaiser CA (1999) Competition between glutathione and protein thiols for disulphide-bond formation. Nat Cell Biol 1: 130–135 [DOI] [PubMed] [Google Scholar]
- Darby NJ, Creighton TE (1995) Characterization of the active site cysteine residues of the thioredoxin-like domains of protein disulphide isomerase. Biochemistry 34: 16770–16780 [DOI] [PubMed] [Google Scholar]
- Dias-Gunasekara S, Gubbens J, van Lith M, Dunne C, Williams JA, Kataky R, Scoones D, Lapthorn A, Bulleid NJ, Benham AM (2005) Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1beta. J Biol Chem 280: 33066–33075 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep 6: 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82: 70–77 [DOI] [PubMed] [Google Scholar]
- Frand AR, Kaiser CA (1998) The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell 1: 161–170 [DOI] [PubMed] [Google Scholar]
- Freedman RB (1995) The formation of protein disulphide bonds. Curr Opin Struct Biol 5: 85–91 [DOI] [PubMed] [Google Scholar]
- Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A (2003) The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem 270: 2228–2235 [DOI] [PubMed] [Google Scholar]
- Gilbert HF (1989) Catalysis of thiol/disulphide exchange: single-turnover reduction of protein disulphide-isomerase by glutathione and catalysis of peptide disulphide reduction. Biochemistry 28: 7298–7305 [DOI] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182: 319–326 [DOI] [PubMed] [Google Scholar]
- Gross E, Kastner DB, Kaiser CA, Fass D (2004) Structure of Ero1p, source of disulphide bonds for oxidative protein folding in the cell. Cell 117: 601–610 [DOI] [PubMed] [Google Scholar]
- Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, Fass D (2006) Generating disulphides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA 103: 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansens A, van Duijn E, Braakman I (2002) Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science 298: 2401–2403 [DOI] [PubMed] [Google Scholar]
- Jessop CE, Bulleid NJ (2004) Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J Biol Chem 279: 55341–55347 [DOI] [PubMed] [Google Scholar]
- Jessop CE, Chakravarthi S, Garbi N, Hammerling GJ, Lovell S, Bulleid NJ (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J 26: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause G, Holmgren A (1991) Substitution of the conserved tryptophan 31 in Escherichia coli thioredoxin by site-directed mutagenesis and structure–function analysis. J Biol Chem 266: 4056–4066 [PubMed] [Google Scholar]
- Kulp MS, Frickel EM, Ellgaard L, Weissman JS (2006) Domain architecture of protein-disulphide isomerase facilitates its dual role as an oxidase and an isomerase in Ero1p-mediated disulphide formation. J Biol Chem 281: 876–884 [DOI] [PubMed] [Google Scholar]
- Lundstrom J, Holmgren A (1993) Determination of the reduction–oxidation potential of the thioredoxin-like domains of protein disulphide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 32: 6649–6655 [DOI] [PubMed] [Google Scholar]
- Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R (2001) Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J 20: 6288–6296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, Cabibbo A, Sitia R (2000) Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J Biol Chem 275: 23685–23692 [DOI] [PubMed] [Google Scholar]
- Pollard MG, Travers KJ, Weissman JS (1998) Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol Cell 1: 171–182 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2002) Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol 3: 836–847 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2006) Disulphide transfer between two conserved cysteine pairs imparts selectivity to protein oxidation by Ero1. Mol Biol Cell 17: 2256–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta 1783: 549–556 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA (2007) Modulation of cellular disulphide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129: 333–344 [DOI] [PubMed] [Google Scholar]
- Thorpe C, Coppock DL (2007) Generating disulphides in multicellular organisms: emerging roles for a new flavoprotein family. J Biol Chem 282: 13929–13933 [DOI] [PubMed] [Google Scholar]
- Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS (2000) Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290: 1571–1574 [DOI] [PubMed] [Google Scholar]
- Xiao R, Wilkinson B, Solovyov A, Winther JR, Holmgren A, Lundstrom-Ljung J, Gilbert HF (2004) The contributions of protein disulphide isomerase and its homologues to oxidative protein folding in the yeast endoplasmic reticulum. J Biol Chem 279: 49780–49786 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–S3