Mechanism of alcohol-induced oxidative stress and neuronal injury (original) (raw)
. Author manuscript; available in PMC: 2009 Dec 1.
Abstract
Neuro-cognitive deficits, neuronal injury, and neurodegeneration are well documented in alcoholics, yet the underlying mechanisms remain elusive. Oxidative damage of mitochondria and cellular proteins intertwines with the progression of neuroinflammation and neurological disorders initiated by alcohol abuse. Here, we present the evidence that metabolism of ethanol in primary human neurons by alcohol dehydrogenase (ADH) or cytochrome P450-2E1 (CYP2E1) generates reactive oxygen species (ROS) and nitric oxide (NO) via induction of NADPH/xanthine oxidase (NOX/XOX) and nitric oxide synthase (NOS) in human neurons. The acetaldehyde-mediated increase in NOX, XOX, or NOS activity is regulated as a transcriptional rather than a translational process. Marked increase in the lipid peroxidation product (4-hydroxynonenal) and enhanced ROS generation coincides with decreased neuronal viability and diminished expression of neuronal marker (neurofilaments). Novel quantitative methods of ROS and NO detection help dissect the mechanisms of alcohol-induced neurodegeneration. Uncovering the basic mechanisms of oxidative neuronal injury will serve as the basis for development of new therapies.
Keywords: Human neurons, Alcohol dehydrogenase, Cytochrome P450-2E1, NADPH/xanthine oxidase, Neuronal nitric oxide synthase, Free radicals
Introduction
Millions of alcoholics exhibit neuro-cognitive deficits and neuronal injury associated with neuronal degeneration [1-3]. Although oxidative stress and mitochondrial damage are implicated in tissue injury, the underlying mechanisms of alcohol-induced neurological disorders remain elusive. Oxidative stress-induced mitochondrial damage is involved in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [4,5]. Increased microglia prior to development of brain atrophy [6], enhanced neuroinflammation, and oxidative damage [7] after chronic ethanol (EtOH) administration in animal models support the putative mechanism for neurodegeneration in alcoholics. Postmortem examination of brain tissues from alcoholics shows cerebral edema, neuronal loss, and blood-brain barrier (BBB) dysfunction [8], associating with the increased risk of hemorrhagic and ischemic stroke among alcoholics [9-11]. We observed that alcohol-induced oxidative stress causes BBB dysfunction [12,13] via an activation of myosin light chain kinase (MLCK) with subsequent phosphorylation of MLC and tight junction proteins [14], activation of inositol 1,4,5-triphosphate receptor-gated intracellular Ca2+ release [15], and activation of matrix metalloproteinases by protein tyrosine kinases [16]. Thus, oxidative stress emerges as the underlying cause of BBB damage, neuroinflammation, and neurological diseases.
It is known that during oxidative stress conditions the levels of oxidants are higher than the levels of antioxidants [17,18]. Besides mitochondrial oxidative phosphorylation, the cellular activation of NADPH/xanthine oxidase (NOX/XOX) or nitric oxide synthase and copper/iron-catalyzed Fenton-Weiss-Haber (FWH) reaction of H2O2 [19] also produce oxidants. Alcohol-induced ROS production is believed to be specific to EtOH metabolism by cytochrome P450-2E1 (CYP2E1), which produces H2O2 in addition to acetaldehyde (Ach), while alcohol dehydrogenase (ADH) mediated produces only Ach. Interaction of H2O2 with copper/iron produces ROS during EtOH oxidation by alcohol-inducible liver microsomal cytochromes P-450 enzymes [19,20]. Cederbaum and colleagues showed that metabolism of EtOH by CYP2E1 causes oxidative liver damage by increased ROS levels [21,22] and by reduction of glutathione and superoxide dismutase activity [23,24].
While oxidative stress as an underlying cause of many neurological diseases is well documented [25,26], the role of alcohol-induced oxidative stress in the CNS is largely unknown. Recent high-throughput neuroproteomic studies of protein expression profiles indicated increased levels of ammonia and ROS in the CNS of alcoholics [27]. Chronic alcohol administration resulted in microencephaly and neuronal loss in juvenile mice [28] and impacted behavior [29] through modulation of neuronal nitric oxide synthase, suggesting the involvement of oxidative stress. Protective effects of vitamin E on neuronal loss against alcohol-induced oxidative product in neonatal rat hippocampus [30] and decrease in glutathione level in fetal cortical neurons suggest the role of alcohol-mediated oxidative stress in the CNS [31].
Chronic administration of an alcohol diet enhanced CYP2E1 protein expression in rat brain tissues [32,33]. Upadhya and colleagues demonstrated the constitutive expression of CYP2E1 protein in neurons within cerebral cortex, Purkinje, or granule cell layers of cerebellum, and induction of CYP2E1 protein level in the cortex of rat or human brain after chronic ethanol consumption [34]. We demonstrated the induction of CYP2E1 activity after EtOH exposure in primary human brain endothelial cells paralleling elevated ROS production and BBB dysfunction [12,13,15]. BBB dysfunction could be important to neurological disorders in the context of FWH reaction within the endothelium and copper deficiency in the brain. Copper accumulation in brain endothelial cells increases the rate of FWH reaction and prevents the transport of essential metal ions into the brain cells (astrocytes and neurons) as observed in Menkes’ disease [35]. Although accumulation of iron in livers [36] and copper in kidneys/heart [37] has been reported in rats fed chronic alcohol diets, changes in metal ion concentrations in alcoholic brains are currently unknown. Here, we hypothesize that metabolism of EtOH by ADH and CYP2E1 generates ROS and NO in human neurons due to activation of NOX/XOX and inducible nitric oxide synthase (iNOS) by Ach. We observed that an induction of CYP2E1 activity parallels increased ROS and NO production in neurons after EtOH treatment. High levels of lipid peroxidation product, 4-hydroxynonenal, indicating cellular oxidative damage are accompanied by diminished expression of neuronal markers (neurofilaments) and enhanced neuronal death.
Materials and methods
Human neuronal isolation
Primary cortical neurons are isolated from human fetal brain tissue (elective abortus specimens) routinely obtained from our neural tissue core facility and are cultured as described [38]. The tissues are obtained in full compliance with the ethical guidelines of both the National Institutes of Health and the University of Nebraska. Briefly, we incubated dissociated tissue with 0.25% trypsin for 30 min, neutralized with 10% fetal bovine serum, and further dissociated by triturating. Resulting single-cell suspension was cultured on poly-D-lysine-coated plates in neurobasal media containing 0.5 mM glutamine, 50 μg/ml penicillin and streptomycin, and supplemental B27. Neuronal purity (>75%) was assessed by antibodies (Abs) to neuronal markers, microtubule-associated protein 2 (MAP-2, Boehringer, 1:50) and neurofilament (NF: Dako, 1:100), and astrocytic marker, glial fibrillary acidic protein, GFAP (Dako, 1:100).
Preparation of cell lysates
Prior to experiments, neurons were cultured for 14 days in 96-well plates (20,000 cells/well), 6-well plates (1 million cells/well), or T75 cm2 flask (5 million cells/flask). For all activity assays, neuronal cell pellets collected from EtOH/Ach treatment (17.5 mM EtOH or 10 μM Ach) with/without specific inhibitor for 48 h were sonicated in 50 mM Tris-HCl, pH 7.5, containing 1 mM EDTA, 150 mM NaCl, and 20% glycerol after scraping off adherent cells without proteolytic enzymes. Cytosolic proteins (10-40 μg/replicate) collected from sonicated samples at 10,000 g centrifugation were used for activity assays.
Cytotoxicity assay
Neuronal cell viability was determined by fluorescence-based Live/Dead assay (Invitrogen Corporation, Carlsbad, CA) following the manufacturer’s instructions. Briefly, primary cortical neurons were cultured for 14 days on poly-D-lysine-coated 96-well plates followed by treatment with test compounds for 48 h in culture. After two washes with phosphate-buffered saline (PBS), cells were incubated with 2 μM calcein-AM and 4 μM ethidium homodimer (EthD-1) for 20 min at room temperature. Enzymatic conversion of the cell-permeable calcein AM to the fluorescent calcein determined the live cells. Cell death was identified by increased fluorescence resulting from the entry of EthD-1 across damaged cell membranes, and binding to nucleic acids. Using a fluorescence plate reader (Molecular Devices, Sunnyvale, CA), fluorescent calcein was detected at 490 nm excitation and 515 nm emission, while fluorescent EthD-1 was detected at 528 nm excitation and 617 nm emission. The results are represented as percentage of live cells.
ADH activity
Protein lysates centrifuged at 35,000 rpm (105,000 g) for 1 h at 4 °C were used for ADH and CYP2E1 activity assays. The high-speed cytosolic fractions (supernatants) were used for ADH activity assay, while the microsomal fractions in phosphate-buffered saline were used for CYP2E1 activity assay. ADH catalytic activity was assayed by formation of reduced nicotinamide adenine dinucleotide (NADH) from NAD oxidation as described [39]. Briefly, 50 μl of cytosolic protein (100 μg protein) was added to a 430-μl reaction mixture containing 0.5 M Tris-HCl, 0.01 M dithiothreitol, and 0.5 M EtOH in 1.5 ml disposable cuvette. A 20 μl of 90 mM NAD was added to prewarmed cuvettes, and changes in optical density were read at 340 nm using the time-drive kinetics for 10 min at 30-s intervals. ADH activity was calculated from nmol of NADH formed/h/ml, and the specific activity of ADH was expressed as nanomoles per hour per milligram cellular protein.
CYP2E1 activity
CYP2E1 activity was determined by hydroxylation of _p_-nitrophenol to 4-nitrocatechol as described [12]. Briefly, a 50-μl reaction mixture containing 0.8 mM _p_-nitrophenol and 2.0 mM β-NADP (Sigma Aldrich, St. Louis, MO) was added to 50 μl of microsomal protein on ice in 1.5-ml microcentrifuge tubes. Blank tubes of identical compositions were prepared except that 20% TCA was added prior to the reaction mixture on ice. After 1 h incubation at 37 °C in a water bath shaker, addition of 30 μl of 20% TCA terminated the reactions. Samples were centrifuged for 10 min at 14,000 rpm, and to the supernatants was added 10 μl of 10 N NaOH for color development. Absorbance was read at 540 nm in the 96-well Vmax kinetic reader (Molecular Devices, Sunnyvale, CA). Using 4-nitrocatechol as internal standard, CYP2E1 activity was calculated and expressed as nanomoles per milligram protein.
ROS detection
ROS levels in neurons were detected by dichlorofluorescein diacetate assay (DCF-DA; Molecular Probes, Eugene, OR) as described [40]. Human primary neuronal cells cultured in 96-well plates (20,000 cells/well) for 14 days were loaded with DCF-DA (10 μM) for 40 min at 37 °C in 200 μl of cell culture media without phenol red in the presence or absence of specific inhibitors. After washing off the excess DCF-DA, neuronal cells were stimulated with test compounds with/without specific inhibitor for 30-120 min in an ELISA plate reader. The fluorescence intensity was then read at differential time points at excitation 488 nm and emission at 525 nm. A standard curve was generated with 6.25, 12.5, 25, 50, and 100 μM acetaldehyde. Results were expressed as mean relative fluorescence units per micromole of Ach after subtracting blank values (media alone) from samples and standard values. The effect of EtOH/Ach (17.5 mM EtOH or 10 μM Ach) on NOX/XOX-mediated ROS production was determined by the use of specific inhibitor to NOX (200 μM, apocynin) or XOX (200 μM, allopurinol). Expression of NOX/XOX protein was determined by Western blot analyses as described previously [14].
NO detection
NO levels in human neuronal culture were detected by the diaminofluorescein-2 diacetate assay (Molecular Probes) as described [41]. Removal of the acetate group from DAF-2DA by intracellular esterases produces a highly fluorescent DAF detected at excitation 488 nm and emission at 515 nm. Briefly, human neurons cultured in 96-well plates (20,000 cells/well) for 14 days were loaded with DAF-2DA (10 μM) for 40 min at 37 °C in 200 μl of cell culture media without phenol red with or without specific inhibitor. After removing the excess DCF-DA, neurons were stimulated with test compounds with/without specific inhibitor for 30-120 min, followed by differential time point fluorescence readings at excitation 488 nm and emission at 515 nm. A standard curve was generated with 1, 5, 10, 20, 50, and 100 μM SNAP (_S_-nitroso-_N_-acetylpenicillamine). Results were expressed as mean relative fluorescence units per micromole SNAP after subtracting blank values (media alone) from samples and standard values. The effect of EtOH/Ach (17.5 mM EtOH or 10 μM Ach) on iNOS induction in neurons was determined by NO production using the iNOS-specific inhibitor, L-NAME (50 μM, N _G_-nitro-L-arginine methyl ester). The novelty of NO or ROS detection assay described in the present studies is the inclusion of a positive standard curve for each assay.
Immunofiuorescent staining
Primary human neurons cultured in glass coverslips (0.4 million cells/well in 12-well plate) for 14 days were washed with PBS, dried, and fixed in absolute methanol/acetone (1:1) solution for 20 min at -20 °C. Following 0.1% Triton X lyses, neurons are blocked with 3% bovine serum albumin for 10 min at 4 °C in PBS, and then immunolabeled with antibody directed against ADH (mouse IgG, 1:100; Lifespan Bioscience, Seattle, WA) or CYP2E1 (rabbit IgG, 1:100; Sigma-Aldrich, St. Louis, MO) for 1 h at 24 °C. Secondary antibody conjugated with Alexa-488 for ADH and Alexa-520 for CYP2E1 (Molecular Probes) was probed against respective primary antibody for 1 h at 24 °C. All immunocytochemical stainings for marker proteins were mounted in Immunomount (Molecular Probes) and analyzed by a fluorescent microscope (Eclipse 80i Nikon microscope, Melville, NY) connected to a color DigFire digital camera (Optronics, Goleta, CA).
Real-time PCR
Primary human neurons cultured in 6-well plates for 14 days were preincubated with actinomycin D (100 ng/ml) or cyclohexamide (10 μg/ml) for 15 min prior to EtOH/Ach (17.5 mM EtOH or 10 μM Ach) treatment for 24 h. Cell pellets were used for protein or RNA extraction. For Western blot analyses, cell lysate proteins were extracted with ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5) containing 20% glycerol, 5 mM MgCl2, 0.5 mM DTT, and protease inhibitor cocktail, while total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA) for NOX and XOX mRNA level analyses. Isolated RNA was reverse-transcribed with random hexamers (Promega, Madison, WI). Real-time quantitative PCR was performed with cDNA using an ABI PRISM 7000 sequence detector (Applied Biosystems, Foster City, CA). NOX2, XOX (gene symbol XDH), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were analyzed using genespecific primers and TaqMan probes that are available as TaqMan gene expression assays Hs00166163_m1, Hs00166010_m1 (for NOX2, XOX), and Hs02758991_g1 (GAPDH), respectively (Applied Biosystems). Gene expression was normalized to human GAPDH used as an endogenous control.
Statistical analysis
Results were expressed as mean values (±SD), and a value of P<0.05 was considered significant. Statistical significance was assessed by two-way ANOVA analyses with Newman-Keuls posttest for multiple comparisons.
Results
The idea that alcohol abuse causes neuronal injury is well accepted; however, its underlying mechanisms are still poorly understood. Previously, we demonstrated that EtOH metabolism in primary human brain endothelial cells resulted in ROS generation and intracellular calcium release leading to BBB dysfunction and enhanced leukocyte migration across the BBB. We hypothesized that EtOH metabolism in primary human neurons causes ROS/NO production leading to neurodegeneration. To verify this idea, we determined the dose- and time-dependent neurotoxic effects of EtOH, Ach, or _S_-nitroso-_N_-acetylpenicillamine (an NO donor) on neuronal cell culture. Using broad range concentrations of EtOH (10-100 mM), Ach (10-100 μM), or SNAP (10-100 μM), we observed that EtOH higher than 20 mM or Ach/SNAP higher than 20 μM adversely affects cell viability as detected by live and dead assay after a 24-h treatment period. For each batch of primary neurons isolated from human fetal tissue, MAP-2 or α-tubulin immunostainings assessed the purity of neurons in cultured neuronal cells demonstrating 75-85% of cells positive for neuronal markers.
Human neurons express EtOH-metabolizing enzymes, CYP2E1 and ADH
We next evaluated the catalytic activity, protein content, and expression of the EtOH-metabolizing enzymes, CYP2E1 or ADH, in primary human neurons after exposure to 17.5 mM EtOH (corresponding to 0.08%, legal alcohol limit in blood) for 24 h with/without 4-methylpyrazole (4-MP, inhibitor of CYP2E1/ADH). EtOH exposure resulted in a 2.8-fold induction of CYP2E1 activity in human neurons (Fig. 1A) paralleling a 1.5-fold increase in CYP2E1 protein content (Figs. 1B and C) or an enhanced CYP2E1 protein expression/localization (Figs. 1D and E) compared with respective controls. The magnitude of CYP2E activity induction by EtOH in human neurons (62 nmol/mg protein) was lower than that of human brain endothelial cells (200 nmol/mg protein). Although EtOH did not significantly induce ADH activity or protein content, it is important to note that ADH activity (>9 nmol/mg), protein content, and immunofluorescent staining for ADH expression were detected in primary human neurons (Figs. 2A-E). These results suggested that human neurons could potentially metabolize EtOH via CYP2E1/ADH, leading to reactive EtOH metabolite production.
Fig. 1.
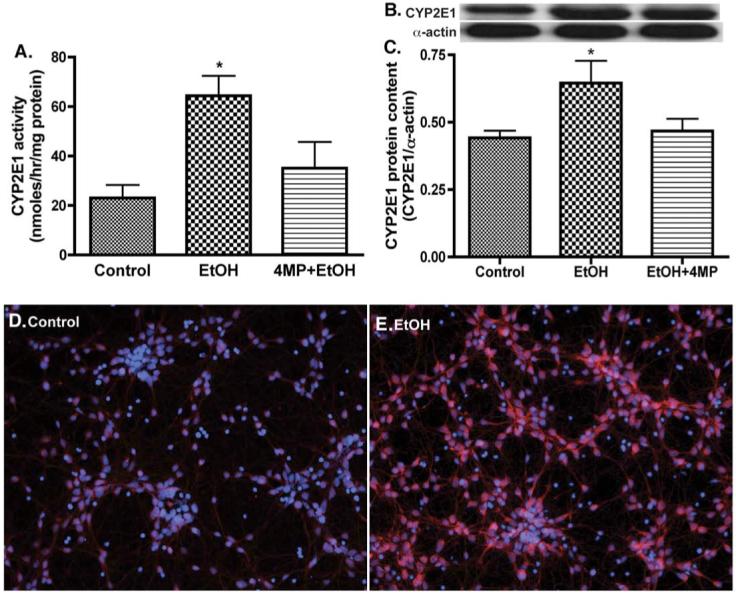
EtOH induces CYP2E1 activity and protein expression in primary human neurons. Microsomal protein fraction from neurons was assayed for (A) CYP2E1 activity, (B) immunoreactive bands of CYP2E1 and actin, and (C) CYP2E1 protein content. Results were expressed as mean values (±SD; _n_=3), and presented as nmol/h/mg protein, or as ratio of CYP2E1 to that of actin-immunoreactive intensity. *Indicates statistical significance (P<0.01) compared with control. Primary neurons after exposure to 17.5 mM EtOH for 24 h were also analyzed for CYP2E1 protein expression: (D) control (E) EtOH. Immunofluorescence stains are DAPI (blue) and CYP2E1 (red). Original magnification ×20. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
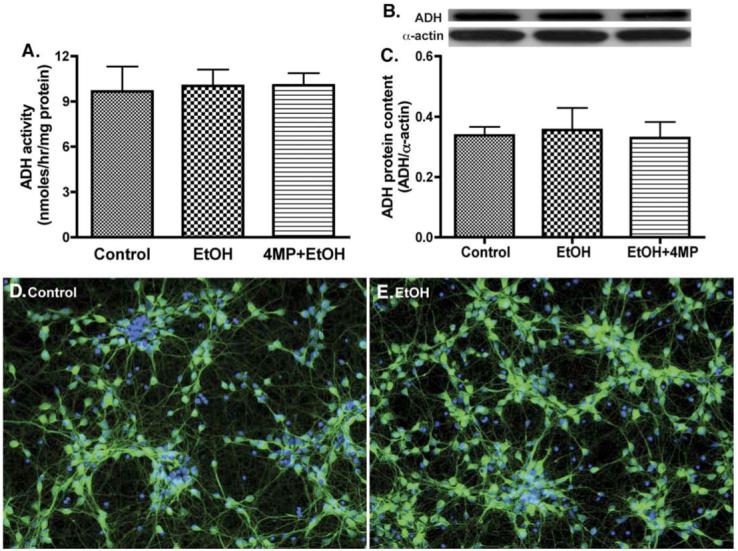
Primary human neurons express ADH activity and protein. Cytosolic protein fraction from neurons was assayed for (A) ADH activity, (B) immunoreactive bands of ADH and actin, and (C) ADH protein content. Results were expressed as mean values (±SD; _n_=3), and presented as nmol/h/mg protein, or as ratio of ADH to that of actin-immunoreactive intensity. *Indicates statistical significance (P<0.01) compared with control. Neuronal immunofluorescent stainings of (D) control and (E) EtOH. DAPI (blue), and ADH (green). Original magnification ×20. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
EtOH/Ach activated ROS producing pathways in neurons
In order to correlate the induction of CYP2E1/ADH activity with reactive metabolite production, we detected ROS levels in cultured neurons after EtOH/Ach exposure with or without 4-MP, DDC (diethydithiocarbamate; inhibitor of superoxide dismutase), or ALC (acetyl-L-carnitine; antioxidant). Increase in ROS production (by 68%) after EtOH treatment was significantly inhibited by 4-MP (Fig. 3A), suggesting that alcohol metabolism resulted in ROS generation, in part, via a Fenton-Weiss-Haber (FWH) reaction as direct EtOH metabolite (see schematic pathway). Inhibition of superoxide dismutase (by DDC) further enhanced the EtOH-induced ROS level (P<0.05), indicating a key role of SOD regulating the oxidants levels. Interestingly, Ach exposure also led to a 68% increase in ROS levels, suggesting the potential involvement of Ach in ROS generation. Since Ach is not an ROS donor, and Ach metabolism is not capable of producing ROS, increased levels of reactive species might be due to Ach-mediated activation of NOX or XOX. In order to address whether Ach increases ROS level via NOX/XOX activation (besides FWH reactions), we studied the dose-dependent effects of Ach on ROS production in cultured neurons using the NOX inhibitor apocynin (APC) or the XOX inhibitor allopurinol (AP). Our results indicated dose-dependent increments of 38-69% in ROS production by Ach stimulation (5-50 μM) in human neurons, and AP/APC individually or in combination significantly inhibited (27-43%, P<0.03) the increased ROS production caused by 50 μM Ach (Fig. 3B). Taken together, these findings suggest that EtOH metabolism by ADH/CYP2E1 generated ROS and Ach, and Ach subsequently activated NOX and XOX, exacerbating oxidative stress level in the CNS.
Fig. 3.
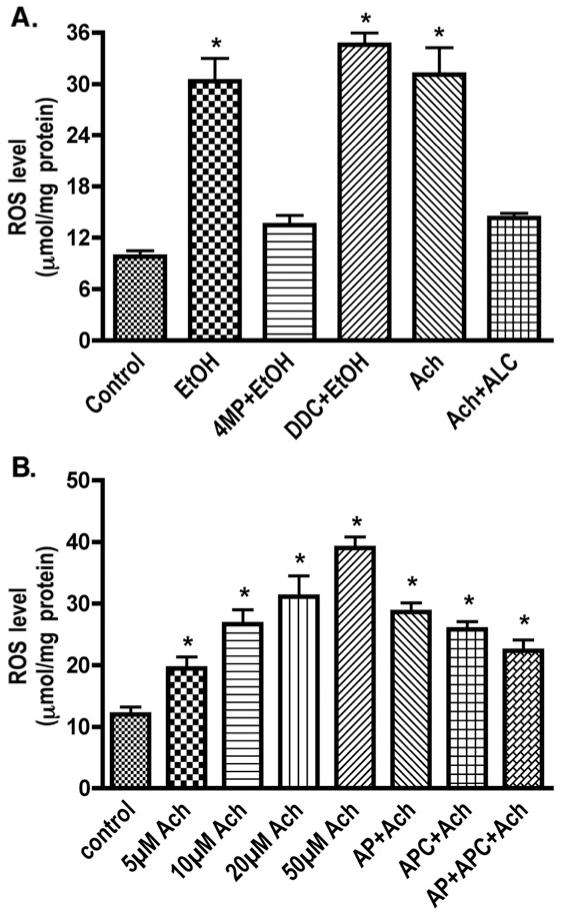
Treatment of neurons with 17.5 mM EtOH or Ach significantly increases ROS production detected by DCF-DA assays using 96-well Fluoroblock plates. (A) Effects of EtOH and specific inhibitors (B) Ach dose-dependent ROS production via NOX/XOX induction. Results were expressed as mean values (±SD; _n_=3). *Indicates statistical significance (P<0.01) compared with controls. APC (apocynin, NOX inhibitor), AP (allopurinol, XOX inhibitor), 4MP (4-methylpyrazole, ADH/CYP2E1 inhibitor), ALC (acetyl-L-carnitine, mitochondrial fatty acid transporter, neurotransmitter, and antioxidant), DDC (diethyldithiocarbamate, SOD inhibitor).
EtOH/Ach increased NOX/XOX protein and mRNA levels
In order to address whether activation of the NOX/XOX pathway by EtOH/Ach resulted in ROS generation, we analyzed the changes in NOX/XOX protein contents in cell lysates derived from neuronal cultures after 48 h EtOH/Ach exposure in the presence or absence of cyclohexamide (Chx; protein synthesis inhibitor) or actinomycin D (ACD; inhibitor of RNA synthesis). Western blot analyses revealed that EtOH or Ach respectively increased the NOX protein contents by 34 or 43%, while XOX levels were elevated by 16 or 20% compared with respective controls (Figs. 4A-D). Pretreatment of neurons with ACD effectively diminished the Ach-induced increase in NOX (43%) or XOX protein levels (20%) to the basal level, while Chx had minimal effects on NOX (P = 0.041), suggesting that EtOH/Ach modulated the transcription of NOX/XOX mRNA level rather than translation. To confirm this observation, we examined the changes in NOX/XOX mRNA levels by real-time PCR in RNA extracts from primary neurons treated with EtOH/Ach in the presence or absence of Chx or ACD for 48 h. EtOH/Ach exposure increased the NOX mRNA levels by 47 or 51%, respectively, compared with control, and ACD significantly inhibited this Ach-mediated increase in NOX mRNA levels by 25% (Fig. 5A). Similarly,treatment of neurons with EtOH/Ach up-regulated XOX mRNA level by 292 or 463%, respectively, compared with control (Fig. 5B), and ACD inhibited 66% of the Ach-mediated increase in XOX mRNA levels. These results suggest that the transcription process modulated the EtOH/Ach-induced increases in NOX/XOX activity and protein contents in neurons.
Fig. 4.
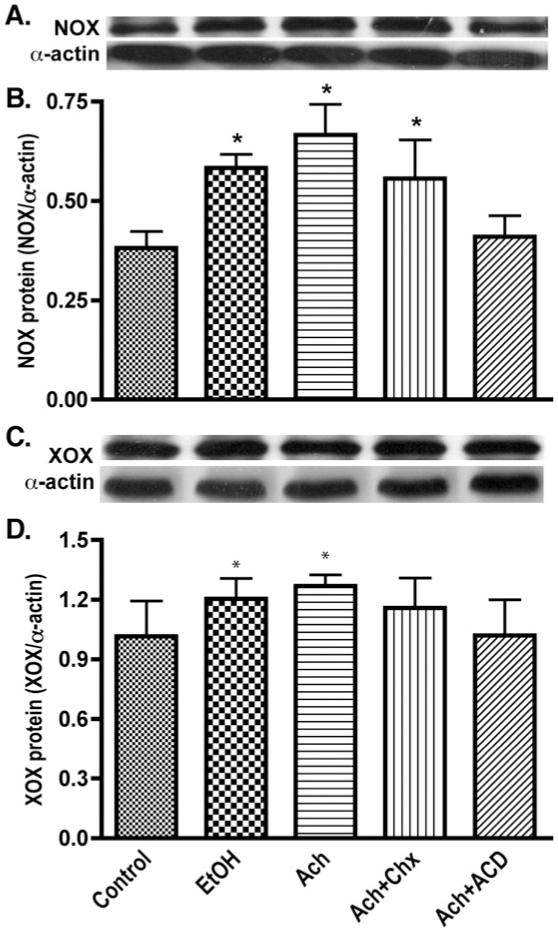
EtOH/Ach elevates NOX and XOX protein levels in human neurons. Neuronal protein extracts derived from EtOH/Ach (17.5 mM/10 μM) treatment for 48 h were assayed for changes in NOX or XOX protein content. (A) Immunoreactive bands of NOX and actin, (B) NOX protein content, (C) immunoreactive bands of XOX and actin, and (D) XOX protein content. Results were expressed as mean values (±SD; _n_=3), and presented as ratio of NOX or XOX to that of actin-immunoreactive bands. *Indicates statistical significance (P<0.01) compared with controls. Cyclohexamide (Chx; protein synthesis inhibitor), and actinomycin D (ACD; inhibitor of RNA synthesis).
Fig. 5.
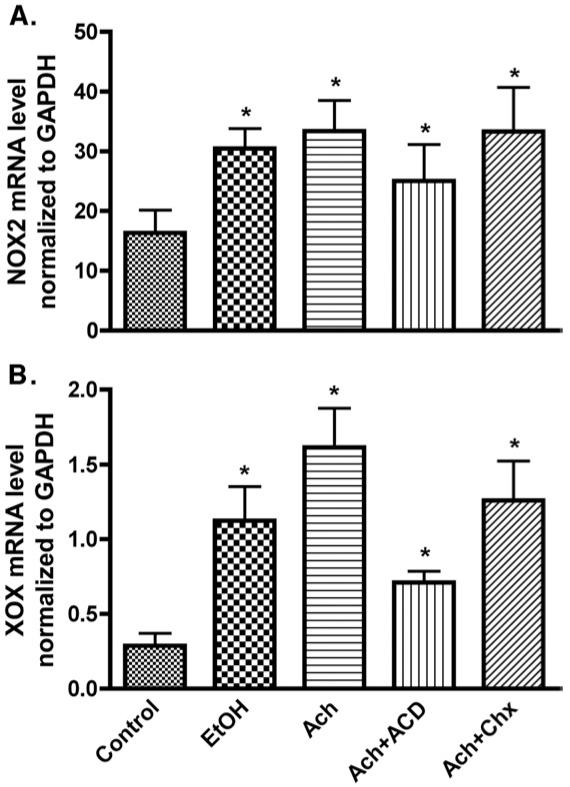
EtOH/Ach up-regulates mRNA levels of NOX and XOX in human neurons. RNA extracts derived from EtOH/Ach treatment for 48 h were quantified by real-time PCR techniques. (A) mRNA levels of NOX, and (B) mRNA levels of XOX. Results were normalized to GAPDH and expressed as mean values (±SD; _n_=3). *Indicates statistical differences (P<0.01) compared with control. Chx (protein synthesis inhibitor, 10 μg/ml), and ACD (inhibitor of RNA synthesis, 100 ng/ml).
EtOH and Ach stimulated NO induction in neurons
In addition to ROS production, EtOH/Ach can potentially activate the nitric oxide synthase (NOS) pathway generating NO in neurons. The nNOS in neurons is usually present in its constitutive form, and therefore produces a minimal level of NO in a calcium-dependent manner, serving as an effective neurotransmitter. Although clearly detectable, the insignificant changes in 160-kDa nNOS protein content that we observed here did not reflect the significant increase in NO production (47-57%) after EtOH/Ach exposure (Figs. 6A-C), suggesting that enhanced NO level was not triggered by nNOS stimulation. Thus, we investigated the role of calcium-independent cytokine inducible NOS in neuronal cell cultures after treatment with EtOH/Ach in the presence or absence of the iNOS-specific inhibitor L-NAME. Our results demonstrated that 45% increase in low molecular weight (10 kDa) iNOS protein content after EtOH/Ach exposure correlated with the 47-57% increase in NO levels (Figs. 6D and E), and augmented NO production was effectively inhibited by L-NAME, similar to the findings in flap vessels or in rat renal endothelial membrane compartments [42,43]. We also detected two other iNOS-immunoreactive bands (molecular weights of 135 and 200 kDa) both significantly up-regulated by EtOH/Ach exposure but to a lesser extent than 10-kDa species (data not shown). These results suggested that exposure of human neurons with alcohol- or Ach-induced iNOS activity led to enhance production of NO in the CNS.
Fig. 6.
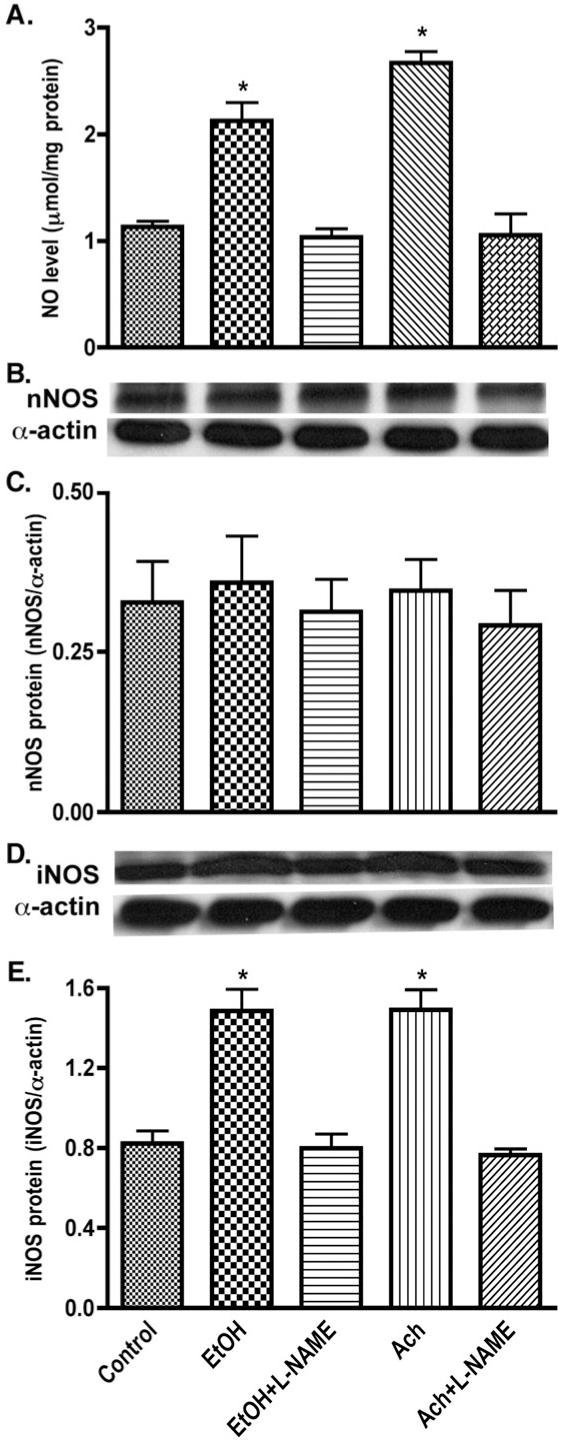
Enhanced NO production due to iNOS induction by EtOH/Ach exposure. (A) NO level was detected by diaminofluorescein-2 diacetate (DAF-2DA) assay in 96-well plate cultured neurons. Neuronal lysates protein (20 μg/well) was used for Western blot analyses. (B) Immunoreactive bands of nNOS and actin protein, (C) nNOS protein content, (D) immunoreactive bands of iNOS and actin protein, and (E) iNOS protein content. Results were expressed as mean values (±SD; _n_=3), and presented as μmol/mg cellular protein (for NO), or as ratio of nNOS or iNOS to that of actin-immunoreactive bands. *Indicates statistical significance (P<0.01) compared with controls. L-NAME (100 μM, _N_G-nitro-L-arginine methyl ester, NOS inhibitor).
EtOH/Ach induced oxidative damage in neurons
In order to correlate the EtOH-induced ROS production with oxidative damage and neuronal injury, we detected the marker for lipid peroxidation product 4-hydroxynonenal (4-HNE), and neuronal injury marker protein neurofilament. Treatment of neurons with EtOH/Ach for 72 h significantly elevated the level of 4-HNE by 43 or 48%, respectively, in neurons compared with control (Figs. 7A and B). Interestingly, this increase in 4-HNE levels in neurons after exposure to EtOH/Ach paralleled a 40% decrease in the light-chain neurofilament protein (70 kDa) or a 36% decrease in neuronal viability as compared with respective controls (Figs. 7C-E). Pretreatment of 4-MP, AP+APC, or ALC prevented the EtOH/Ach-induced increase in 4-HNE, and decrease in neurofilament protein or neuronal death. These results suggest that EtOH/Ach-induced neuronal injury can be respectively reversed by restoring mitochondrial function (by ALC), or by inhibiting the activation of NOX/XOX (by AP+APC), or by inhibiting alcohol metabolism (by 4-MP). The potential mechanisms of alcohol-induced mitochondrial impairment for producing excess ROS and the putative role of ROS for initiation of neuroinflammation are currently under investigation, thus will be described in a future communication.
Fig. 7.
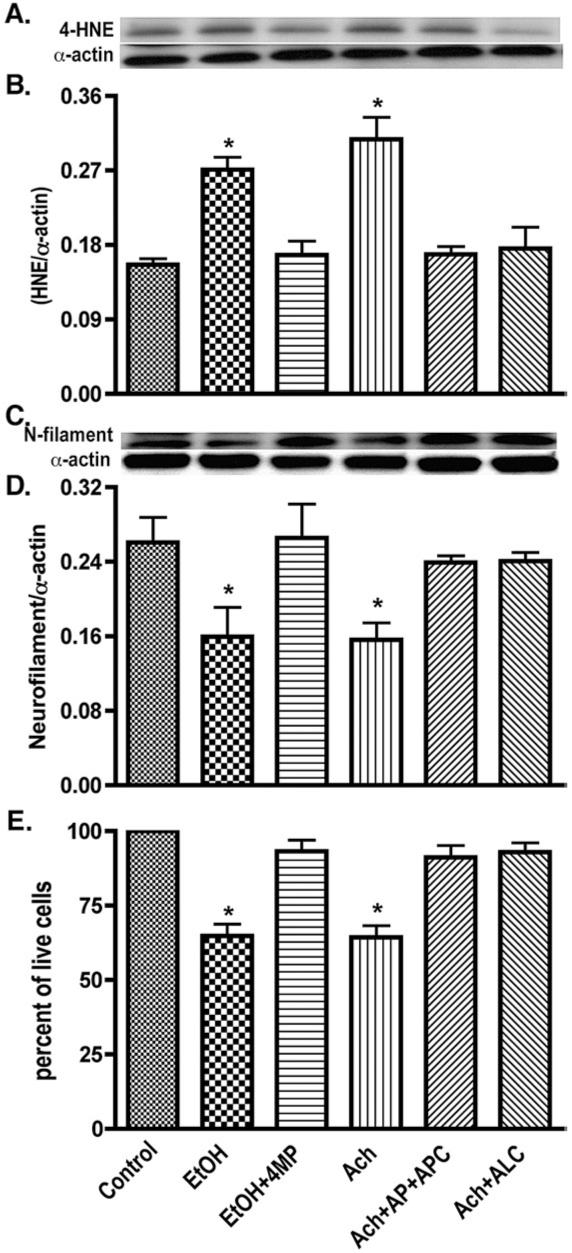
EtOH/Ach causes oxidative damage and neuronal injury. Lysate protein (20 μg/well) derived from EtOH/Ach (17.5 mM/10 μM) treatment for 72 h was analyzed by Western blot for changes in protein content. (A) Immunoreactive bands of 4-HNE and actin, (B) 4-HNE protein content, (C) immunoreactive bands of neurofilament and actin, and (D) neurofilament protein content. (E) Neuronal Live/Dead assay determines the loss of neurons from EtOH/Ach (17.5 mM/10 μM) treatment for 72 h. Results were expressed as ratio of 4-HNE or neurofilaments to that of actin bands, or percentage of live cells for neuronal loss (mean values ±SD; _n_=3). *Indicates statistical significance (P<0.01) compared with controls.
Discussion
Our findings point to a novel pathway of neurodegeneration associated with alcohol abuse stemming from alcohol-induced oxidative stress. We found that both alcohol-metabolizing enzymes (ADH and CYP2E1) are active in human neurons. While ADH is modestly expressed, EtOH exposure significantly up-regulates the CYP2E1 activity and protein content in primary human neuronal cultures. The present study demonstrates that EtOH metabolism by these enzymes up-regulates the production of ROS and NO via the induction of NOX/XOX and iNOS activities by Ach. Induction of NOX/XOX and iNOS activities and protein expression by Ach is modulated through a transcriptional regulation resulting in enhanced formation of ROS, NO, and ONOO- in the CNS. The sensitive methods detecting ROS and NO allow dissection of mechanisms of alcohol-induced oxidative damage and neurodegeneration. The biological source of oxidants and their role as signaling molecules are extensively described in recent reviews [41,44]. We propose that chronic oxidative stress conditions initiate oxidative neuronal injury leading to neurodegeneration as illustrated in Fig. 8.
Fig. 8.
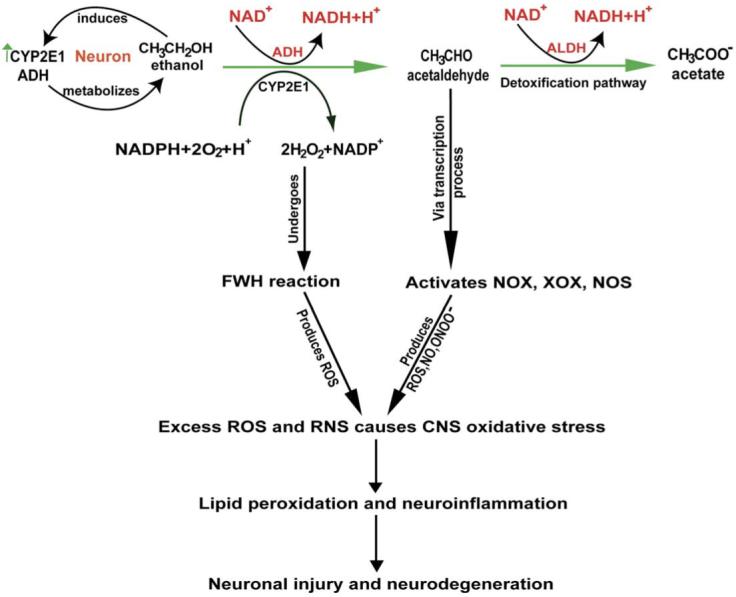
Alcohol-induced activation of oxidative pathways in the CNS leading to neurodegeneration.
We demonstrated before that CYP2E1 and ADH expressed in human brain endothelial cells generate ROS after EtOH exposure [12]. Here, we present evidence that primary human neurons show 3-fold induction of CYP2E1 activity after EtOH treatment, and CYP2E1 activation parallels increased ROS and NO production. These data complement the findings that ADH mRNA level was detected in human brain tissue [45]. Our results suggest that brain cells including neurons can metabolize alcohol, thereby contributing to elevated oxidative stress commonly observed in alcoholics. These findings also support the idea that EtOH metabolism results in end-organ injury. Although the tonic inhibitory current mediated by neuronal GABA-A receptor α1/δ subunit partnership is highly sensitive to low EtOH concentration [46], the molecular site of action or the binding site of the EtOH on receptor protein is unknown. It is generally accepted that direct effects of alcohol (nonmetabolized EtOH) include alteration of neuronal lipid bilayer membrane fluidity and permeability that considerably affect the ion channel conductance such as passive potassium channel [47,48]. It has been shown in neurovascular components that it is the reactive EtOH metabolites that act as the signaling molecules (second messengers) in triggering the activation of cell signaling pathways [15].
We propose that alcohol-induced ROS production derived from FWH reaction via the CYP2E1-mediated EtOH metabolism is modest. It appears that ROS and NO are derived mostly from aftermath activation of NOX, XOX, or NOS by EtOH metabolite, resulting in exacerbation of oxidative stress in the CNS. This claim is supported by the findings that Ach dose dependently increases the ROS levels in neuronal culture, and APC (NOX inhibitor) or AP (XOX inhibitor) reduces the Ach-induced ROS production, suggesting a direct involvement of the NOX/XOX activation pathway. Interestingly, activation of NOX/XOX by EtOH/Ach seems to be regulated by a transcriptional factor because actinomycin D (RNA synthesis inhibitor) effectively reduces both mRNA and protein levels of NOX/XOX. Since we used the p47phox peptide sequence for NOX mRNA analysis, we propose that tyrosine phosphorylation of p47phox initiated the downstream stimulation of transcription factor with a subsequent activation of NOX at the cytoplasmic membrane, similar to protein tyrosine kinase-mediated activation of matrix metalloproteinases [13,16]. It is plausible because the complex enzyme NOX is active only when the cytosolic components of the regulatory subunits p47phox or p67phox protein become translocated to the plasma membrane on activation, which forms a p47phox-p22phox or p47phox-gp91 complex with the membrane subunits. Such a molecular mechanism of NOX activation by serine phosphorylation of p47phox [49,50] and that of XOX activation [51,52] after angiotensin II stimulation was shown in endothelial cells. We also note that ROS production by alcohol-induced activation of NOX appears to be more prominent than that of XOX activation.
EtOH/Ach exposure minimally affects nNOS protein levels in primary human neurons. Physiologic NO concentrations released by nNOS serve as essential neurotransmitters and an integral part of guanylate cyclase activation via NO signaling. In the nNOS knockout mice, alcohol-induced impairment and deficiency of nNOS caused microencephaly and neuronal loss [28] and neuro-cognitive deficits [29]. The present findings attempt to explain this mechanistic switch in physiologic conditions of chronic alcoholism. We hypothesize that occasional low doses of alcohol stimulate nNOS (not inducible) producing adequate levels of NO to maintain this proper physiologic function. On the contrary, high dose or chronic exposure (even at low-dose) induces iNOS in the CNS, and an excess amount of NO suppresses the proper physiological functions. The relevance of these data is supported by the findings that NOS induction was detected in cerebellar cortical neurons of alcoholics [53]. Similarly, reactive EtOH metabolites (ROS, Ach), depending on the concentrations, can have opposite effects within the active self-organizing biological system. At low concentrations, these reactive species act as activating molecules; whereas, at high concentrations, these oxidants act as transducers of oxidative stress and neurodegenerative agents.
Marked increase in lipid peroxidation product (4-HNE) and decrease of neuron-specific neurofilaments support the notion that the peroxidation process of cellular protein and disruption of neuronal cytoskeleton could be initial steps in alcohol-associated neurodegeneration. Clinical trials for many neurological diseases target the role of antioxidants, and it is imperative to understand the mechanisms leading to induction of oxidative stress and the nature of oxidative damage resulting in progressive neurodegeneration. Thus, our data on how alcohol (drug of abuse and potent stimulant) initiates the activation of biochemical pathways and the cell signaling leading to neuronal injury will serve as the basis for future therapeutic interventions.
Acknowledgments
The authors appreciate the excellent administrative support from Ms. Robin Taylor. This work was supported in part by NIH grants (AA016403, AA015913, and AA017398).
Abbreviations
Ach
acetaldehyde
ADH
alcohol dehydrogenase
ALC
acetyl-L-carnitine
AP
allopurinol
APC
apocynin
BBB
blood-brain barrier
CYP2E1
cytochrome P450-2E1
DAF-2DA
diaminofluorescein-2 diacetate
DCF-DA
dichlorofluorescein 2-diacetate
EtOH
ethanol
4-HNE
4-hydroxynenonal
iNOS
inducible nitric oxide synthase
L-NAME
N _G_-nitro-L-arginine methyl ester
4-MP
4-methylpyrazole
NO
nitric oxide
NOX
NADPH oxidase
PBS
phosphate-buffered saline
ROS
reactive oxygen species
SNAP
_S_-nitroso-_N_-acetylpenicillamine
XOX
xanthine oxidase
References
- [1].Parsons OA. Neurocognitive deficits in alcoholics and social drinkers: a continuum? Alcohol. Clin. Exp. Res. 1998;22:954–961. [PubMed] [Google Scholar]
- [2].Zeigler DW, Wang CC, Yoast RA, Dickinson BD, McCaffree MA, Robinowitz CB, et al. The neurocognitive effects of alcohol on adolescents and college students. Prev. Med. 2005;40:23–32. doi: 10.1016/j.ypmed.2004.04.044. [DOI] [PubMed] [Google Scholar]
- [3].Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J. Neuropathol. Exp. Neurol. 1998;57:101–110. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- [4].Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- [5].Maracchioni A, Totaro A, Angelini DF, Di Penta A, Bernardi G, Carri MT, et al. Mitochondrial damage modulates alternative splicing in neuronal cells: implications for neurodegeneration. J. Neurochem. 2007;100:142–153. doi: 10.1111/j.1471-4159.2006.04204.x. [DOI] [PubMed] [Google Scholar]
- [6].Riikonen J, Jaatinen P, Rintala J, Porsti I, Karjala K, Hervonen A. Intermittent ethanol exposure increases the number of cerebellar microglia. Alcohol Alcohol. 2002;37:421–426. doi: 10.1093/alcalc/37.5.421. [DOI] [PubMed] [Google Scholar]
- [7].Potula R, Haorah J, Knipe B, Leibhart J, Chrastil J, Heilman D, et al. Alcohol abuse enhances neuroinflammation and impairs immune responses in an animal model of human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 2006;168:1335–1344. doi: 10.2353/ajpath.2006.051181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pratt OE, Rooprai HK, Shaw GK, Thomson AD. The genesis of alcoholic brain tissue injury. Alcohol Alcohol. 1990;25:217–230. doi: 10.1093/oxfordjournals.alcalc.a044995. [DOI] [PubMed] [Google Scholar]
- [9].Reynolds KLB, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. JAMA. 2003;289:579–588. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- [10].Regan TJ. Alcohol and the cardiovascular system. JAMA. 1990;264:377–381. [PubMed] [Google Scholar]
- [11].Hillbom M, Kaste M. Alcohol abuse and brain infarction. Ann. Med. 1990;22:347–352. doi: 10.3109/07853899009147918. [DOI] [PubMed] [Google Scholar]
- [12].Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005;78:1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- [13].Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007;101:566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- [14].Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol. Clin. Exp. Res. 2005;29:999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- [15].Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor IP3R-gated intracellular calcium release. J. Neurochem. 2007;100:324–336. doi: 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- [16].Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008;56:78–88. doi: 10.1002/glia.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goldstein S, Merenyi G. The chemistry of peroxynitrite: implications for biological activity. Methods Enzymol. 2008;436:49–61. doi: 10.1016/S0076-6879(08)36004-2. [DOI] [PubMed] [Google Scholar]
- [18].Battino M, Bullon P, Wilson M, Newman H. Oxidative injury and inflammatory periodontal diseases: the challenge of anti-oxidants to free radicals and reactive oxygen species. Crit. Rev. Oral Biol. Med. 1999;10:458–476. doi: 10.1177/10454411990100040301. [DOI] [PubMed] [Google Scholar]
- [19].Ingelman-Sundberg M, Johansson I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochromes P-450. J. Biol. Chem. 1984;259:6447–6458. [PubMed] [Google Scholar]
- [20].Winston GW, Cederbaum AI. Evidence for two ethanol oxidizing pathways in reconstituted mixed-function oxidase systems. Pharmacol. Biochem. Behav. 1983;18(Suppl 1):189–194. doi: 10.1016/0091-3057(83)90170-3. [DOI] [PubMed] [Google Scholar]
- [21].Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- [23].Bai J, Cederbaum AI. Overexpression of CYP2E1 in mitochondria sensitizes HepG2 cells to the toxicity caused by depletion of glutathione. J. Biol. Chem. 2006;281:5128–5136. doi: 10.1074/jbc.M510484200. [DOI] [PubMed] [Google Scholar]
- [24].Kessova IG, Cederbaum AI. Mitochondrial alterations in livers of Sod1-/- mice fed alcohol. Free Radic. Biol. Med. 2007;42:1470–1480. doi: 10.1016/j.freeradbiomed.2007.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- [26].Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE. 2007;2:e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Matsumoto H, Matsumoto I. Alcoholism: protein expression profiles in a human hippocampal model. Expert Rev. Proteomics. 2008;5:321–331. doi: 10.1586/14789450.5.2.321. [DOI] [PubMed] [Google Scholar]
- [28].Bonthius DJTG, Karacay B, Mahoney J, Hutton A, McKim R, Pantazis NJ. Deficiency of neuronal nitric oxide synthase (nNOS) worsens alcohol-induced microencephaly and neuronal loss in developing mice. Brain Res. Dev. Brain Res. 2002;138:45–59. doi: 10.1016/s0165-3806(02)00458-3. [DOI] [PubMed] [Google Scholar]
- [29].Spanagel R, Siegmund S, Cowen M, Schroff KC, Schumann G, Fiserova M, et al. The neuronal nitric oxide synthase gene is critically involved in neuro-behavioral effects of alcohol. J. Neurosci. 2002;22:8676–8683. doi: 10.1523/JNEUROSCI.22-19-08676.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Marino MD, Aksenov MY, Kelly SJ. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int. J. Dev. Neurosci. 2004;22:363–377. doi: 10.1016/j.ijdevneu.2004.04.005. [DOI] [PubMed] [Google Scholar]
- [31].Maffi SK, Rathinam ML, Cherian PP, Pate W, Hamby-Mason R, Schenker S, et al. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. J. Neurosci. Res. 2008;86:1064–1076. doi: 10.1002/jnr.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anandatheerthavarada HK, Shankar SK, Bhamre S, Boyd MR, Song BJ, Ravindranath V. Induction of brain cytochrome P-450IIE1 by chronic ethanol treatment. Brain Res. 1993;601:279–285. doi: 10.1016/0006-8993(93)91721-4. [DOI] [PubMed] [Google Scholar]
- [33].Kapoor N, Pant AB, Dhawan A, Dwievedi UN, Gupta YK, Seth PK, et al. Differences in sensitivity of cultured rat brain neuronal and glial cytochrome P450 2E1 to ethanol. Life Sci. 2006;79:1514–1522. doi: 10.1016/j.lfs.2006.04.023. [DOI] [PubMed] [Google Scholar]
- [34].Upadhya SC, Tirumalai PS, Boyd MR, Mori T, Ravindranath V. Cytochrome P4502E (CYP2E) in brain: constitutive expression, induction by ethanol and localization by fluorescence in situ hybridization. Arch. Biochem. Biophys. 2000;373:23–34. doi: 10.1006/abbi.1999.1477. [DOI] [PubMed] [Google Scholar]
- [35].Zatta P, Frank A. Copper deficiency and neurological disorders in man and animals. Brain Res. Rev. 2007;54:19–33. doi: 10.1016/j.brainresrev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- [36].Kohgo Y, Ohtake T, Ikuta K, Suzuki Y, Hosoki Y, Saito H, et al. Iron accumulation in alcoholic liver diseases. Alcohol. Clin. Exp. Res. 2005;29:189S–193S. doi: 10.1097/01.alc.0000189274.00479.62. [DOI] [PubMed] [Google Scholar]
- [37].Brzóska MMM-JJ, Jurczuk M, Gaazyn-Sidorczuk M. Cadmium turnover and changes of zinc and copper body status of rats continuously exposed to cadmium and ethanol. Alcohol Alcohol. 2002;37:213–221. doi: 10.1093/alcalc/37.3.213. [DOI] [PubMed] [Google Scholar]
- [38].Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, et al. Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox. Res. 2005;7:183–192. doi: 10.1007/BF03036448. [DOI] [PubMed] [Google Scholar]
- [39].Clemens DL, Halgard CM, Miles RR, Sorrell MF, Tuma DJ. Establishment of a recombinant hepatic cell line stably expressing alcohol dehydrogenase. Arch. Biochem. Biophys. 1995;321:311–318. doi: 10.1006/abbi.1995.1400. [DOI] [PubMed] [Google Scholar]
- [40].Haorah J, Heilman D, Diekmann C, Osna N, Donohue TM, Jr., Ghorpade A, et al. Alcohol and HIV decrease proteasome and immunoproteasome function in macrophages: implications for impaired immune function during disease. Cell. Immunol. 2004;229:139–148. doi: 10.1016/j.cellimm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- [41].Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007;43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kuo YR, Wang FS, Jeng SF, Lutz BS, Huang HC, Yang KD. Nitrosoglutathione improves blood perfusion and flap survival by suppressing iNOS but protecting eNOS expression in the flap vessels after ischemia/reperfusion injury. Surgery. 2004;135:437–446. doi: 10.1016/j.surg.2003.07.006. [DOI] [PubMed] [Google Scholar]
- [43].Al-Nimri MA, Komers R, Oyama TT, Subramanya AR, Lindsley JN, Anderson S. Endothelial-derived vasoactive mediators in polycystic kidney disease. Kidney Int. 2003;63:1776–1784. doi: 10.1046/j.1523-1755.2003.00913.x. [DOI] [PubMed] [Google Scholar]
- [44].Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- [45].Westerlund M, Galter D, Carmine A, Olson L. Tissue- and species-specific expression patterns of class I, III, and IV Adh and Aldh 1 mRNAs in rodent embryos. Cell Tissue Res. 2005;322:227–236. doi: 10.1007/s00441-005-0038-7. [DOI] [PubMed] [Google Scholar]
- [46].Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABA (A) receptor subunit partnership with high sensitivity to ethanol. Nat. Neurosci. 2007;10:40–48. doi: 10.1038/nn1813. [DOI] [PubMed] [Google Scholar]
- [47].Adermark L, Lovinger DM. Ethanol effects on electrophysiological properties of astrocytes in striatal brain slices. Neuropharmacology. 2006;51:1099–1108. doi: 10.1016/j.neuropharm.2006.05.035. [DOI] [PubMed] [Google Scholar]
- [48].Chu B, Dopico AM, Lemos JR, Treistman SN. Ethanol potentiation of calcium-activated potassium channels reconstituted into planar lipid bilayers. Mol. Pharmacol. 1998;54:397–406. doi: 10.1124/mol.54.2.397. [DOI] [PubMed] [Google Scholar]
- [49].Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 2003;278:12094–12100. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- [50].Ribe D, Sawbridge D, Thakur S, Hussey M, Ledent C, Kitchen I, et al. Adenosine A(2A) receptor signaling regulation of cardiac NADPH oxidase activity. Free Radic. Biol. Med. 2008;44(7):1433–1442. doi: 10.1016/j.freeradbiomed.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Landmesser U, Spiekermann S, Preuss C, Sorrentino S, Fischer D, Manes C, et al. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007;27:943–948. doi: 10.1161/01.ATV.0000258415.32883.bf. [DOI] [PubMed] [Google Scholar]
- [52].Kayyali US, Donaldson C, Huang H, Abdelnour R, Hassoun PM. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J. Biol. Chem. 2001;276:14359–14365. doi: 10.1074/jbc.M010100200. [DOI] [PubMed] [Google Scholar]
- [53].Konovko O, Yu E, Morozov YE, Kalinichenko G, Dyuzen V, Motavkin PA. Induction of NO-synthase and acetaldehyde dehydrogenase in neurons of human cerebellar cortex during chronic alcohol intoxication. J. Bull. Exp. Biol. Med. 2004;137:211–214. doi: 10.1023/b:bebm.0000028142.86579.50. [DOI] [PubMed] [Google Scholar]