Detection of JC Virus DNA Fragments but Not Proteins in Normal Brain Tissue (original) (raw)
. Author manuscript; available in PMC: 2009 Jan 29.
Published in final edited form as: Ann Neurol. 2008 Oct;64(4):379–387. doi: 10.1002/ana.21443
Abstract
Objective
Progressive multifocal leukoencephalopathy (PML) is a fatal demyelinating disease of the white matter affecting immunocompromised patients that results from the cytolytic destruction of glial cells by the human neurotropic JC virus (JCV). According to one model, during the course of immunosuppression, JCV departs from its latent state in the kidney and after entering the brain, productively infects and destroys oligodendrocytes. The goal of this study was to test the hypothesis that JCV may reside in a latent state in a specific region of the brains of immunocompetent (non-PML) individuals without any neurological conditions.
Methods
Gene amplification was performed together with immunohistochemistry to examine the presence of JCV DNA sequences and expression of its genome in five distinct regions of the brain from seven immunocompetent non-PML individuals.
Results
Although no viral proteins were expressed in any of these cases, fragments of the viral DNA were present in various regions of normal brain. Laser-capture microdissection showed the presence of JCV DNA in oligodendrocytes and astrocytes, but not in neurons.
Interpretation
The detection of fragments of viral DNA in non-PML brain suggests that JCV has full access to all regions of the brain in immunocompetent individuals. Thus, should the immune system become impaired, the passing and/or the resident virus may gain the opportunity to express its genome and initiate its lytic cycle in oligodendrocytes. The brain as a site of JCV latency is a possibility.
The human polyomavirus, JC virus (JCV), infects greater than 80% of the human population by early adulthood and remains in a persistent state throughout life.1-3 Under certain pathological conditions, when the immune system is severely impaired, JCV becomes reactivated and its productive replication in oligodendrocytes causes the fatal demyelinating disease, progressive multifocal leukoencephalopathy (PML).4,5 Once a rare disease of white matter seen in patients with lymphoproliferative and myeloproliferative disease,5 PML has been seen more frequently in acquired immune deficiency syndrome patients.6,7 More recently, it was shown that manipulation of immune cells, both T and B cells, by specific monoclonal antibodies that impair trafficking to the brain may promote JCV replication and the development of PML.8 The histopathological hallmarks of PML include demyelinated areas in the subcortical white matter of the brain, eosinophilic intranuclear inclusions in the infected oligodendrocytes, and giant bizarre astrocytes with lobulated hyperchromatic nuclei.9
Similar to its well-studied family member, SV40, the genome of JCV consists of closed, circular, double-stranded DNA of 5130 nucleotides in size10 with 3 distinct domains: early, late, and control regions (Fig 1). The early region is responsible for expression of a series of proteins, called T antigens, which possess regulatory functions on the viral replication cycle and affect several cellular processes including cell cycle, apoptosis, and others.11 The late region is responsible for expression of three classes of structural proteins, VP1, VP2, and VP3, which constitute the capsid of the virus, and an auxiliary agnoprotein with an important role in virus morphogenesis and host function.12 The control region has bidirectional regulatory activity that initiates transcription of the viral early and late genes, and encompasses the origin of viral DNA replication.4 Activation of the early promoter results in expression of T antigen, which by recruiting host factors, triggers subsequent events in viral lytic infection including stimulation of viral late gene expression and initiation of DNA replication leading to virion formation and the cytolytic destruction of host cells.4 Although productive replication of the viral genome occurs in oligodendrocytes, a class of glial cells that are responsible for the production of the myelin sheath, expression of viral proteins, and abortive replication of the virus has also been detected in another class of glial cells, that is, astrocytes.13-15 In one study, JCV has been detected in neuronal cells of a clinical sample, a rare event that is not common in PML.16
Fig 1.
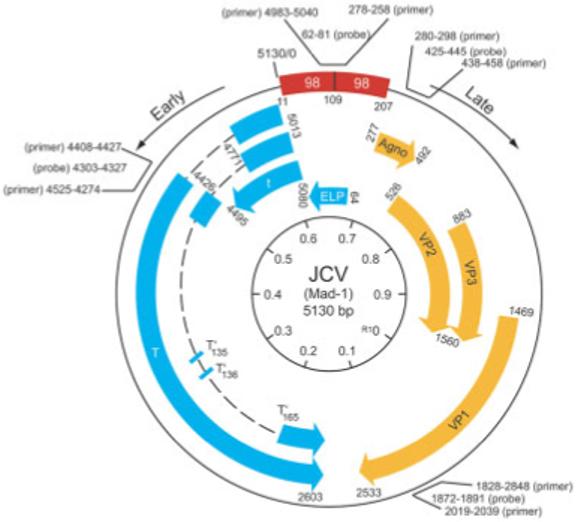
Structural organization of JC virus (JCV) genome. The bidirectional control region of Mad-1 composed of a 98bp tandem repeat, the early genome with the ability to express several proteins, T-antigen family, the early leader protein (ELP), and the late region with four distinct open-reading frames for the regulatory agnoprotein, and capsid proteins VP1, VP2, and VP3 are shown. The positions of the primers and probes for amplification of the various regions of the JCV genome are shown.
Although viral replication and active infection occur in the brain, early speculation led to the belief that, in a latent state, JCV resides mainly in the kidney.17 However, some studies have pointed to the presence of JCV in normal brain18-21; detection of JCV in B cells and tonsil also led to the assumption that the virus may persist in circulating B cells.22 In this report, we utilized gene amplification combined with immunohistochemical techniques to evaluate the presence of JCV DNA and expression of its early and late proteins in five distinct areas of brains from non-PML immunocompetent individuals. Our results show that although segments of the viral DNA are detectable in various regions of the brain, no viral proteins are expressed in the normal brain. Furthermore, laser-capture microdissection (LCM) indicated that DNA sequences are present in astrocytes and oligodendrocytes, and hence not derived solely from B cells.
Subjects and Methods
Specimens and DNA Extraction
Samples were obtained from the Department of Pathology at Hahnemann Hospital consisting of seven human immunodeficiency virus type 1-negative individuals deceased from non-neurological causes with the following clinical histories: Case 1 (52-year-old man) had diabetic nephropathy; Case 2 (46-year-old-man) had acute cardiac infarction; Case 3 (78-year-old woman) had pneumonia; Case 4 (66-year-old man) had chronic renal insufficiency; Case 5 (62-year-old woman) had hypertension and chronic renal failure; Case 6 (33-year-old woman) had an accident; and Case 7 (69-year-old man) had chronic cardiac failure. All autopsies were performed within 12 hours after death. Gross histopathological examination of the brain was always performed, and no evidence for neuropathological alterations, including inflammation or areas of demyelination, was noticed. Each brain was dissected into five specific portions representative of frontal cortex, basal ganglia, hippocampus, pons, and cerebellum. The samples had been fixed in 10% buffered formalin and embedded in paraffin. Ten- and 4μm sections (as specified) were obtained using a dedicated microtome for this study and fresh, disposable blades for each sample. DNA was extracted using the QiAmp DNA mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
DNA Detection and Analysis
DNA amplification was performed using four sets of primers: Pep1 and Pep2 (nucleotides 4255-4274 and 4408-4427, respectively) for detection of the early DNA sequence (T antigen); VP2 and VP3 (nucleotides 1828-1848 and 2019-2039, respectively) for detection of the late DNA sequence corresponding to VP1 gene; Agno1 and Agno2 (nucleotides 279-298 and 438-458, respectively) for detection of the late region corresponding to the Agnoprotein gene; and NCRR1 and NCRR2 (nucleotides 4993-5004 and 258-279, respectively) for detection of the control region. Amplification of the T-antigen, agnoprotein, and VP1 regions was performed in a 50μl reaction containing 200ng of template in Failsafe Buffer B, Failsafe Taq polymerase (Epicenter, Madison, WI) and 500nM of each primer. Amplification of the control region was conducted in Buffer A. After denaturation at 94°C for 5 minutes, 35 cycles of denaturation at 94°C for 45 seconds, annealing for 45 seconds, and extension at 72°C for 45 seconds, a final extension step of 7 minutes at 72°C was allowed. Annealing temperatures were 55°C for Pep primers, 54°C for VP primers, 57°C for Agno primers, and 56°C for NCRR primers. Fifteen microliters of the polymerase chain reaction (PCR) products were separated by 1.3% agarose gel electrophoresis. For Southern blot analysis, the gels with the PCR products were depurinated (0.2M HCl for 15 minutes), denaturated (1.5M NaCl, 0.5M NaOH for 15 minutes), and neutralized (1.5M NaCl, 0.5M Tris-Cl for 15 minutes), and then transferred overnight to a nylon membrane (Hybond-N; Amersham, Buckinghamshire, UK). After ultraviolet cross-linking, the membranes were hybridized in Ultrahyb buffer (Ambion, Austin, TX) overnight at 42°C with 1 × 107cpm/ml γ-[32P] end-labeled oligonucleotide probe. Nucleotides homologous to the following JCV-specific sequences were used as probes: T-antigen probe (Pep primers) (nucleotides 4303-4327); VP probe (nucleotides 1827-1891); Agno probe (nucleotides 1872-1891); CR probe (NCRR primers) (nucleotides 62-81). For DNA sequence analysis, PCR products from the amplification of the control region visible on the gel through ultraviolet illumination were excised and purified with QiA-quick (Qiagen, Valenica, CA) PCR purification kit according to the manufacturer’s instructions. DNA sequencing was performed using ABI Prism 3730x/DNA analyzer (Applied Biosystems, Foster City, CA). For amplification of DNA from the housekeeping gene, GAPDH, the following primers were used: 5′’ TTC TCC CCATTC CGT CTT CC 3′’ and 3′’ GTA CAT GGT ATT CAC CAC CC 5′’.
Immunohistochemistry and Laser-Capture Microdissection
Sections of 4μm in thickness were obtained from the paraffin blocks containing the samples. All the samples were processed for routine hematoxylin and eosin staining for histological analysis. Immunohistochemistry was performed using an avidin-biotin-peroxidase complex following the manufacturer’s protocol (Vectastain Elite ABC Peroxidase Kit; Vector Laboratories, Burlingame, CA). For this purpose, all the slides were deparaffinized by immersion in xylenes. Rehydration of the tissue was then performed by subsequent immersion in descending grades of ethanol up to water, followed by nonenzymatic antigen retrieval, through heating the section to 95°C in 0.01M citrate buffer (pH 6.0) for 30 minutes in a vacuum oven. Slides were allowed to cool down and then incubated for 30 minutes in 3% H2O2/methanol, for endogenous peroxidase quenching. The slides were then blocked with 5% normal horse or goat serum and incubated overnight at room temperature with the primary antibodies in a humidified chamber. The primary antibodies utilized in this study included SV40 T antigen that cross-reacts with JCV T antigen (1:100 dilution; Oncogene Science Biomarker Group, Cambridge, MA), VP1 (1:100 dilution; kindly provided by Dr Walter Atwood, Brown University), and Agnoprotein (1:2,000 dilution; developed in our laboratory). Incubation with the secondary antibody followed by the avidin-biotin-peroxidase complex was allowed for 1 hour each. Finally, development with diaminobenzidine for 3 to 5 minutes and counterstaining with hematoxylin was performed. For LCM, 4μm-thick sections were labeled by immunohistochemistry for neurofilaments (SMI 312; 1:2,000 dilution; Sternberger Monoclonals, Baltimore, MD), myelin basic protein (MBP; 1:500 dilution; Roche, Nutley, NJ), and glial fibrillary acidic protein (GFAP; 1:100 dilution; Dako, Carpinteria, CA), as described earlier. The sections were dehydrated, cleared with xylene, and air-dried for 24 hours. Afterward, LCM was performed under direct microscopic visualization by laser heating of a thermoplastic film mounted on optically transparent CapSure HS LCM caps (Arcturus Engineering, Mountainview, GA). The PixCell II LCM System (Arcturus Engineering) was set to the following parameters: 15μm spot size, 40mW power, 3.0-millisecond duration. A total of 100 cells of each kind (neurons, astrocytes and oligodendrocytes) were captured by focal melting of the membrane through a carbon dioxide laser-pulse activation. DNA isolation was performed using the Arcturus PicoPure DNA extraction kit according to the manufacturer’s instructions (Arcturus Engineering).
Results and Discussion
First, we searched for the JCV early genome in our clinical samples. The positions of the primers used for amplification of the early gene (T antigen) and the representative data showing the results from gene amplification in two cases are shown in Figures 1 and 2A, respectively. The integrity of the DNA preparations were examined by amplification of segments from a housekeeping gene, GAPDH (see Fig 2A). Results from all seven cases are summarized in Table 1. As seen, although no DNA sequence corresponding to JCV early region was detected in two cases (Cases 1 and 2), all other cases showed the presence of the early DNA sequence in one or more regions of the brain. No specific region of the brain exhibited the consistent presence of JCV genomic sequences. Results from immunohistochemical labeling did not show evidence for the expression of T antigen in any region of the brain from these cases. Figure 2C illustrates a representative result from immunolabeling of one of the cases (Case 4) together with a result from parallel labeling of a brain sample from a PML patient for JCV T antigen (see Fig 2B). The search for the detection of other human polyomaviruses, including SV40 and BKV by gene amplification, showed no evidence for their presence in any region of the brain from these cases (data not shown).
Fig 2.
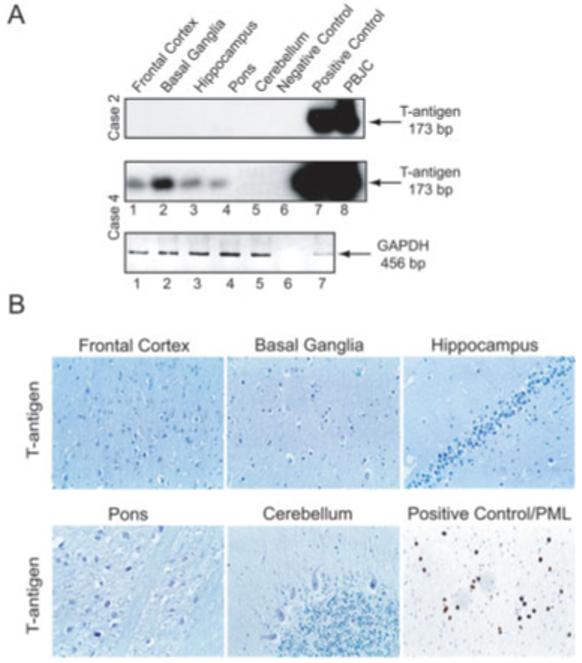
Detection of JC virus (JCV) DNA sequence corresponding to the early genome. (A) Representative Southern blot analysis of amplified DNA from two cases using primers that are specific for the JCV early gene and the housekeeping gene, GAPDH. Negative and positive controls demonstrate results from reactions containing no DNA and DNA from brains of patients with progressive multifocal leukoencephalopathy (PML), respectively. Lane 8 represents gene amplification using pBJC, a plasmid containing JCV DNA as a template. Lane 7 in the bottom panel corresponding to GAPDH demonstrates gene amplification using DNA from the human astrocytic cell line, U-87MG. (B) Representative results from immunohistochemical labeling for detection of JCV early protein, T-antigen in the various regions of non-PML brain (Case 4) and in a section of a brain from a PML patient where T antigen is present in the nuclei of infected oligodendrocytes.
Table 1. Summary of the Results from JCV Early Gene Amplification in All Five Regions of the Seven Cases.
| Case | Frontal Cortex | Basal Ganglia | Hippocampus | Pons | Cerebellum |
|---|---|---|---|---|---|
| 1 | - | - | - | - | - |
| 2 | - | - | - | - | - |
| 3 | + | + | + | + | - |
| 4 | + | + | + | + | - |
| 5 | + | + | + | + | + |
| 6 | - | - | - | + | + |
| 7 | - | - | - | - | + |
A similar approach was utilized for the detection of the JCV late DNA sequence that corresponds to VP1 and expression of the capsid protein, VP1. The position of the region of VP1 that was selected for amplification is shown in Figure 1, and Figure 3A illustrates results from gene amplification in Cases 4 and 7. Table 2 summarizes the results from all cases. As seen in this table, with the exception of Cases 1, 2, and 5, the VP1 DNA sequence was detected in basal ganglia, hippocampus, and pons of the remaining cases. Again, results from immunohistochemistry showed no evidence for the presence of VP1 in Case 7 (see Fig 3B) or in any of other cases (data not shown). As expected, VP1 was detected in oligodendrocytes of brain tissue from a case of PML.
Fig 3.
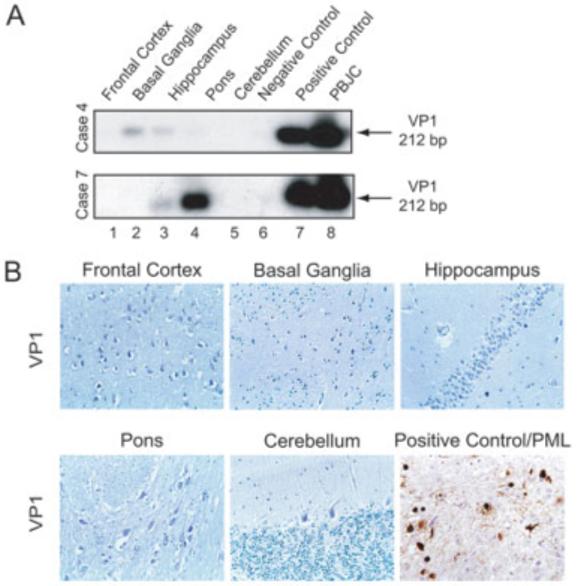
Detection of DNA sequence corresponding to VP1 in brain. (A) Representative results from polymerase chain reaction/Southern blot from two cases. (B) Representative immunohistochemical evaluation for detection of VP1 expression in the various regions of non-PML brain, as well as in demyelinated areas from a brain of a PML patient. VP1 was detected only in the oligodendrocytes of PML samples.
Table 2. Summary of Gene Amplification Directed Toward Detection of the VP1 DNA Sequence in the Various Regions of Non-PML Brains.
| Case | Frontal Cortex | Basal Ganglia | Hippocampus | Pons | Cerebellum |
|---|---|---|---|---|---|
| 1 | - | - | - | - | - |
| 2 | - | - | - | - | - |
| 3 | - | + | - | + | - |
| 4 | - | + | + | - | - |
| 5 | - | - | - | - | - |
| 6 | - | - | + | - | - |
| 7 | - | - | + | + | - |
With the exception of Case 4, the DNA sequences from the late region of the JCV genome responsible for expression of agnoprotein (see Fig 1) were detected in the various regions of all cases (Fig 4A, Table 3). Results from immunohistochemistry ruled out expression of agnoprotein in these samples (see Fig 4B). The JCV-infected oligodendrocytes from PML samples served as a positive control and showed the perinuclear cytoplasmic presence of agnoprotein.
Fig 4.
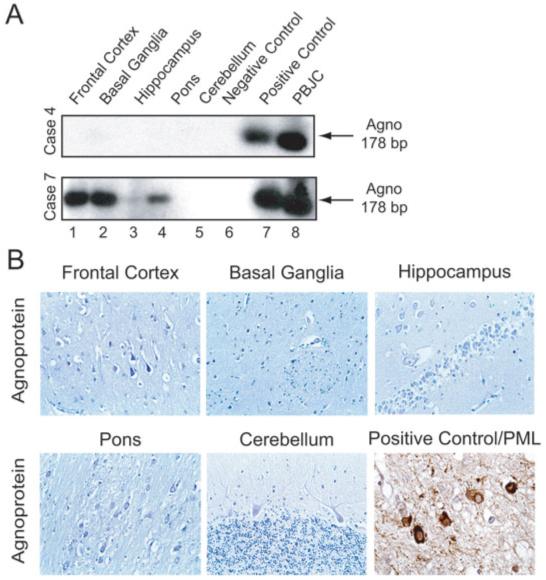
Detection of DNA sequence corresponding to a region of JC virus (JCV) that encodes agnoprotein. (A) Representative results from gene amplification in two cases. (B) Immunohistochemical labeling of brain from non-PML and PML showing perinuclear accumulation of agnoprotein in oligodendrocytes from PML, but not non-PML specimens.
Table 3. Summary of Results from PCR Amplification of Agnoprotein Gene in Brain from Non-PML Samples.
| Case | Frontal Cortex | Basal Ganglia | Hippocampus | Pons | Cerebellum |
|---|---|---|---|---|---|
| 1 | + | - | + | + | + |
| 2 | - | - | + | - | - |
| 3 | + | + | + | - | - |
| 4 | - | - | - | - | - |
| 5 | + | - | + | + | + |
| 6 | + | + | - | + | - |
| 7 | + | + | - | + | - |
Examination of brain tissue for the presence of the JCV control region demonstrated its presence in all regions of Cases 2 and 3 (Fig 5A), and in the cerebellum of Case 4 (see Table 4). Results from DNA sequence analysis of the amplified DNA from Case 2 showed the presence of JCVMad4 with a single point mutation (C→T) positioned on the late side of the 79bp region (see Fig 5B).
Fig 5.
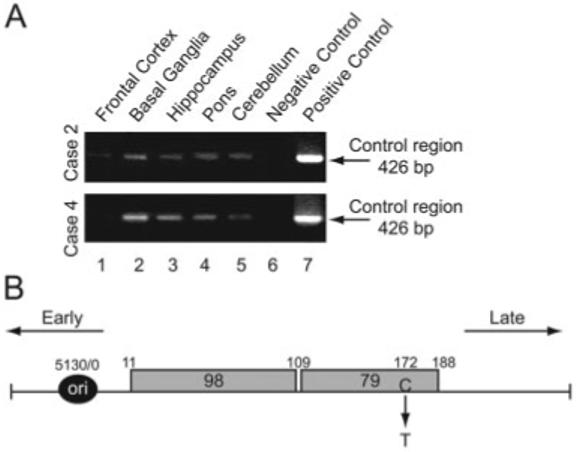
Detection of DNA sequence corresponding to the JC virus (JCV) control region in non-progressive multifocal leukoencephalopathy (non-PML) (non-PML) brain. (A) Results from polymerase chain reaction (PCR) amplification of the control region of JCV in two cases. The negative control illustrates results from amplification with no template DNA, and the positive control shows gene amplification using pBJC plasmid DNA. (B) Structural organization of the JCV control region and the nucleotide composition of amplified JCV DNA in normal brain. The position of a single nucleotide mutation is shown.
Table 4. Summary of Results from PCR Amplification of Agnoprotein Gene in Brain from Non-PML Samples.
| Case | Frontal Cortex | Basal Ganglia | Hippocampus | Pons | Cerebellum |
|---|---|---|---|---|---|
| 1 | - | - | - | - | - |
| 2 | + | + | + | + | + |
| 3 | + | + | + | + | + |
| 4 | - | + | + | + | + |
| 5 | - | - | - | - | - |
| 6 | - | - | - | - | - |
| 7 | - | - | - | - | - |
To identify the cell type(s) that harbors JCV DNA, we used LCM where GFAP, neurofilament (SM 312), and MBP were used as cellular markers for identifying astrocytes, neurons, and oligodendrocytes, respectively. Approximately 100 GFAP-, SMI 312-, and MBP-positive cells from the frontal cortex of Case 4 were excised (Figs 6A-C), and after DNA extraction, incubated with JCV-specific probes for amplification of the viral early genome. Results from gene amplification showed the presence of JCV DNA in oligodendrocytes and astrocytes, but not neurons from the normal brain (see Fig 6D).
Fig 6.
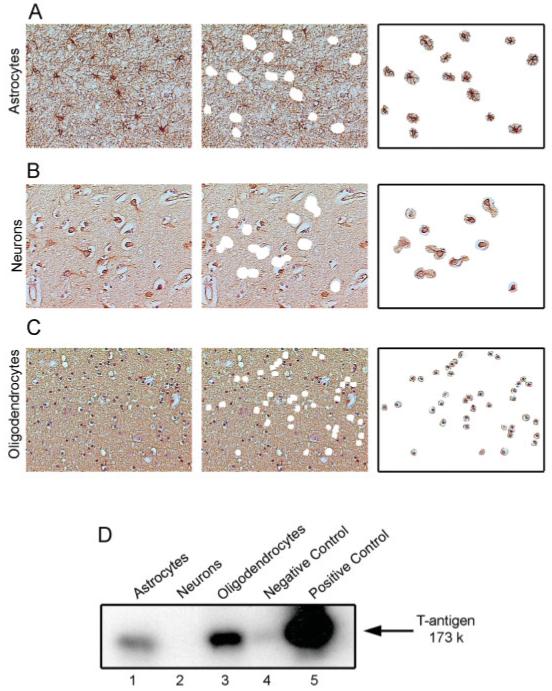
Laser-capture microdissection of frontal cortex from normal brain. Selection of astrocytes (A), neurons (B), and oligodendrocytes (C) was guided by immunohistochemical detection of GFAP, neurofilament (left panels) and MBP. After the thermoplastic film is removed, the tissue left behind after laser capture shows punched holed in the receiving section (middle panels). Removal of the cells is verified by microscopic visualization prior to DNA preparation (right panels) D. Gene amplification followed by Southern blot by hybridization using a pair of primers that recognizes sequences of the JCV early gene.
Our results presented in this study verify the presence of JCV DNA fragments, but not expression of its genome in various regions of brain from immunocompetent, non-PML individuals with no neurological disease. Recent reports on the reactivation of JCV in multiple sclerosis patients who received natalizumab (Tysabri), a humanized monoclonal antibody against α4β1 integrin that blocks trafficking of T cells across the blood-brain barrier,23-25 revived the questions related to the mechanism involved in reactivation of JCV and the sites of JCV latency in humans.26 This clinical observation suggests that JCV may be present in the brain but is kept in check by immune cells. Thus, a disturbance in the status of the immune cells that affects their surveillance of the brain may result in reactivation of JCV and the development of PML. As a first step to examine this hypothesis, we sought to investigate the presence of JCV and expression of its proteins in the various regions of the brain obtained from individuals with no neurological disorders. Furthermore, we used LCM to search for JCV sequences in specific cell types, that is, oligodendrocytes, astrocytes, and neurons from normal brain tissue. Here, we demonstrate that JCV DNA is detectable in multiple regions of the brain including the frontal cortex, basal ganglia, hippocampus, pons, and cerebellum obtained from non-PML individuals. This broad distribution suggests that JCV may be unrestricted in its ability to circulate in the brain, yet may not be able to express its genome in host tissue and cells. Our results show that, with one exception (ie, basal ganglia in Case 3), none of the specimens that was tested harbored all major regions of the JCV genome. Our observation implies that during its passage through or within the brain through hematogenous spread and/or virally infected immune cells, most likely B cells, fragments of viral DNA either after integration in the host genome and/or as free episomal DNA molecules can persist in specific brain cells. This observation raises the question of whether the brain can be considered as a site of latency for JCV despite the fact that the intact viral genome capable of completing its replication cycle is not frequently detected. Results from clinical studies showing the rare occurrence of PML in even high-risk patients such as those with advanced acquired immune deficiency syndrome (4-8%) and infrequent detection of full-length JCV genome (1/7 cases) may provide an explanation why only a small population of severely immunosuppressed individuals who are initially infected with JCV experience the development of PML. Thus, one may conclude that a small subset of brain cells that carry the intact JCV genome may serve as a site of latency for JCV.
According to an earlier model, PML develops on reactivation of the JCV archetype form (JCVCY) that persistently infects the kidney and has a more distinct regulatory region than the strain of the virus that causes PML (JCVMad).27,28 During the course of immunosuppression, rearrangement of the viral control region results in conversion of JCVCY to JCVMad, which has the ability to replicate in oligodendrocytes and astrocytes, and causes PML.29 There are several issues that invite close reevaluation of this model. For example, JCVMad has not been routinely obtained in kidney of normal or immunocompetent patients.27,28 In addition, JCVMad has been consistently isolated from brain lesions of PML patients,30 whereas JCVCY is routinely detected in kidney.27,28 Furthermore, neither JCVCY nor intermediary forms suggesting the conversion of JCVCY to JCVMad have been isolated from brain, or any other tissue. Thus, there is no evidence connecting the two forms of JCV that have distinct tissue tropism. Here we propose an alternative model that excludes the involvement of JCVCY in the development of PML. According to this model, JCVMad is constantly circulating in the human population throughout life and passes through the brain without being able to productively replicate because of suppression of its expression by the immune system. Once the immune system is impaired, because of illness or specific immunotherapy, the circulating and/or resident JCVMad gains the opportunity to express its early genome, which leads to a lytic infection cycle in oligodendrocytes. By considering immunosurveillance as a key factor responsible for JCV gene expression and replication, one may anticipate detection of immune cells in the normal brain sections. Our results, however, showed no evidence for the presence of T cells in any region of the tested brain sections, suggesting that immune regulation of JCV may be a complicated event involving both direct and indirect pathways.
The detection of JCV DNA in the cerebellum of normal brain is of particular interest in light of recent evidence on the presence of JCV DNA sequences and expression of the viral nonstructural proteins in primitive neuroectodermal tumors, some of which arise from the cerebellum.31-33 Of note, in primitive neuroectodermal tumor studies, only viral T antigen, which has oncogenic activity,11 and agnoprotein, which dysregulates the cell cycle and DNA repair,34,35 have been detected. The lack of viral capsid proteins in these JCV-positive tumor cells rules out the possibility of productive replication of JCV in the tumor cells. Thus, one can envision a model in which transient physiological changes in normal individuals that permit expression of nonstructural viral protein by the integrated DNA fragment results in the accumulation of the oncogenic protein, T antigen, in brain cells. Under these circumstances, T antigen, by associating with tumor suppressors and the other cellular proteins that control proliferation, promotes uncontrolled cell division and stimulates tumor formation in the absence of viral replication. Detection of the JCV DNA fragments in brains from the non-PML individuals and the absence of the viral protein invites further investigation to assess the mechanisms involved in immunosuppression of the JCV genome in healthy individuals and determine the factors that initiate viral gene expression in a subpopulation of immunosuppressed patients.
Acknowledgments
This work was supported by the NIH (NINDS) (P01 NS30916, P01 NS36466, K.K.).
We thank past and present members of the Department of Neuroscience at Temple University for their insightful discussion, and sharing ideas and reagents. We thank Dr T. Sweet for her advice in developing primers, and Drs M. White and J. Gordon for critical reading of the manuscript. We thank C. Schriver for editorial assistance.
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Padgett BL, Walker DL. Prevalence of antibodies in human sera against JC virus, an isolate from a case of progressive multifocal leukoencephalopathy. J Infect Dis. 1973;127:467–470. doi: 10.1093/infdis/127.4.467. [DOI] [PubMed] [Google Scholar]
- 2.Walker DL, Padgett BL. The epidemiology of human polyomaviruses. Prog Clin Biol Res. 1983;105:99–106. [PubMed] [Google Scholar]
- 3.Lundstig A, Dillner J. Serological diagnosis of human polyoma-virus infection. Adv Exp Med Biol. 2006;577:96–101. doi: 10.1007/0-387-32957-9_7. [DOI] [PubMed] [Google Scholar]
- 4.Khalili K, Gordon J, White MK. The polyomavirus, JCV, and its involvement in human disease. Adv Ex Med Biol. 2006;577:274–287. doi: 10.1007/0-387-32957-9_20. [DOI] [PubMed] [Google Scholar]
- 5.Hou J, Major EO. Progressive multifocal leukoencephalopathy: JC virus induced demyelination in the immune compromised host. J Neurovirol. 2000;6(suppl 2):S98–S100. [PubMed] [Google Scholar]
- 6.Berger JR, Major EO. Progressive multifocal leukoencephalopathy. Semin Neurol. 1999;19:193–200. doi: 10.1055/s-2008-1040837. [DOI] [PubMed] [Google Scholar]
- 7.Berger JR. Progressive multifocal leukoencephalopathy in acquired immuno-deficiency syndrome: explaining the high incidence and disproportionate frequency of the illness relative to other immunosuppressive conditions. J Neurovirol. 2003;9(suppl 1):38–41. doi: 10.1080/13550280390195261. [DOI] [PubMed] [Google Scholar]
- 8.Berger JR. Progressive multifocal leukoencephalopathy. Curr Neurol Neurosci Rep. 2007;7:461–469. doi: 10.1007/s11910-007-0072-9. [DOI] [PubMed] [Google Scholar]
- 9.Del Valle L, Piña-Oviedo S. HIV disorders of the brain: pathology and pathogenesis. Front Biosci. 2006;11:718–732. doi: 10.2741/1830. [DOI] [PubMed] [Google Scholar]
- 10.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51:458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White MK, Khalili K. Polyomaviruses and human cancer: molecular mechanisms underlying patterns of tumorigenesis. Virology. 2004;324:1–16. doi: 10.1016/j.virol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 12.Khalili K, White MK, Sawa H, et al. The agnoprotein of polyomaviruses: a multifunctional auxiliary protein. J Cell Physiol. 2005;204:1–7. doi: 10.1002/jcp.20266. [DOI] [PubMed] [Google Scholar]
- 13.Boldorini R, Cristina S, Vago L, et al. Ultrastructural studies in the lytic phase of progressive multifocal leukoencephalopathy in AIDS patients. Ultrastruct Pathol. 1993;17:599–609. doi: 10.3109/01913129309027796. [DOI] [PubMed] [Google Scholar]
- 14.Boldorini R, Cristina S, Vago L, et al. Anatomo-pathological features of JCV infection in patients with acquired immunodeficiency syndrome (AIDS). Histological, immunohistochemical, and ultrastructural study including the in situ hybridization technique of 54 AIDS autopsies. Pathologica. 1993;85:17–30. [PubMed] [Google Scholar]
- 15.Del Valle L, Croul S, Morgello S, et al. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- 16.Du Pasquier RA, Corey S, Margolin DH, et al. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology. 2003;61:775–782. doi: 10.1212/01.wnl.0000081306.86961.33. [DOI] [PubMed] [Google Scholar]
- 17.Chesters PM, Heritage J, McCance DJ. Persistence of DNA sequences of BK virus and JC virus in normal human tissues and in diseased tissues. J Infect Dis. 1983;147:676–684. doi: 10.1093/infdis/147.4.676. [DOI] [PubMed] [Google Scholar]
- 18.Elsner C, Dorries K. Evidence of human polyomavirus BK and JC infection in normal brain tissue. Virology. 1992;191:72–80. doi: 10.1016/0042-6822(92)90167-n. [DOI] [PubMed] [Google Scholar]
- 19.Greenlee JE, Clawson SA, Carney HC, O’Neill FJ. Detection of JC virus early region sequences of brains of individuals without progressive multifocal leukoencephalopathy. Ann Neurol. 2005;28(S9):S61. [Google Scholar]
- 20.Mori M, Aoki N, Shimada H, et al. Detection of JC virus in the brains of aged patients without progressive multifocal leukoencephalopathy by the polymerase chain reaction and Southern hybridization analysis. Neurosci Lett. 1992;141:151–155. doi: 10.1016/0304-3940(92)90883-9. [DOI] [PubMed] [Google Scholar]
- 21.White FA, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66:5726–5734. doi: 10.1128/jvi.66.10.5726-5734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monaco MC, Atwood WJ, Gravell M, et al. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol. 1996;70:7004–7012. doi: 10.1128/jvi.70.10.7004-7012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 24.Langer-Gould A, Atlas SW, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 25.van Assche G, van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 26.Khalili K, White MK, Lublin F, et al. Reactivation of JC virus and development of PML in patients with multiple sclerosis. Neurology. 2007;68:985–990. doi: 10.1212/01.wnl.0000257832.38943.2b. [DOI] [PubMed] [Google Scholar]
- 27.Bofill-Mas S, Clemente-Casares P, Major EO, et al. Analysis of the excreted JC virus strains and their potential oral transmission. J Neurovirol. 2003;9:498–507. doi: 10.1080/13550280390218887. [DOI] [PubMed] [Google Scholar]
- 28.Rossi A, Delbue S, Mazziotti R, et al. Presence, quantitation and characterization of JC virus in the urine of Italian immunocompetent subjects. J Med Virol. 2007;79:408–412. doi: 10.1002/jmv.20829. [DOI] [PubMed] [Google Scholar]
- 29.Martin JD, King DM, Slauch JM, Frisque RJ. Differences in regulatory sequences of naturally occurring JC virus variants. J Virol. 1985;53:306–311. doi: 10.1128/jvi.53.1.306-311.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisque RJ, White FA. The molecular biology of JC virus, causative agent of progressive multifocal leukoencephalopathy. In: Roos RP, editor. Molecular neurovirology. Humana Press; Totowa, NJ: 1992. pp. 25–158. [Google Scholar]
- 31.Khalili K, Krynska B, Del Valle L, et al. Medulloblastomas and the human neurotropic polyomavirus, JCV. Lancet. 1999;353:1152–1153. doi: 10.1016/s0140-6736(99)00357-8. [DOI] [PubMed] [Google Scholar]
- 32.Krynska B, Del Valle L, Croul SE, et al. Detection of human neurotropic JC virus DNA sequence and expression of the viral oncogenic protein in pediatric medulloblastomas. Proc Natl Acad Sci USA. 1999;96:11519–11524. doi: 10.1073/pnas.96.20.11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Del Valle L, Gordon J, Enam S, et al. Expression of human neurotropic polyomavirus JCV late gene product AGNO protein in human medulloblastoma. J Natl Cancer Inst. 2001;94:267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- 34.Darbinyan A, Siddiqui KM, Slonina D, et al. Role of JC virus agnoprotein in DNA repair. J Virol. 2004;78:8593–8600. doi: 10.1128/JVI.78.16.8593-8600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darbinyan A, White MK, Akan S, et al. Alterations of DNA damage repair pathways resulting from JCV infection. Virology. 2007;364:73–86. doi: 10.1016/j.virol.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]