Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 1.
Published in final edited form as: Nat Genet. 2009 Jul 13;41(8):879–881. doi: 10.1038/ng.416
Abstract
We conducted meta-analyses of genome-wide association studies (GWAS) for atrial fibrillation (AF) in participants from five community-based cohorts. Meta-analyses of 896 prevalent (15,768 referents) and 2,517 incident (21,337 referents) AF cases identified a novel locus for AF (ZFHX3, rs2106261, risk ratio [RR]=1.19; _P_=2.3×10−7), an association that was replicated in the German AF Network (odds ratio=1.44; _P_=1.6×10−11). Combining the discovery and replication results, rs2106261 was significantly associated with AF (RR=1.25; _P_=1.8×10−15).
Keywords: atrial fibrillation, epidemiology, genetics, polymorphism, single nucleotide
With increasing longevity of individuals in developed countries, late-onset chronic cardiovascular diseases such as AF have become important public health problems. AF is an electrical disorder of the heart’s upper chambers characterized by an irregular heart rhythm. The overall lifetime risk of AF is almost 25% in the U.S. and Europe1,2. Furthermore, the incidence of AF is increasing over time; in the U.S. it is projected that up to 15.9 million individuals may be affected by 20503. The growing number of individuals with AF is of concern because of its association with significantly increased risks of stroke, heart failure, and death4.
AF is a complex disease with many etiologies, including cardiovascular disease and its risk factors. Families demonstrating Mendelian inheritance of AF have been reported, most frequently in individuals with lone AF (early-onset AF without structural heart disease)5. Recently it was reported that even for typical forms of AF, individuals with an affected relative are at higher risk of AF6. Moreover, a GWAS identified single nucleotide polymorphisms (SNPs) in the chromosome 4q25 region that are associated with increased AF risk7.
We hypothesized that additional common genetic variation contributes to the development of AF. We conducted and combined meta-analyses of prevalent AF and incident AF, using existing GWAS data from the _C_ohorts for _H_eart and _A_ging _R_esearch in _G_enomic _E_pidemiology (CHARGE) AF Consortium. CHARGE included the following five community-based cohorts8: Age, Gene/Environment Susceptibility Reykjavik Study (AGES); Atherosclerosis Risk in Communities (ARIC); Cardiovascular Health Study (CHS); Framingham Heart Study (FHS); and Rotterdam Study (RS). Genotyping inclusion criteria were unbiased towards AF as genotyping was performed as a core effort for numerous phenotypes in each cohort. Study design and genotyping features are in Supplementary Tables 1 and 2. Genotypes for more than 2.5 million SNPs were imputed within each study using reference genotype data and linkage disequilibrium patterns from the HapMap CEU population.
Our community-based participants were middle-aged to elderly, with mean ages at DNA collection from 57 (ARIC) to 76 (AGES) years (Supplementary Table 3). To assess potential population stratification, we computed genomic inflation factors (λ) of meta-analyses results: λ was 1.005 for prevalent AF, 1.014 for incident AF, and 1.026 for combined prevalent-incident AF (Supplementary Table 2 provides λ by cohort and analysis). The observed versus expected P value distributions (quantile-quantile plots) and Manhattan plots of log10 P values for separate prevalent and incident AF analyses are displayed in Supplementary Figures 1 and 2.
We prespecified genome-wide significance as P<5×10−8, corresponding to significance at 5% adjusting for approximately one million independent tests as estimated in HapMap samples of European ancestry. To prioritize follow-up genotyping, we required SNPs to have P<4×10−7 (corresponding to one expected false positive per GWAS), and that at least six of nine analyses (out of four prevalent and five incident AF analyses) contributed results for the SNP, to reduce possible false-positives due to poor imputation.
The quantile-quantile plot and Manhattan plot of the meta-analysis of combined prevalent and incident AF are depicted in Supplementary Figure 3. We replicated the previously reported chromosome 4 locus7 (rs17042171, _P_=6.0×10−27; Table 1, Fig. 1a), which was approximately 150 kb telomeric from the transcription factor PITX2.
Table 1.
Summary of CHARGE atrial fibrillation genome-wide association meta-analysis signals with P ≤ 4×10−7 and German AFNET replication analysis
| Locus | Prevalent AFanalysis | Incident AFanalysis | Combined analysis of prevalent and incident AF896 prevalent 15,768 non-cases2,517 incident cases and 21,337 non cases | German AFNET2,145 cases, 4,073 control_s_ | Meta-analysisCHARGE Community-AFand German AFNET results | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPnearby gene | Chromosomeposition | Minor/majorallele | MinorallelefrequencyCHARGE/GermanAFNET | Oddsratio | P value | Hazardratio | P value | Range ofObserved/expectedvarianceratios | Overall_Beta_± s.e. | Relativeriska | Meta _P_value | Heterogeneity_P_ valueb | Supportingsignalsc | Overall_B_±s.e. | Oddsratio | P value | OverallBeta± s.e. | Relativerisk | P value |
| rs17042171dPITX2 | 4111927736 | A/C | 0.1220.156 | 1.59 | 3.1×10−11 | 1.40 | 8.3×10−18 | 0.96–1.0 | 0.37±0.03 | 1.45 | 6.0×10−27 | 0.01 | 75 | 0.90±0.06 | 2.46 | 6.9×10−51 | 0.50±0.03 | 1.65 | 3.9 x10−63 |
| rs2106261_ZFHX_3 | 1671609121 | T/C | 0.1740.192 | 1.33 | 9.0×10−6 | 1.14 | 7.9×10−4 | 0.66–1.0 | 0.17±0.03 | 1.19 | 2.3×10−7 | 0.01 | 7 | 0.36±0.05 | 1.44 | 1.6×10−11 | 0.23±0.03 | 1.25 | 1.8×10−15 |
| rs17375901_MTHFR_ | 111775103 | T/C | 0.0530.058 | 1.41 | 8.5×10−4 | 1.30 | 1.2×10−5 | 0.81–1.0 | 0.29±0.05 | 1.33 | 4.6×10−8 | 0.45 | 8 | 0.04±0.09 | 1.04 | 0.68 | 0.23±0.05 | 1.26 | 5.9×10−7 |
Figure 1. Regional association plots for signal loci on chromosomes 4, 16 and 1.
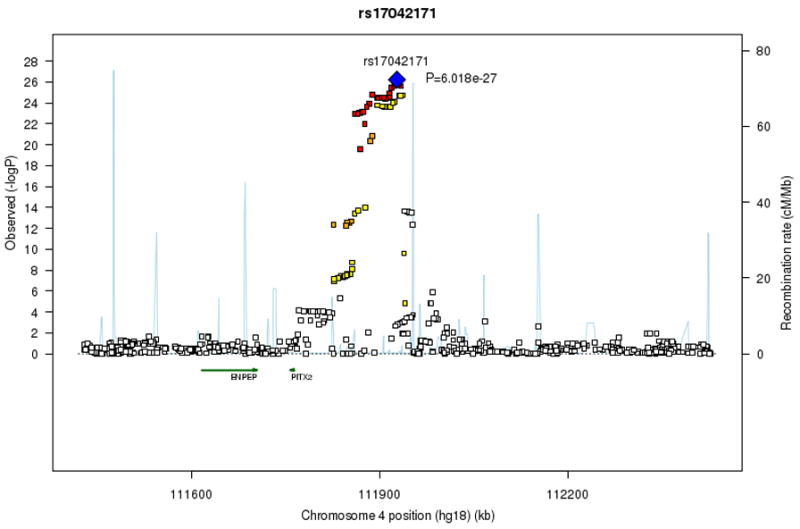
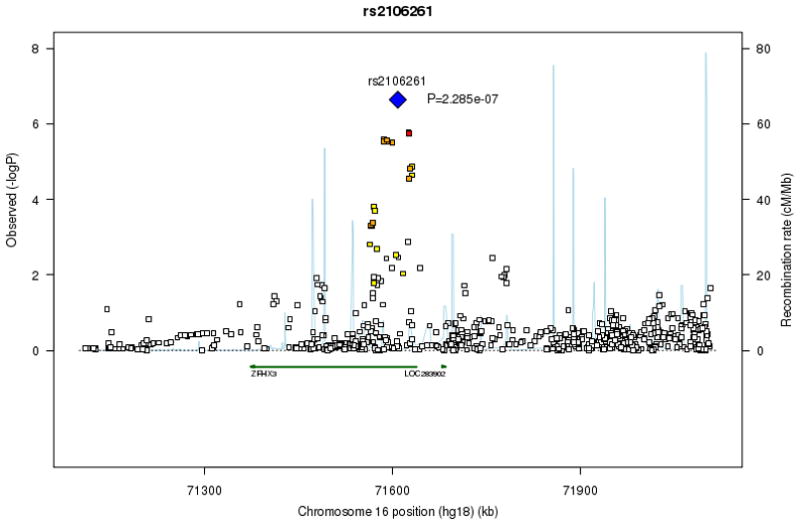
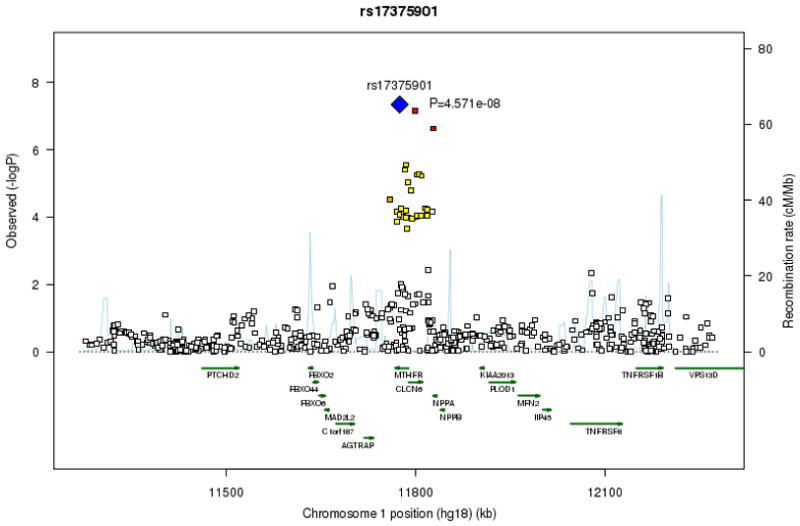
At each SNP location (genomic position, NCBI Build 36) we plot the log10 P value from combined analysis of incident and prevalent AF. Symbol colors indicate the strength of linkage disequilibrium derived from CEU HapMap build 22: strong (red, r2≥0.8) moderate (orange, 0.5≤r2<0.8) weak (yellow, 0.20≤r2<0.5) and low (white, r2<0.2). Estimated recombination rates are represented by pale blue lines and gene annotations by dark green arrows.
SNP rs2106261 on chromosome 16q22, located in an intronic region of transcription factor _ZFHX_3 (previously known as ATBF1), showed suggestive evidence of association (Table 1, combined prevalent-incident _P_=2.3×10−7, Fig. 1b). Results were consistent in the separate prevalent (_P_=9.0×10−6) and incident (_P_=7.9×10−4) AF analyses (Supplementary Table 4 provides cohort-specific estimates). We replicated the association between SNP rs2106261 and AF in a large independent cohort, the German AFNET consisting of 2,145 cases and 4,073 controls (odds ratio=1.44, _P_=1.6×10−11; Table 1). In a meta-analysis of the results from the discovery (CHARGE community AF) and replication (German AFNET) studies, rs2106261 was significantly associated with AF (RR 1.25, _P_= 1.8×10−15; Table 1). ZFHX3 appears to regulate myogenic9 and neuronal differentiation10. ZFHX3 has been reported to be a tumor suppressor gene in multiple cancers11, and recently SNPs in ZFHX3 have been associated with susceptibility to Kawasaki Disease12. Although the function of ZFHX3 in cardiac tissue is unknown, it is expressed in mouse13 hearts.
Another significant association signal was on chromosome 1p36 within MTHFR (rs17375901, _P_=4.6×10−8), which encodes 5,10-methylenetetrahydrofolate reductase. The association with the MTHFR locus was not confirmed in independent subjects from the AFNET cohort (Table 1). The initial MTHFR finding may be a false positive result. However, the region may merit further investigation because MTHFR is in linkage disequilibrium with the atrial natriuretic peptide gene (Fig. 1c); a NPPA frameshift mutation has been described in a family with AF14.
We acknowledge several study limitations. Although our findings were generally consistent, we observed some between-analysis heterogeneity in effect sizes (_P_=0.01), possibly arising from variation in cohort participant characteristics, duration and etiology of AF, low study-specific precision, subtle locus-specific population stratification, and population differences in underlying haplotype structure. Population stratification at a larger scale did not appear to have a substantial impact on our findings as we did not observe inflation of the genomic control factors in the study-specific analyses or the meta-analyses. We note that for the previously validated PITX2 locus we observed between-study heterogeneity. Thus, heterogeneity appears to be a general feature of even the strongest genome-wide findings for AF, and remains to be addressed in follow up studies. In addition, our findings may not be generalizable to other races/ethnicities. It also was not possible to perform a pooled analysis using participant specific data given the restrictions imposed by the Institutional Review Boards at some study sites. Furthermore, there is a potential for survival bias in the prevalent AF analysis if the variant is associated with both AF onset and lethality; in this situation individuals who died shortly after AF onset might not survive until DNA collection. Nonetheless, a moderate association was present in prevalent, incident, and combined AF meta-analyses for both the validated chromosome 4q25 and the novel chromosome 16q22 loci. Another limitation is that beyond single SNPs, our study did not analyze patterns of haplotypes, and thus complex haplotype associations may not have been captured in this study. However, our use of imputation to the HapMap does leverage available linkage disequilibrium information. Finally, we recognize that we likely have identified variants in linkage disequilibrium with causal variants rather than the specific functional variants; the pathophysiology by which locus variation contributes to AF risk remains unknown.
Our study has multiple strengths. We included five community-based cohorts, with large numbers of cases, whose participants were not selected for phenotypic characteristics, thereby enhancing the generalizability of our findings. The robustness of the chromosome 16q22 result is strengthened by its documentation in samples ascertained with different study designs including case-control and cohort studies.
In summary, by examining GWAS data for AF in five community-based cohorts we replicated the previously reported association with chromosome 4q25 variants and we identified a novel locus on chromosome 16 in a gene encoding the transcription factor _ZFHX_3. We provided confirmatory support for the novel ZFHX3 finding by replicating our findings in a large independent study of AF. Further studies are needed to elucidate functional variants and mechanisms by which the novel 16q22 locus predisposes to AF.
URLS
AGES, http://www.hjarta.is/english/ages
ARIC, http://www.cscc.unc.edu/aric/
CHS, http://www.chs-nhlbi.org/
FHS, http://www.framinghamheartstudy.org/about/index.html
RS, http://www.epib.nl/ergo.htm
BIMBAM, http://stephenslab.uchicago.edu/software.html
EIGENSTRAT, http://genepath.med.harvard.edu/~reich/Software.htm
GenABLE and ProbABEL (http://mga.bionet.nsc.ru/~yurii/ABEL/)
HapMap, http://hapmap.org
MACH v1.0.15/16 (all others; http://www.sph.umich.edu/csg/abecasis/MaCH/index.html)
PLINK http://pngu.mgh.harvard.edu/purcell/PLINK/
Supplementary Material
1
Acknowledgments
AGES: The Age, Gene/Environment Susceptibility Reykjavik Study has been funded by NIH contract N01-AG-12100, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament).
ARIC: The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, N01-HC-55022, R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
CHS: The Cardiovascular Health Study research reported in this article was supported by contract numbers N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, and grant numbers U01 HL080295, R01 HL087652, and R01 HLO88456 from the National Heart, Lung, and Blood Institute, with additional contribution from the National Institute of Neurological Disorders and Stroke. DNA handling and genotyping was supported in part by National Center for Research Resources grant M01RR00069 to the Cedars-Sinai General Clinical Research Center Genotyping core and National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center. A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/pi.htm.
FHS: This research was conducted using data and resources from the Framingham Heart Study of the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine based on analyses by Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. This work was supported by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195) and its contract with Affymetrix, Inc for genotyping services (Contract No.N02-HL-6-4278). A portion of this research utilized the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center. The Framingham Heart Study research was supported by NIH grants 1R01HL092577-01A1 (PTE, EJB); HL076784, AG028321 (EJB); 6R01-NS 17950 (PAW); T32 HL007575 (SAL); R01 HL093328 and RO1 HL 080124 (RSV); Deane Institute for Integrative Research in Atrial Fibrillation and Stroke (PTE). The Deutsche Forschungsgemeinschaft (German Research Foundation) Research Fellowship SCHN 1149/1-1 supported Gutenberg Heart Study investigator RBS’ FHS research.
AFNET/KORA: German National Genome Research Network NGFN 01GS0499 and 01GS0838 (SK), NGFN 01GR0803 (AP), NGFN 01GR0103 (TM), German Federal Ministry of research BMBF 01EZ0874 (AP), German Competence Network on AF (AF-Net) 01 GI 0204/N (SK, HEW), Leducq Foundation 07-CVD 03 (SK), LMU Excellence Initiative (SK), KORA is supported by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria.
RS: The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University Rotterdam; The Netherlands Organization for Scientific Research; The Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly; The Netherlands Heart Foundation; the Ministry of Education, Culture and Science; the Ministry of Health Welfare and Sports; the European Commission; and the Municipality of Rotterdam. Support for genotyping was provided by The Netherlands Organization for Scientific Research (NWO) (175.010.2005.011, 911.03.012) and Research Institute for Diseases in the Elderly (RIDE). This study was supported by The Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) project nr. 050-060-810.
Statistical analyses K.M.R., D.E.A., A.P., C.V.N., A.V.S., A.D., R.B.D., T.L., G.B.E., T.A., M.L., K.W., A.K., C.M.v.D., K.L.L.; Informatics A.V.S., T.A., K.L.L.; Data Preparation T.A., K.W., A.C., K.L.L., V.G.; Sample collection and phenotype collection E.J.B., C.V.N., A.V.S., R.B.S., E.B., M.F.S., J.H., E.Z.S., T.J.W., G.E., D.L., B.M.P., A.M.C, A.H., R.S.V., S.K.A., P.A.W., S.R.H. V.G., A.A., S.K.; Genotyping A.V.S., M.F.S., T.A., G.E., J.I.R., V.G., Manuscript writing E.J.B., K.M.R., D.E.A., S.A.L., K.L.L., V.G.; Review and revision of the manuscript E.J.B., K.M.R., D.E.A., A.P., C.V.N., A.V.S.,R.B.S., J.C.B., E.B., A.D., S.A.L., T.L., G.B.E., J.H., T.A., C.N.-C., M.G.L., K.D.M., E.Z.S., F.R., T.J.W., G.E., D.L., B.M.P., A.M.C, A.H., R.S.V., T.B.H., J.I.R., W.H.L.K., B.H.C.S., L.J.L., N.L.S., A.C., A.G.U., N.S., A.K., C.M.v.D., K.L.L., S.R.H., V.G., A.A.,, S.K., P.T.E., J.C.W. Intellectual input E.J.B., K.M.R., D.E.A., R.B.S., M.G.L., K.L.L., S.R.H., S.K, P.T.E., J.C.W.; Study design E.J.B., K.M.R., D.E.A., A.P., C.V.N., T.L., M.G.L., G.E., B.M.P., A.H., R.S.V., T.B.H., B.H.C.S., L.J.L., A.G.U., N.S., K.L.L., S.R.H., S.K., P.T.E., J.C.W. Study interpretation E.J.B., K.M.R., C.V.N., R.B.S., J.C.B., E.B., T.L., R.S.V., W.H.L.K., K.L.L., S.R.H., A.A., S.K., P.T.E., Study coordination E.J.B., J.C.B.,G.E., S.R.H., S.K., P.T.E., Study funding E.J.B., A.P., E.B., B.M.P., T.B.H., L.J.L., P.A.W., V.G., S.K., P.T.E.
Abbreviations
AF
atrial fibrillation
λ
genomic inflation factor
CHARGE
Cohorts for Heart and Aging Research in Genomic Epidemiology
AGES
Age, Gene/Environment Susceptibility Reykjavik Study
ARIC
Atherosclerosis Risk in Communities
CHS
Cardiovascular Health Study
FHS
Framingham Heart Study
RS
Rotterdam Study
GWAS
genome-wide association study
RR
risk ratio
SNP
single nucleotide polymorphism
Footnotes
COMPETING INTERESTS STATEMENT
Aravinda Chakravarti is a paid member of the Scientific Advisory Board of Affymetrix, a role that is managed by the Committee on Conflict of Interest of the Johns Hopkins University School of Medicine.
References
- 1.Lloyd-Jones DM, et al. Circulation. 2004;110:1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 2.Heeringa J, et al. Eur Heart J. 2006;27:949–953. doi: 10.1093/eurheartj/ehi825. [DOI] [PubMed] [Google Scholar]
- 3.Miyasaka Y, et al. Circulation. 2006;114:119–125. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- 4.Stewart S, et al. Am J Med. 2002;113:359–364. doi: 10.1016/s0002-9343(02)01236-6. [DOI] [PubMed] [Google Scholar]
- 5.Ellinor PT, et al. Hum Gene t. 2005;118:179–184. doi: 10.1007/s00439-005-0034-8. [DOI] [PubMed] [Google Scholar]
- 6.Fox CS, et al. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 7.Gudbjartsson DF, et al. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 8.Psaty BM, et al. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry FB, et al. J Biol Chem. 2001;276:25057–25065. doi: 10.1074/jbc.M010378200. [DOI] [PubMed] [Google Scholar]
- 10.Jung CG, et al. Development. 2005;132:5137–5145. doi: 10.1242/dev.02098. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, et al. Nat Genet. 2005;37:407–412. doi: 10.1038/ng1528. [DOI] [PubMed] [Google Scholar]
- 12.Burgner D, et al. PLoS Genet. 2009;5:e1000319. doi: 10.1371/journal.pgen.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ido A, et al. Gene. 1996;168:227–231. doi: 10.1016/0378-1119(95)00740-7. [DOI] [PubMed] [Google Scholar]
- 14.Hodgson-Zingman DM, et al. N Engl J Med. 2008;359:158–165. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1