Structural characterization of α-synuclein in an aggregation prone state (original) (raw)
Abstract
The relation of α-synuclein (αS) aggregation to Parkinson's disease has long been recognized, but the pathogenic species and its molecular properties have yet to be identified. To obtain insight into the properties of αS in an aggregation-prone state, we studied the structural properties of αS at acidic pH using NMR spectroscopy and computation. NMR demonstrated that αS remains natively unfolded at lower pH, but secondary structure propensities were changed in proximity to acidic residues. The ensemble of conformations of αS at acidic pH is characterized by a rigidification and compaction of the Asp and Glu-rich C-terminal region, an increased probability for proximity between the NAC-region and the C-terminal region and a lower probability for interactions between the N- and C-terminal regions.
Keywords: Parkinson's disease, neurodegeneration, aggregation, protein structure
Introduction
The pathological hallmark of Parkinson's disease and several other neurodegenerative disorders is the deposition of intracytoplasmic neuronal inclusions termed Lewy bodies.1 The major component of Lewy bodies are amyloid fibrils of the protein α-synuclein (αS)2 and locus triplication causing an increased dosage of the αS gene potentiates Parkinson's disease.3 Mutations of αS associated with familial Parkinson's disease (A30P, A53T, and E46K) have an increased aggregation propensity in vitro, in agreement with aggregation of αS into fibrillar Lewy bodies in vivo.4,5 Recent reports suggest that the mature fibrils have a protective function. Instead, soluble aggregation intermediates may constitute the major toxic species.6 As the loss of αS seems to have minimal effects on development,7 the pathogenic effects of the aggregation intermediates are attributed to a toxic gain-of function.
αS is a 140-amino acid protein of unknown function. The amino acid sequence of αS is characterized by the amphipathic N-terminus (residues 1–60), showing the consensus repeats KTKEGV and involved in lipid binding, the highly hydrophobic self-aggregating sequence known as NAC (residues 61–95), which is presumed to initiate fibrillation, and the acidic C-terminal region (residues 96–140), rich in Pro, Asp, and Glu residues and critical for blocking rapid αS filament assembly [Fig. 1(A)].6 Structurally, αS belongs to the class of natively unfolded proteins that adopts an ensemble of conformations with no rigid secondary structure in solution.8 However, at neutral pH transient long-range interactions are present that stabilize a closed conformation.9,10 The mechanisms underlying the structural transition from the innocuous, monomeric conformations of αS to its neurotoxic forms are unclear.
Figure 1.
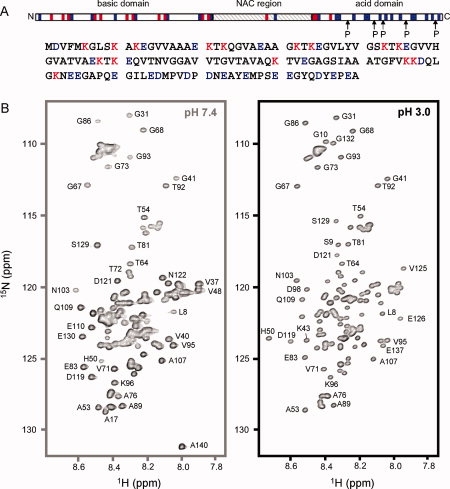
The aggregation-prone state of αS at acidic pH is natively unfolded. A: Primary sequence of αS. αS consists of three regions based on the distribution of charged residues. Residues that are positively and negatively charged at pH 7.4 are color-coded red and blue, respectively. B: 1H-15N HSQC spectra of αS at pH 7.4 (left) and pH 3.0 (right). Both spectra show only a limited dispersion of chemical shifts typical of a natively unfolded protein. Selected cross peaks are labeled with their assignment.
It has been hypothesized that a pathological decrease in pH occurs in neurons affected by neurodegenerative diseases resulting in apoptosis.11 It has also been suggested that lowering the reduction in the overall net charge of proteins accompanying lowering of the pH can increase the aggregation propensity.12 Although selectively maintaining in vivo affected cells at a slightly alkaline pH is probably not a viable therapeutic strategy for the treatment of neurodegenerative disorders, studying variable pH in vitro may give insights into in vivo factors that increase the aggregation propensity of αS. Indeed, previous studies suggested that αS converts into a partially folded intermediate conformation at pH 3.0 and that conditions of low pH correlate with enhanced kinetics of fibrillation.12 Both nucleation and growth rate constants of fibril-formation were increased under acidic conditions, with a maximum in the nucleation rate at pH 3.0, suggesting that the partially folded intermediate of αS is an important species on the fibril-forming pathway.12,13
Here, we characterized the secondary and tertiary structure propensities of αS at acidic pH, the conditions in which previous studies suggested an aggregation-prone partially intermediate conformation of αS,12 at single-residue resolution using NMR spectroscopy and computation.
Results and Discussions
The aggregation-prone state of αS at acidic pH is natively unfolded
To study with single-residue resolution, the structure of the aggregation-prone intermediate state of αS, which was suggested on the basis of CD, ANS, and SAXS measurements,12 we used multidimensional NMR spectroscopy. NMR resonances in the 1H-15N HSQC spectrum of αS at pH 3.0 were sharp and showed only a limited dispersion of chemical shifts (∼1.2 ppm in the proton dimension) [Fig. 1(B)], indicating that the aggregation-prone state of αS at pH 3.0 is natively unfolded. In addition, SAXS measurements had shown that αS is monomeric at pH 3.0.12
Secondary structure propensities in αS at acidic pH are changed in the proximity to Glu and Asp residues
Since 15N chemical shifts are sensitive to many changes in the chemical environment such as pH, temperature and ionic strength, 13C backbone chemical shifts were needed to determine secondary structure propensities in αS at pH 3.0. Figure 2(A) shows the secondary structure propensities estimated by the program SSP14 on the basis of experimental Cβ, Cα, CO, HN, and N chemical shifts observed in αS at pH 3.0. Positive and negative SSP scores indicate propensity for formation of helical and β-structure, respectively. Along the primary sequence, stretches of 5–10 residues with positive and negative SSP scores alternate. The largest stretch of residual helical structure was observed for residues 14–27, as reported previously at pH 7.4.15 However, almost all SSP values are <20% indicating that no rigid elements of regular secondary structure are formed in the aggregation-prone state of αS at pH 3.0 [Fig. 2(A)], in agreement with circular dichroism that suggested only limited secondary structure in αS at pH 3.0.12 Comparison of Cα chemical shifts observed at pH 3.0 and pH 7.4 shows that, as expected, mostly the Cα resonances of acidic residues shift upon lowering the pH (Supporting Information Figure 1). In addition, differences in SSP scores and 3J(HNHα) scalar coupling values indicate an increased propensity for helical or turn-like structure for residues 11–19 and 41–56 [Fig. 2(B,C)].
Figure 2.
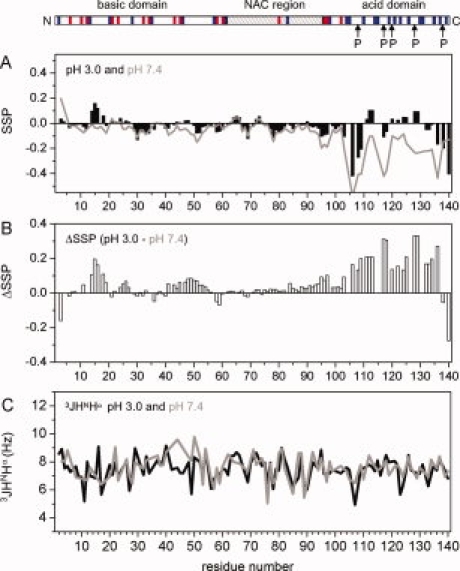
Secondary structure propensities of αS at acidic pH. A: SSP analysis using Cα and Cβ chemical shifts. Positive SSP values are indicative of residual helical structure, negative values of residual β-structure. Note that all Glu and Asp residues were excluded in this analysis. B: Difference in SSP scores at pH 3.0 and pH 7.4. Most difference values are positive suggesting a preferentially population of helical and turn conformations at pH 3.0, in particular in the C-terminal region. C: 3J(HNHα) scalar couplings at pH 3.0 (black) and pH 7.4 (gray). On top, the region organization of αS is shown. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.\]
The C-terminal region of αS is more rigid at acidic pH than at neutral pH
The largest differences in Cα chemical shifts and SSP scores at pH 3 and pH 7.4 were observed in the Asp and Glu rich C-terminal region [Fig. 2(A,B)]. Whereas at pH 7.4, residues in the C-terminal region preferentially populate extended structure,16,17 propensity for extended structure is reduced at pH 3.0 and some residue stretches show a tendency to form helical structure. In agreement with a higher propensity for helical and turn conformations, the 3J(HNHα) scalar couplings of Ala107, Glu123, and Tyr133 were decreased by 1.6, 1.8, and 0.6 Hz down to values of 4.9, 5.7, and 6.3 Hz, respectively [Fig. 2(C)].
Signal intensities observed in a 2D HSQC spectrum for the 40 C-terminal residues are on average lower than for those observed in the N-terminal region [Fig. 3(A)]. In addition, steady-state heteronuclear 15N{1H}-NOE values of residues 118–132 are higher at pH 3.0 than at pH 7.4 [Fig. 3(B)], indicating a reduced flexibility of the C-terminal region at pH 3.0. At the same time, chemical exchange is present in this region, as suggested by the difference in 15N R2 and R1ρ spin relaxation rates [Fig. 3(C)].
Figure 3.
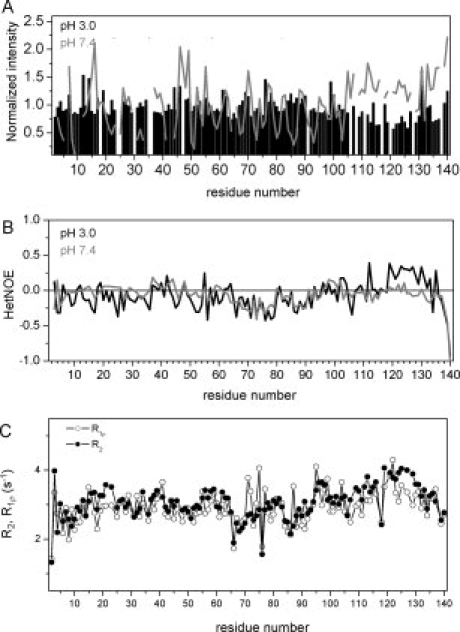
The C-terminal region of αS is more rigid at acidic pH. A: Comparison of normalized peak intensities observed in 1H-15N HSQC spectra of αS at pH 3.0 and pH 7.4. At pH 7.4, the peak intensities observed for residues in the C-terminal region are higher than those observed for residues located in the N-terminal region. At pH 3.0, however, residues 105–130 and 134–137 have lower peak intensities than residues in the N-terminal region, indicating that residues in the C-terminal region are less flexible at pH 3.0. B: Steady-state heteronuclear 15N{1H}-NOEs of αS at pH 7.4 (gray) and at pH 3.0 (black). B: 15N R2 and R1ρ spin relaxation rates of backbone resonances of αS at pH 3.0.
Residual dipolar couplings are exquisitely sensitive to bond vector orientation and can be measured in a weakly aligned protein, for which the large internuclear dipolar interactions no longer average to zero. At pH 3.0, residues 100–140 are uncharged and electrostatic interactions will be strongly altered, suggesting that the RDC profile observed in αS will be changed. Indeed, whereas large positive NH RDCs were observed in the C-terminal region at pH 7.4 in agreement with previous measurements [Fig. 4(A)],9 both positive and negative RDCs were observed at pH 3.0. The RDC profile is characterized by three residue stretches with a length of approximately 10 residues each and starting at residue Glu105. Within each fragment positive NH RDCs were observed in the N-terminal part, whereas negative NH RDCs were observed at the end [Fig. 4(B)]. Sign inversion of RDCs might be due to changes in the local alignment tensor,18 and due to formation of helical19 or turn conformation,20 in which the H—N internuclear vector is parallel to the long axis of this segment. Taken together, the NMR data demonstrate that the C-terminal region of αS is more rigid at pH 3.0 than at pH 7.4. Most likely are the formation of short stretches of helical and turn conformation in acidic conditions.
Figure 4.
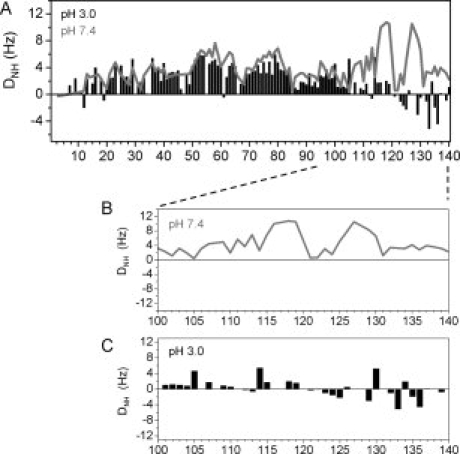
Residual dipolar couplings probe structural changes in C-terminal region. A: One-bond H—N RDCs of αS at pH 7.4 (gray line) and at pH 3.0 (black bars) as a function of residue number. B, C: One-bond H—N RDCs of residues 100–140 in detail.
The network of transient long-range interactions is changed at acidic pH
To support the compaction of the C-terminal region of αS at pH 3.0, we estimated the hydrodynamic radius _R_h using pulsed-field gradient NMR experiments.21 _R_h values of about 32 ± 1 Å were obtained for full-length αS at both pH 7.4 and pH 3.0 (Fig. 5). The values are between that of a random coil and a globular conformation expected for the 140-residue protein αS, in agreement with the closed conformation reported previously at neutral pH.9 In addition, we determined the _R_h values for a fragment of αS that does not contain the C-terminal region, αS(1–108), and for a peptide comprising only residues 105–136 of αS, αS(105–136). For αS(1–108), the _R_h values were 23 ± 1 Å and 26 ± 1 Å at pH 7.4 and pH 3.0, respectively, and for αS(105–136) 17 ± 2 Å and 12 ± 3 Å, respectively (Fig. 5). Thus, whereas the N-terminal 108 residues are more extended at acidic pH, the Asp and Glu-rich C-terminal region is more compact than at neutral pH, in agreement with the increased ordering observed by NMR chemical shifts, scalar couplings, spin relaxation measurements, and RDCs (Figs. 2–4). For both αS(1–108) and αS(105–136), the hydrodynamic radius changed by 4–5 Å upon lowering the pH to 3.0, but as the change is in the opposite direction, no significant change in _R_h was observed for full-length αS. In contrast, values of the radius of gyration as estimated by SAXS suggested a compaction of full-length αS at pH 3.0.12 We currently do not know the reason for this difference, but it could be due to differences in buffer condition or temperature. However, the development of the beginnings of a packed core, as suggested by a Kratky plot analysis of the SAXS data at pH 3.0,12 is in agreement with the compaction of the C-terminal region observed by pulsed-field gradient NMR.
Figure 5.
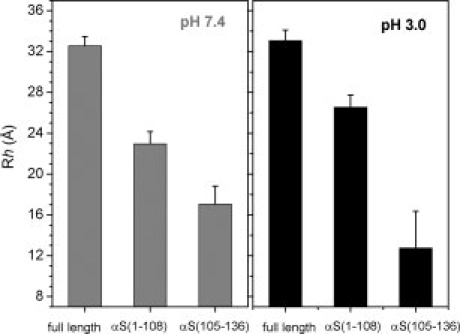
The C-terminal region of αS is more compact at acidic pH. A: Hydrodynamic radius values _R_h of full-length αS, αS(1–108), and αS(105–136) as estimated by pulsed-field gradient NMR. The left panel shows _R_h values at pH 7.4, the right panel at pH 3.0. Error bars are based on two independent measurements.
To probe changes in the long-range structural order with single-residue resolution, we used paramagnetic enhancement of nuclear spin relaxation by nitroxide spin labels.22 A nitroxide radical causes broadening of the NMR signals of nearby protons, an effect that extends as far as 25 Å from modified cysteine residues. For attachment of the MTSL label, we used previously prepared single cysteine mutants of αS (A18C, A90C, and A140C),9 together with one new variant (A76C). Comparison of the paramagnetic relaxation enhancement (PRE) profiles of αS at pH 3.0 and pH 7.4 showed [Fig. 6(A) and Supporting Information Figure 2]: (1) PRE broadening observed for residues of the C-terminal region, when the spin label is attached to A18C, was weakened at pH 3.0; (2) PRE broadening induced by A76C-MTSL and A90C-MTSL in the C-terminal region was much stronger at acidic pH; (3) PRE broadening induced by A90C-MTSL extended further towards the N-terminus at pH 3.0; and (4) attachment of MTSL to A140C resulted in strong PRE broadening of the NMR signals of residues 98–140, whereas less signal attenuation was observed in the N-terminal region when compared with the data at pH 7.4.
Figure 6.
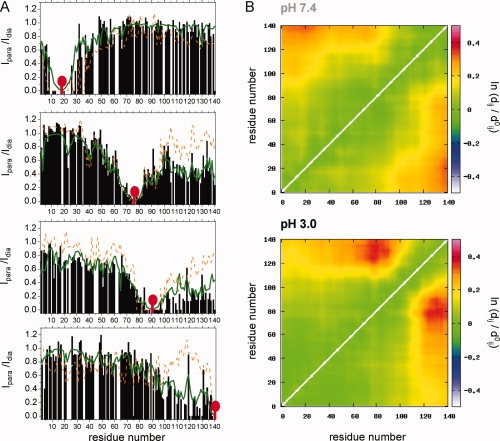
Transient long-range interactions in αS. A: Comparison of experimental PRE profiles (black bars for pH 3.0, orange dashed line for pH 7.4) with the PRE profiles calculated from the FM simulation at pH 3.0 (green line). The nitroxide label MTSL (red mark) was attached to the single-cysteine mutants A18C, A76C, A90C, and A140C (from top to bottom). B: Long-range contacts in the representative ensemble as a function of pH. Contacts are plotted as log(〈_dij〉/〈dij,_ref〉), where _dij,_ref refers to the distance in the reference ensemble of 10,000 structures and d ij refers to the distance in the selected ensemble.
Ensembles of structures that are in agreement with the two data sets support the interpretation of the raw data [Fig. 6(B)]. At pH 3.0, the N- and C-termini have a decreased probability to be in proximity. In contrast, a higher probability for spatial proximity is seen between the NAC region and residues 120–140 at pH 3.0. Thus, whereas the C-terminal half of αS becomes more compact at pH 3, the N—C terminal interaction seems to be weakened, in agreement with its electrostatic nature.23 The overall dimensions of the ensemble of structures are not significantly changed, with hydrodynamic radius values of 30.7 and 30.5 Å as predicted by HydroPRO24 at pH 7.4 and pH 3.0, respectively. Moreover, no change in the overall dimensions of the ensemble of structures was detected by pulsed field gradient NMR measurements (Fig. 5).
In conclusion, NMR spectroscopy demonstrated that the intermediate conformation of αS at acidic pH, which was suggested to be an important species on the fibril-forming pathway of αS,12 is an ensemble of natively unfolded conformations. The ensemble of conformations of αS at acidic pH is characterized by a rigidification and compaction of the Asp and Glu-rich C-terminal region, an increased probability for proximity between the NAC-region and the C-terminal region and a lower probability for interactions between the N- and C-terminal regions (Fig. 7). The structural changes allow different hypotheses. First, a release of transient long-range interactions between the N- and C-terminal region can contribute to the increased nucleation rate constants of fibril-formation as observed for αS at acidic pH.12 Alternatively, the differences in aggregation might be related to the increased contacts between the C-terminus and the middle region of αS. A shift in contacts from the N-terminus to the more hydrophobic amyloid-forming region may promote self-association and thus increase aggregation. At the same time, the impact of the changes in transient long-range interactions on aggregation at acidic pH might only be a minor contribution, which is dominated by the strong reduction in negative charge in the C-terminal region at acidic pH.25
Figure 7.
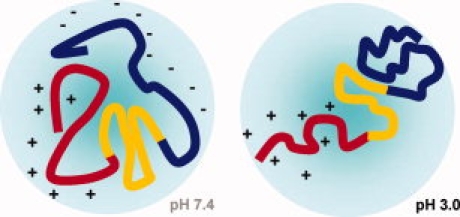
Schematic representation of the conformational changes that occur in αS, when the pH is lowered to pH 3.0.
Materials and Methods
NMR samples contained ∼0.1 m_M_ 15N- or 15N/13C-labelled αS in 20 m_M_ Na acetate, 100 m_M_ NaCl, pH 3.0. NMR experiments were recorded on Bruker Avance 600, 700, and 800 MHz NMR spectrometers at 15°C. PRE effects were measured from the peak intensity ratios between two 2D 15N-1H HSQC spectra with a paramagnetic sample and a diamagnetic sample, respectively.
Flexible-meccano, was used to sample the conformational space available to disordered proteins in terms of explicit ensembles of molecules.26 Unbiased conformational ensembles of 10,000 structures were calculated, and effective relaxation rates for each conformer calculated in the presence of the four spin probes. Relaxation rates for each conformer are described using the model free formalism introduced for interpretation of 1H-1H dipole–dipole cross relaxation (nOe) interactions,27 and recently adapted to the calculation of PRE by Clore et al.28 The correlation time for the electron-nuclear interaction τc was set to 4 ns, in agreement with previous studies.22 The average relaxation rate was calculated over all individual backbone conformers. Experimental values for the intrinsic transverse relaxation rates15 were used to account for the diamagnetic contribution to the spin relaxation rate.
Subensembles were then selected on the basis of agreement with respect to the experimental data using a genetic algorithm. Simulations indicated that for a molecule of this size with four spin labels, ensembles of 80 structures presented a reasonable description of the conformational behavior while avoiding over-fitting. A more detailed description of this algorithm is in preparation. The identical procedure was applied for both samples, and contact maps showing a logarithmic comparison between the selected ensemble and the original unbiased ensemble are used to compare the data.
A detailed description of Materials and Methods is available in the Supporting Information.
Acknowledgments
The authors thank Carlos Bertoncini for help with NMR measurements in the initial stages of the project, and Pinar Karpinar and Christian Griesinger for useful discussions.
References
- 1.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. Alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 4.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 6.Lashuel HA, Lansbury PT., Jr Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q Rev Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 7.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 8.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 9.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 11.Harguindey S, Reshkin SJ, Orive G, Arranz JL, Anitua E. Growth and trophic factors, pH and the Na+/H+ exchanger in Alzheimer's disease, other neurodegenerative diseases and cancer: new therapeutic possibilities and potential dangers. Curr Alzheimer Res. 2007;4:53–65. doi: 10.2174/156720507779939841. [DOI] [PubMed] [Google Scholar]
- 12.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 13.Morris AM, Finke RG. Alpha-synuclein aggregation variable temperature and variable pH kinetic data: a re-analysis using the Finke-Watzky 2-step model of nucleation and autocatalytic growth. Biophys Chem. 2009;140:9–15. doi: 10.1016/j.bpc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synucleimplications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bussell R, Jr, Eliezer D. Residual structure and dynamics in Parkinson's disease-associated mutants of alpha-synuclein. J Biol Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 16.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 17.Kim HY, Heise H, Fernandez CO, Baldus M, Zweckstetter M. Correlation of amyloid fibril beta-structure with the unfolded state of alpha-synuclein. Chembiochem. 2007;8:1671–1674. doi: 10.1002/cbic.200700366. [DOI] [PubMed] [Google Scholar]
- 18.Skora L, Cho MK, Kim HY, Becker S, Fernandez CO, Blackledge M, Zweckstetter M. Charge-induced molecular alignment of intrinsically disordered proteins. Angew Chem Int Ed Engl. 2006;45:7012–7015. doi: 10.1002/anie.200602317. [DOI] [PubMed] [Google Scholar]
- 19.Mohana-Borges R, Goto NK, Kroon GJ, Dyson HJ, Wright PE. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J Mol Biol. 2004;340:1131–1142. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Mukrasch MD, Markwick P, Biernat J, Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J Am Chem Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 21.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 22.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 23.Bernado P, Bertoncini CW, Griesinger C, Zweckstetter M, Blackledge M. Defining long-range order and local disorder in native alpha-synuclein using residual dipolar couplings. J Am Chem Soc. 2005;127:17968–17969. doi: 10.1021/ja055538p. [DOI] [PubMed] [Google Scholar]
- 24.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43:16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- 26.Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RW, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruschweiler R, Roux B, Blackledge M, Griesinger C, Karplus M, Ernst RR. Influence of rapid intramolecular motion on NMR cross-relaxation rates—a molecular-dynamics study of antamanide in solution. J Am Chem Soc. 1992;114:2289–2302. [Google Scholar]
- 28.Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against 1H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]