An Integrated Genomic Analysis of Human Glioblastoma Multiforme (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 11.
Published in final edited form as: Science. 2008 Sep 4;321(5897):1807. doi: 10.1126/science.1164382
Abstract
Glioblastoma multiforme (GBM) is the most common and lethal type of brain cancer. To identify the genetic alterations in GBMs, we sequenced 20,661 protein coding genes, determined the presence of amplifications and deletions using high-density oligonucleotide arrays, and performed gene expression analyses using next-generation sequencing technologies in 22 human tumor samples. This comprehensive analysis led to the discovery of a variety of genes that were not known to be altered in GBMs. Most notably, we found recurrent mutations in the active site of isocitrate dehydrogenase 1 (IDH1) in 12% of GBM patients. Mutations in IDH1 occurred in a large fraction of young patients and in most patients with secondary GBMs and were associated with an increase in overall survival. These studies demonstrate the value of unbiased genomic analyses in the characterization of human brain cancer and identify a potentially useful genetic alteration for the classification and targeted therapy of GBMs.
Malignant gliomas are the most frequent and lethal cancers originating in the central nervous system. The most biologically aggressive subtype is glioblastoma multiforme (GBM) [World Health Organization (WHO) grade IV astrocytoma], a tumor associated with a dismal prognosis (1). The current standard of care for GBM patients—surgical resection followed by adjuvant radiation therapy and chemotherapy with the oral alkylating agent temozolomide—produces a median survival of only 15 months (2). Historically, GBMs have been categorized into two groups (“primary” and “secondary”) on the basis of clinical presentation (3). Secondary GBMs are defined as cancers that have clinical, radiologic, or histopathologic evidence of malignant progression from a preexisting lower-grade tumor, whereas primary GBMs have no such history and present at diagnosis as advanced cancers (4). Clinical differences have been re ported between the two groups, with secondary GBMs occurring less frequently (~5% of GBMs) and predominantly in younger patients (median age ~45 years versus ~60 years for primary GBM) (5, 6). The histopathologic findings of primary and secondary GBMs are indistinguishable, and the prognosis does not appear to be different after adjustment for age (5, 6).
Substantial research effort has focused on the identification of genetic alterations in GBMs that might help define subclasses of GBM patients with differing prognoses and/or response to specific therapies (7). Distinctions between the genetic lesions found in primary and secondary GBMs have been made, with TP53 mutations occurring more commonly in secondary GBMs and EGFR amplifications and PTEN mutations occurring more frequently in primary GBMs (6, 8, 9); however, none of these alterations is sufficiently specific to distinguish between primary and secondary GBMs. This issue is further confounded by the possibility that a fraction of GBMs designated as primary tumors may follow a sequence of genetic events similar to that of secondary lesions but not come to clinical attention until malignant progression to a GBM has occurred.
The comprehensive elucidation of genetic alterations in GBMs could provide novel targets that might be used for diagnostic, prognostic, or therapeutic purposes as well as to identify subgroups of patients that preferentially respond to particular targeted therapies. The determination of the human genome sequence and improvements in sequencing and bioinformatic technologies have recently permitted genome-wide sequence analyses in human cancers. We have previously studied the genomes of 11 breast and 11 colorectal cancers by determining the sequence of the more than 18,000 Consensus Coding Sequence (CCDS) and Reference Sequence (RefSeq) genes (10, 11). Here, we have analyzed 20,661 protein coding genes in 22 human GBM samples. To complement these sequencing data, we have also performed a genome-wide analysis of focal copy number alterations, including amplifications and homozygous deletions, using high-density oligonucleotide microarrays on the same GBM tumors. Finally, we have examined the expression profiles of these same samples using serial analysis of gene expression (SAGE) and next-generation sequencing technologies.
Sequencing strategy
We extended our previous sequencing strategy for identification of somatic mutations to include 23,219 transcripts from 20,661 genes (fig. S1). These included 2783 additional genes from the Ensembl databases that were not present in the CCDS or RefSeq databases analyzed in the previous studies (10, 11). In addition, we redesigned polymerase chain reaction (PCR) primers for regions of the genome that (i) were difficult to PCR amplify in previous studies or (ii) were found to share substantial identity with other human or mouse sequences. The combination of these new, redesigned, and existing primers sequences resulted in a total of 208,311 primer pairs (table S1) that were successfully used for sequence analysis of the coding exons of these genes.
Twenty-two GBM samples (table S2) were selected for PCR sequence analysis, consisting of 7 samples extracted directly from patient tumors and 15 samples passaged in nude mice as xenografts. In the first stage of this analysis, called the Discovery Screen, the primer pairs were used to amplify and sequence 175,471 coding exons and adjacent intronic splice donor and acceptor sequences in the 22 GBM samples and in one matched normal sample. The data were assembled for each amplified region and evaluated using stringent quality criteria (12), resulting in successful amplification and sequencing of 95.0% of targeted amplicons in the 22 tumors (Table 1). A total of 689 Mb of sequence data was generated in this fashion. The amplicon traces were analyzed using automated approaches to identify changes in the tumor sequences that were not present in the reference sequences of each gene. Alterations present in the normal control sample and in single nucleotide polymorphism (SNP) databases were then removed from further analyses. The remaining sequence traces of potential alterations were visually inspected to remove false-positive mutation calls generated by the automated software. All exons containing putative mutations were then reamplified and sequenced in both the affected tumor and the matched normal DNA sample. This process allowed us to confirm the presence of the mutation in the tumor sample and determine whether the alteration was somatic (i.e., tumor-specific) or was present in the germ-line. All putative somatic mutations were examined computationally and experimentally to confirm that the alterations did not arise through the aberrant coamplification of related gene sequences (12).
Table 1.
Summary of genomic analyses.
| Sequencing analysis | |
|---|---|
| Number of amplicons attempted | 219,229 (100%) |
| Number of amplicons passing quality control* | 208,311 (95%) |
| Fraction of bases in passing amplicons with PHRED > 20 | 98.3% |
| Number of genes analyzed | 20,661 |
| Number of transcripts analyzed | 23,219 |
| Number of exons analyzed | 175,471 |
| Total number of nucleotides successfully sequenced | 689,071,123 |
| Number of somatic mutations identified (n = 22 samples) | 2,325 |
| Number of somatic mutations (excluding Br27P) | 993 |
| Missense | 622 |
| Nonsense | 43 |
| Insertion | 3 |
| Deletion | 46 |
| Duplication | 7 |
| Splice site or UTR | 27 |
| Synonymous | 245 |
| Average number of sequence alterations per sample | 47.3 |
| Copy number analysis | |
| Total number of SNP loci assessed for copy number changes | 1,069,688 |
| Number of copy number alterations identified (n = 22 samples) | 281 |
| Amplifications | 147 |
| Homozygous deletions | 134 |
| Average number of amplifications per sample | 6.7 |
| Average number of homozygous deletions per sample | 6.1 |
Analysis of sequence alterations
Analysis of the identified somatic mutations revealed that one tumor (Br27P), from a patient previously treated with radiation therapy and temozolomide, had 17 times as many alterations as any of the other 21 patients (table S3). The mutation spectrum of this sample was also dramatically different from those of the other GBM patients (12) and was consistent with previous observations of a hypermutation phenotype in glioma samples of patients treated with temozolomide (13, 14). After removing Br27P from consideration, we found that 685 genes (3.3% of the 20,661 genes analyzed) contained at least one nonsilent somatic mutation. The vast majority of these alterations were single-base substitutions (94%), whereas the others were small insertions, deletions, or duplications (Table 1). The 993 somatic mutations were observed to be distributed relatively evenly among the 21 remaining tumors (table S3), with a mean of 47 mutations per tumor, representing 1.51 mutations per Mb of GBM tumor genome sequenced. The six DNA samples extracted directly from patient tumors had smaller numbers of mutations than those obtained from xenografts, likely because of the masking effect of nonneoplastic cells in the former. It has previously been shown that cell lines and xenografts provide the optimal template DNA for cancer genome sequencing analyses (15) and that they faithfully represent the alterations present in the original tumors (16). Both the total number and the frequency of sequence alterations in GBMs were substantially smaller than the number and frequency of such alterations observed in colorectal or breast cancers and slightly less than in pancreatic cancers (10, 11, 17). The most likely explanation for this difference is the reduced number of cell generations in glial cells before the onset of neoplasia (18).
We further evaluated a set of 21 mutated genes identified in the Discovery Screen in a second screen, called a Prevalence Screen, comprising an additional 83 GBMs with well-documented clinical histories (table S2). The 21 genes selected were mutated in at least two Discovery Screen tumors and had mutation frequencies of >10 mutations per Mb of tumor DNA sequenced. Nonsilent somatic mutations were identified in 16 of these 21 genes in the additional tumor samples (table S4). The mutation frequency of all analyzed genes in the Prevalence Screen was 23 mutations per Mb of tumor DNA, markedly increased from the overall mutation frequency in the Discovery Screen of 1.5 mutations per Mb (P < 0.001). Additionally, the observed ratio of nonsilent to silent mutations among mutations in the Prevalence Screen was 14.5:1, substantially higher than the 3.1:1 ratio that was observed in the Discovery Screen (P < 0.001). The increased mutation frequency and higher fraction of nonsilent mutations suggested that genes mutated in the Prevalence Screen were enriched for genes that actively contributed to tumorigenesis.
In addition to the frequency of mutations in a gene, the type of mutation can provide information useful for evaluating its potential role in disease (19). The likely effect of missense mutations can be assessed through evaluation of the mutated residue by evolutionary or structural means. To evaluate missense mutations, we developed an algorithm (LS-MUT) that employs machine learning of 58 predictive features based on evolutionary conservation and the physical-chemical properties of amino acids involved in the alteration (12). About 15% of the missense mutations evaluated were predicted to have a statistically significant effect on protein function when assessed by this method (table S3). We also were able to make structural models of 244 of the 870 missense mutations identified in this study (20). In each case, the model was based on x-ray crystallography or nuclear magnetic resonance spectroscopy of the normal protein or a closely related homolog. This analysis showed that 35 of the missense mutations are located close to a domain interface or substrate-binding site and thus are likely to affect protein function [links to structural models are available in (12)].
Analysis of copy number changes
The same tumors were then evaluated for copy number alterations through genomic hybridization of DNA samples to Illumina SNP arrays containing ~1 million probes (21). We have recently developed a sensitive and specific approach for the identification of focal amplifications resulting in 12 or more copies per nucleus (amplification by a factor of 6 or more compared with the diploid genome) as well as deletions of both copies of a gene (homozygous deletions) using such arrays (22). Unlike larger chromosomal aberrations, such focused alterations can be used to identify underlying candidate genes in these regions.
We identified a total of 147 amplifications (table S5) and 134 homozygous deletions (table S6) in the 22 samples used in the Discovery Screen (Table 1). Although the number of amplifications was similar in samples extracted from patient tumors and those that had been passaged as xenografts, the latter samples allowed detection of a larger number of homozygous deletions (average of 8.0 deletions per sample in the xenografts versus 2.2 per sample in the tumors). These observations are consistent with previous reports that document the difficulty of identifying homozygous deletions in samples containing contaminating normal DNA (23) and highlight the importance of using purified human tumor cells, such as those present in xenografts or cell lines, for genomic analyses.
Integration of sequencing, copy number, and expression analyses
Mutations that arise during tumorigenesis may provide a selective advantage to the tumor cell (driver mutations) or have no net effect on tumor growth (passenger mutations). The mutational data obtained from sequencing and analysis of copy number alterations were integrated to identify GBM candidate cancer genes (_CAN_-genes) that are most likely to be drivers and therefore worthy of further investigation. To determine whether a gene was likely to harbor driver mutations, we compared the number and type of mutations observed (including sequence changes, amplifications, and homozygous deletions) and determined the probability that these alterations would result from passenger mutation rates alone (12) (fig. S1).
The _CAN_-genes, together with their passenger probabilities, are listed in table S7. The _CAN_-genes included several with established roles in gliomas, including TP53, PTEN, CDKN2A, RB1, EGFR, NF1, PIK3CA, and PIK3R1 (24–34). Of these genes, the most frequently altered were CDKN2A (altered in 50% of GBMs); TP53, EGFR, and PTEN (altered in 30 to 40%); NF1, CDK4, and RB1 (altered in 12 to 15%); and PIK3CA and PIK3R1 (altered in 8 to 10%) (Table 2). Overall, these frequencies, which are similar to or in some cases higher than those previously reported, validate the sensitivity of our approach for detecting somatic alterations.
Table 2.
Most frequently altered GBM _CAN-_genes. All _CAN-_genes are listed in table S7.
| Point mutations* | Amplifications† | Homozygous deletions† | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene | No. of tumors | Fraction of tumors (%) | No. of tumors | Fraction of tumors (%) | No. of tumors | Fraction of tumors (%) | Fraction of tumors with any alteration (%) | Passenger probability‡ |
| CDKN2A | 0/22 | 0 | 0/22 | 0 | 11/22 | 50 | 50 | <0.01 |
| TP53 | 37/105 | 35 | 0/22 | 0 | 1/22 | 5 | 40 | <0.01 |
| EGFR | 15/105 | 14 | 5/22 | 23 | 0/22 | 0 | 37 | <0.01 |
| PTEN | 27/105 | 26 | 0/22 | 0 | 1/22 | 5 | 30 | <0.01 |
| NF1 | 16/105 | 15 | 0/22 | 0 | 0/22 | 0 | 15 | 0.04 |
| CDK4 | 0/22 | 0 | 3/22 | 14 | 0/22 | 0 | 14 | <0.01 |
| RB1 | 8/105 | 8 | 0/22 | 0 | 1/22 | 5 | 12 | 0.02 |
| IDH1 | 12/105 | 11 | 0/22 | 0 | 0/22 | 0 | 11 | <0.01 |
| PIK3CA | 10/105 | 10 | 0/22 | 0 | 0/22 | 0 | 10 | 0.10 |
| PIK3R1 | 8/105 | 8 | 0/22 | 0 | 0/22 | 0 | 8 | 0.10 |
Through analysis of additional gene members within cell signaling pathways affected by these genes, we identified alterations of critical genes in the TP53 pathway (TP53, MDM2, and MDM4), the RB1 pathway (RB1, CDK4, and CDKN2A), and the PI3K/PTEN pathway (PIK3CA, PIK3R1, PTEN, and IRS1). These alterations affected pathways in a majority of tumors (64%, 68%, and 50%, respectively), and in all cases but one, mutations within each tumor affected only a single member of each pathway in a mutually exclusive manner (P < 0.05) (Table 3).
Table 3.
Mutations of the TP53, PI3K, and RB1 pathways in GBM samples. Mut, mutated; Amp, amplified; Del, deleted; Alt, altered.
| TP53 pathway | PI3K pathway | RB1 pathway | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumorsample | TP53 | MDM2 | MDM4 | Allgenes | PTEN | PIK3CA | PIK3R1 | IRS1 | Allgenes | RB1 | CDK4 | CDKN2A | Allgenes |
| Br02X | Del | Alt | Mut | Alt | Del | Alt | |||||||
| Br03X | Mut | Alt | Mut | Alt | |||||||||
| Br04X | Mut | Alt | Mut | Alt | Mut | Alt | |||||||
| Br05X | Amp | Alt | Mut | Alt | Del | Alt | |||||||
| Br06X | Del | Alt | |||||||||||
| Br07X | Mut | Alt | Mut | Alt | Del | Alt | |||||||
| Br08X | Del | Alt | |||||||||||
| Br09P | Mut | Alt | Amp | Alt | |||||||||
| Br10P | Mut | Alt | |||||||||||
| Br11P | Mut | Alt | |||||||||||
| Br12P | Mut | Alt | Mut | Alt | |||||||||
| Br13X | Mut | Alt | Del | Alt | |||||||||
| Br14X | Mut | Alt | Del | Alt | |||||||||
| Br15X | Mut | Del | Alt | ||||||||||
| Br16X | Amp | Alt | Amp | Alt | |||||||||
| Br17X | Mut | Alt | Del | Alt | |||||||||
| Br20P | |||||||||||||
| Br23X | Mut | Alt | Del | Alt | |||||||||
| Br25X | Mut | Alt | Del | Alt | |||||||||
| Br26X | Mut | Alt | Del | Alt | |||||||||
| Br27P | Mut | Alt | Amp | Alt | |||||||||
| Br29P | Mut | Alt | |||||||||||
| Fraction of tumors with altered gene/pathway* | 0.55 | 0.05 | 0.05 | 0.64 | 0.27 | 0.09 | 0.09 | 0.05 | 0.50 | 0.14 | 0.14 | 0.45 | 0.68 |
Systematic analyses of functional gene groups and pathways contained within the well-annotated MetaCore database (35) identified enrichment of alterations in a variety of cellular processes in GBMs, including additional members of the TP53 and PI3K/PTEN pathways. Many of the pathways identified were similar to core signaling pathways found to be altered in pancreas, colorectal, and breast tumors, such as those regulating control of cellular growth, apoptosis, and cell adhesion (17, 22, 36). However, several pathways were enriched only in GBMs. These included channels involved in transport of sodium, potassium, and calcium ions, as well as nervous system–specific cellular pathways such as synaptic transmission, transmission of nerve impulses, and axonal guidance (table S8). Mutations in these latter pathways may represent a subversion of normal glial cell processes to promote dysregulated growth and invasion.
Gene expression patterns can inform the analysis of pathways because they can reflect epigenetic alterations not detectable by sequencing or copy number analyses. To analyze the transcriptome of GBMs, we performed SAGE (37, 38) on all GBM samples for which sufficient RNA was available (total of 16 samples), as well as on two independent normal brain RNA controls (table S9). When combined with sequencing-by-synthesis methods (39–42), SAGE provides a highly quantitative and sensitive measure of gene expression. We first used the transcript analysis to help identify previously uncharacterized target genes from the amplified and deleted regions that were revealed by our study. In tables S5 and S6, a candidate target gene could be identified within several of these regions through the use of the mutational as well as transcriptional data. Second, we used the transcript analysis to help identify genes that were differentially expressed in GBMs compared to normal brain. A large number of genes (143) were expressed on average at levels 10 times as high in the GBMs. Among the overexpressed genes, 16 encoded proteins that are predicted to be secreted or expressed on the cell surface, suggesting new opportunities for diagnostic and therapeutic applications. Third, we used expression data to help assess the significance of genes containing missense mutations (table S3). Finally, we assessed whether the gene sets implicated in the pathways enriched for genetic alterations were also altered through expression changes. Notably, the gene sets in these pathways were more highly enriched for differentially expressed genes than the remaining sets (P < 0.001) (12). These expression data thus independently highlight the potential importance of these pathways in the development of GBMs.
High-frequency alterations of IDH1 in GBM
The _CAN_-gene list (table S7) included a number of individual genes that had not previously been linked to GBMs. The most frequently mutated of these genes, IDH1 on chromosome 2q33, encodes isocitrate dehydrogenase 1, which catalyzes the oxidative carboxylation of isocitrate to α-ketoglutarate, resulting in the production of nicotinamide adenine dinucleotide phosphate (NADPH). Of the five isocitrate dehydrogenase proteins encoded in the human genome, at least three are localized to the mitochondria, while IDH1 is localized within the cytoplasm and peroxisomes (43). The IDH1 protein forms an asymmetric homodimer (44) and is thought to play a substantial role in cellular control of oxidative damage through generation of NADPH (45, 46). None of the other IDH genes were found to be genetically altered in our analysis.
IDH1 was somatically mutated in 5 of the 22 GBM tumors in the Discovery Screen. Surprisingly, all 5 had the same heterozygous point mutation, a change of a guanine to an adenine at position 395 of the IDH1 transcript (G395A), leading to the replacement of an arginine with a histidine at amino acid residue 132 of the protein (R132H). In our previous study of colorectal cancers, this same codon was mutated in a single case through alteration of the adjacent nucleotide, resulting in a R132C amino acid change (10). Five GBMs evaluated in our Prevalence Screen were found to have heterozygous somatic R132H mutations, and an additional two tumors had a third distinct somatic mutation affecting the same amino acid residue, R132S (fig. S2 and Table 4). In addition to the Discovery and Prevalence Screen samples, 44 other GBMs were analyzed for IDH1 mutations, revealing six tumors with somatic mutations affecting R132. In total, 18 of 149 GBMs (12%) analyzed had alterations in IDH1. The R132 residue is conserved in all known species and is localized to the substrate binding site, where it forms hydrophilic interactions with the alpha-carboxylate of isocitrate (Fig. 1) (44, 47).
Table 4.
Characteristics of GBM patients with IDH1 mutations
| IDH1 mutation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Patient age(years)* | Sex | RecurrentGBM† | SecondaryGBM‡ | Overall survival(years)§ | Nucleotide | Amino acid | Mutationof TP53 | Mutation of PTEN,RB1, EGFR, or NF1 |
| Br10P | 30 | F | No | No | 2.2 | G395A | R132H | Yes | No |
| Br11P | 32 | M | No | No | 4.1 | G395A | R132H | Yes | No |
| Br12P | 31 | M | No | No | 1.6 | G395A | R132H | Yes | No |
| Br104X | 29 | F | No | No | 4.0 | C394A | R132S | Yes | No |
| Br106X | 36 | M | No | No | 3.8 | G395A | R132H | Yes | No |
| Br122X | 53 | M | No | No | 7.8 | G395A | R132H | No | No |
| Br123X | 34 | M | No | Yes | 4.9 | G395A | R132H | Yes | No |
| Br237T | 26 | M | No | Yes | 2.6 | G395A | R132H | Yes | No |
| Br211T | 28 | F | No | Yes | 0.3 | G395A | R132H | Yes | No |
| Br27P | 32 | M | Yes | Yes | 1.2 | G395A | R132H | Yes | No |
| Br129X | 25 | M | Yes | Yes | 3.2 | C394A | R132S | No | No |
| Br29P | 42 | F | Yes | Unknown | Unknown | G395A | R132H | Yes | No |
| IDH1 mutant patients (_n_=12) | 33.2 | 67% M | 25% | 42% | 3.8 | 100% | 100% | 83% | 0% |
| IDH1 wild- type patients (_n_=93) | 53.3 | 65% M | 16% | 1% | 1.1 | 0% | 0% | 27% | 60% |
Fig. 1.
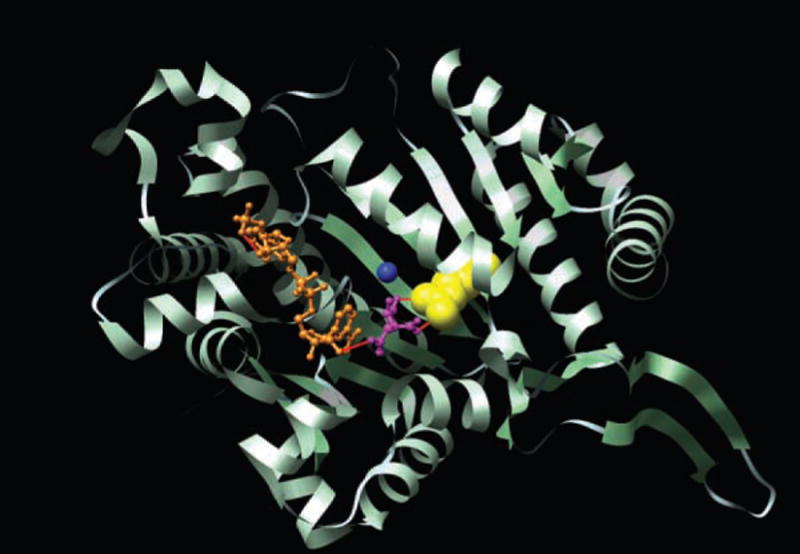
Structure of the active site of IDH1. The crystal structure of the human cytosolic NADP(+)–dependent IDH is shown in ribbon format (PDBID: 1T0L) (44). The active cleft of IDH1 consists of a NADP-binding site and the isocitrate-metal ion-binding site. The alpha-carboxylate oxygen and the hydroxyl group of isocitrate chelate the Ca2+ ion. NADP is colored in orange, isocitrate in purple and Ca2+ in blue. The Arg132 residue, displayed in yellow, forms hydrophilic interactions, shown in red, with the alpha-carboxylate of isocitrate. Displayed image was created with UCSF Chimera software version 1.2422 (50).
Several important observations were made about IDH1 mutations and their potential clinical importance. First, mutations in IDH1 preferentially occurred in younger GBM patients, with a mean age of 33 years for _IDH1_-mutated patients, as opposed to 53 years for patients with wild-type IDH1 (P < 0.001, t test) (Table 4). In patients under 35 years of age, nearly 50% (9 of 19) had mutations in IDH1. Second, mutations in IDH1 were found in nearly all of the patients with secondary GBMs (mutations in 5 of 6 secondary GBM patients, as compared to 7 of 99 patients with primary GBMs) (P < 0.001, binomial test). Third, patients with IDH1 mutations had a significantly improved prognosis, with a median overall survival of 3.8 years as compared to 1.1 years for patients with wild-type IDH1 (Fig. 2) (P < 0.001, log-rank test). Although both younger age and mutated TP53 are known to be positive prognostic factors for GBM patients, this association between IDH1 mutation and improved survival was noted even in the subgroup of young patients with TP53 mutations (P < 0.02, log-rank test).
Fig. 2.
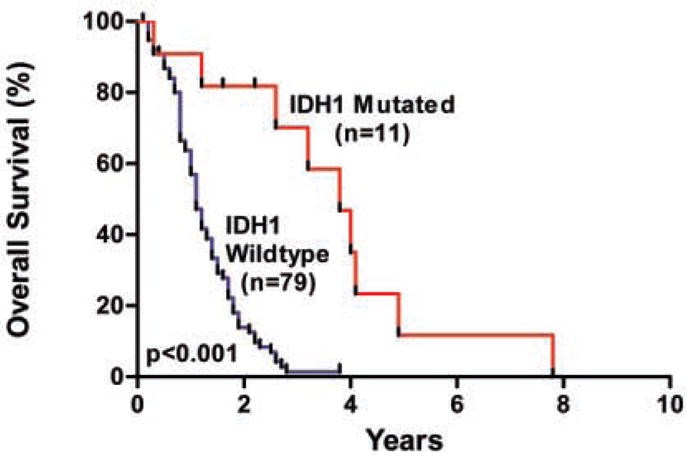
Overall survival according to IDH1 mutation status. The hazard ratio for death among patients with wild-type IDH1 (n = 79), as compared to those with mutant IDH1 (n = 11), was 3.7 (95 percent confidence interval, 2.1 to 6.5; P < 0.001). The median survival was 3.8 years for patients with mutated IDH1, as compared to 1.1 years for patients with wild-type IDH1.
Discussion
The data resulting from this integrated analysis of mutations and copy number alterations have provided a novel view of the genetic landscape of glioblastomas. Like all large-scale genetic analyses, our study has limitations. We did not assess certain molecular alterations, including chromosomal translocations and epigenetic changes. However, our large-scale expression studies should have identified any genes that were differentially expressed through these mechanisms (table S9). Additionally, we focused on copy number changes that were focal amplifications or homozygous deletions, because these have historically been most useful in identifying cancer genes. The array data we have generated can also be analyzed to determine loss of heterozygosity (LOH) or low-amplitude regions of copy number gains, but such changes cannot generally be used to pinpoint new candidate cancer genes. Finally, the samples directly extracted from patient tumors contained small amounts of contaminating normal tissue, which limited our ability to detect homozygous deletions and, to a lesser extent, somatic mutations, in those specific tumors.
Despite these limitations, our study provides a number of important genetic and clinical insights into GBMs. First, it revealed that some of the pathways known to be altered in GBMs affect a larger fraction of genes and patients than previously anticipated. A majority of the tumors analyzed had alterations in genes encoding components of each of the TP53, RB1, and PI3K pathways. The fact that all but one of the cancers with mutations in members of a pathway did not have alterations in other members of the same pathway suggests that such alterations are functionally equivalent in tumorigenesis. Second, these results have identified a variety of new genes and signaling pathways not previously implicated in GBMs (table S7 and S8). Some of these pathways were found to be altered in previous genome-wide analyses of pancreatic, breast, and colorectal cancers and may represent core processes that underlie human tumorigenesis (17, 22, 36). A number of the signaling pathways mutated or altered through expression differences in GBMs appear to be involved in nervous system signaling processes and represent novel and potentially useful aspects of GBM biology.
The comprehensive nature of our study allowed us to identify IDH1 as an unexpected target of genetic alteration in patients with GBM. All mutations in this gene resulted in amino acid substitutions at position 132, an evolutionarily conserved residue located within the isocitrate binding site (44). The recurrent nature of the mutations is reminiscent of activating alterations in oncogenes such as BRAF, KRAS, and PIK3CA. Our speculation that this sequence change is an activating mutation is strengthened by the absence of inactivating changes (e.g., frameshift or stop mutations), the absence of other alterations in key residues of the active site, and the fact that all mutations observed to date were heterozygous (without any evidence of loss of the second allele through LOH). Interestingly, enzymatic studies have shown that in vitro engineered substitution of arginine at residue 132 with a different amino acid (glutamate) than that observed in patients results in a catalytically inactive enzyme, suggesting a critical role for this residue (48). Further biochemical and molecular analyses will be needed to determine the effect of alterations of IDH1 on enzymatic activity and cellular phenotype.
Regardless of the specific molecular consequences of IDH1 alterations, detection of mutations in IDH1 is likely to be clinically useful. Although considerable effort has focused on the identification of characteristic genetic lesions in primary and secondary GBMs, the altered genes identified to date are not optimal for this purpose (5). Our study revealed IDH1 mutation to be a novel and potentially more specific marker for secondary GBM. One hypothesis is that IDH1 alterations identify a biologically specific subgroup of GBM patients, including both patients who would be classified as having secondary GBMs and a subpopulation of primary GBM patients with a similar tumor biology and a more protracted clinical course (Table 4). Interestingly, patients with IDH1 mutations had a very high frequency of TP53 mutation and a very low frequency of mutations in other commonly altered GBM genes (Table 4). Patients with mutated IDH1 also had distinct clinical characteristics, including younger age and a considerably improved clinical prognosis (Table 4). It is conceivable that new treatments could be designed to take advantage of IDH1 alterations in these patients, because inhibition of a different IDH enzyme (IDH2) has recently been shown to result in increased sensitivity of tumor cells to a variety of chemotherapeutic agents (49). In summary, the discovery of IDH1 and other genes previously not known to play a role in human tumors (table S7) validates the utility of genome-wide genetic analysis of tumors in general and opens new avenues of basic and clinical brain tumor research.
References and Notes
- 1.Louis DN, et al. Acta Neuropathol. 2007;114:97. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, et al. N Engl J Med. 2005;352:987. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Scherer H. Am J Cancer. 1940;40:159. [Google Scholar]
- 4.Kleihues P, Ohgaki H. Neuro-oncol. 1999;1:44. doi: 10.1093/neuonc/1.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohgaki H, Kleihues P. Am J Pathol. 2007;170:1445. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohgaki H, et al. Cancer Res. 2004;64:6892. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 7.Mellinghoff IK, et al. N Engl J Med. 2005;353:2012. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 8.Maher EA, et al. Cancer Res. 2006;66:11502. doi: 10.1158/0008-5472.CAN-06-2072. [DOI] [PubMed] [Google Scholar]
- 9.Tso CL, et al. Cancer Res. 2006;66:159. doi: 10.1158/0008-5472.CAN-05-0077. [DOI] [PubMed] [Google Scholar]
- 10.Sjöblom T, et al. Science. 2006;314:268. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Wood LD, et al. Science. 2007;318:1108. [Google Scholar]
- 12.Materials and methods are available as supporting material on Science Online.
- 13.Cahill DP, et al. Clin Cancer Res. 2007;13:2038. doi: 10.1158/1078-0432.CCR-06-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter C, et al. Cancer Res. 2006;66:3987. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winter JM, Brody JR, Kern SE. Cancer Biol Ther. 2006;5:360. doi: 10.4161/cbt.5.4.2552. [DOI] [PubMed] [Google Scholar]
- 16.Jones S, et al. Proc Natl Acad Sci USA. 2008;105:4283. [Google Scholar]
- 17.Jones S, et al. Science. 2008;321:1801. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraus-Ruppert R, Laissue J, Burki H, Odartchenko N. J Comp Neurol. 1973;148:211. doi: 10.1002/cne.901480206. [DOI] [PubMed] [Google Scholar]
- 19.Ng PC, Henikoff S. Nucleic Acids Res. 2003;31:3812. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karchin R. Structural models of mutants identified in glioblastomas. 2008 http://karchinlab.org/Mutants/CAN-genes/brain/GBM.html.
- 21.Steemers FJ, et al. Nat Methods. 2006;3:31. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- 22.Leary RJ, et al. Proc Natl Acad Sci USA. in press. [Google Scholar]
- 23.Cairns P, et al. Nat Genet. 1995;11:210. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 24.Nigro JM, et al. Nature. 1989;342:705. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 25.Li J, et al. Science. 1997;275:1943. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 26.Ueki K, et al. Cancer Res. 1996;56:150. [PubMed] [Google Scholar]
- 27.Wong AJ, et al. Proc Natl Acad Sci USA. 1987;84:6899. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Brain Pathol. 2004;14:372. doi: 10.1111/j.1750-3639.2004.tb00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frederick L, Wang XY, Eley G, James CD. Cancer Res. 2000;60:1383. [PubMed] [Google Scholar]
- 30.Li Y, et al. Cell. 1992;69:275. [Google Scholar]
- 31.Thiel G, et al. Anticancer Res. 1995;15:2495. [PubMed] [Google Scholar]
- 32.Samuels Y, et al. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 33.Broderick DK, et al. Cancer Res. 2004;64:5048. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 34.Gallia GL, et al. Mol Cancer Res. 2006;4:709. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 35.Ekins S, Nikolsky Y, Bugrim A, Kirillov E, Nikolskaya T. Methods Mol Biol. 2007;356:319. doi: 10.1385/1-59745-217-3:319. [DOI] [PubMed] [Google Scholar]
- 36.Lin J, et al. Genome Res. 2007;17:1304. doi: 10.1101/gr.6431107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Science. 1995;270:484. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 38.Saha S, et al. Nat Biotechnol. 2002;20:508. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- 39.Sultan M, et al. Science. 2008;321:956. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 40.Lister R, et al. Cell. 2008;133:523. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Nat Methods. 2008;5:621. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 42.Morin R, et al. Biotechniques. 2008;45:81. doi: 10.2144/000112900. [DOI] [PubMed] [Google Scholar]
- 43.Geisbrecht BV, Gould SJ. J Biol Chem. 1999;274:30527. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, et al. J Biol Chem. 2004;279:33946. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 45.Lee SM, et al. Free Radic Biol Med. 2002;32:1185. doi: 10.1016/s0891-5849(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 46.Kim SY, et al. Mol Cell Biochem. 2007;302:27. doi: 10.1007/s11010-007-9421-x. [DOI] [PubMed] [Google Scholar]
- 47.Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ. Mol Biol Evol. 1998;15:1674. doi: 10.1093/oxfordjournals.molbev.a025894. [DOI] [PubMed] [Google Scholar]
- 48.Jennings GT, Minard KI, McAlister-Henn L. Biochemistry. 1997;36:13743. doi: 10.1021/bi970916r. [DOI] [PubMed] [Google Scholar]
- 49.Kil IS, Kim SY, Lee SJ, Park JW. Free Radic Biol Med. 2007;43:1197. doi: 10.1016/j.freeradbiomed.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF, et al. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Contact information for the authors who directed the major components of this project is as follows: H. Yan, G. J. Riggins (clinical and sample coordination), yan00002@mc.duke.edu, griggin1@jhmi.edu; D. D. Bigner (pathological and clinical review), bigne001@mc.duke.edu; R. Karchin (bioinformatic analysis), karchin@jhu.edu; N. Papadopoulos (gene expression analysis), npapado1@jhmi.edu; G. Parmigiani (statistical analysis), gp@jhu.edu; B. Vogelstein, V. E. Velculescu, K. W. Kinzler (sequencing and copy number analysis), bertvog@gmail.com, velculescu@jhmi.edu, kinzlke@jhmi.edu. We thank N. Silliman, J. Ptak, L. Dobbyn, and M. Whalen for assistance with PCR amplification; D. Lister, L. J. Ehinger, D. L. Satterfield, J. D. Funkhouser, and P. Killela for assistance with DNA purification; T. Sjöblom for assistance with database management; the Agencourt sequencing team for assistance with automated sequencing; and C.-S. Liu and the SoftGenetics team for their assistance with mutation detection analyses. This project was carried out under the auspices of the Ludwig Brain Tumor Initiative and was supported by the Virginia and D. K. Ludwig Fund for Cancer Research, NIH grants CA121113, NS052507, CA43460, CA57345, CA62924, CA09547, 5P50-NS-20023, CA108786, and CA11898, the Pew Charitable Trusts, the Pediatric Brain Tumor Foundation Institute, the Hirschhorn Foundation, Alex’s Lemonade Stand Foundation, the American Brain Tumor Association, the American Society of Clinical Oncology, the Brain Tumor Research Fund and Beckman Coulter Corporation. Under separate licensing agreements between the Johns Hopkins University and Genzyme, Beckman Coulter, and Exact Sciences Corporations, B.V., V.E.V., and K.W.K. are entitled to a share of royalties received by the university on sales of products related to research described in this paper. These authors and the university own Genzyme and Exact Sciences stock, which is subject to certain restrictions under university policy. The terms of these arrangements are managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.