Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes (original) (raw)
Abstract
Neutral nucleotide substitutions occur at varying rates along genomes, and it remains a major issue to unravel the mechanisms that cause these variations and to analyze their evolutionary consequences. Here, we study the role of replication in the neutral substitution pattern. We obtained a high-resolution replication timing profile of the whole human genome by massively parallel sequencing of nascent BrdU-labeled replicating DNA. These data were compared to the neutral substitution rates along the human genome, obtained by aligning human and chimpanzee genomes using macaque and orangutan as outgroups. All substitution rates increase monotonously with replication timing even after controlling for local or regional nucleotide composition, crossover rate, distance to telomeres, and chromatin compaction. The increase in non-CpG substitution rates might result from several mechanisms including the increase in mutation-prone activities or the decrease in efficiency of DNA repair during the S phase. In contrast, the rate of C → T transitions in CpG dinucleotides increases in later-replicating regions due to increasing DNA methylation level that reflects a negative correlation between timing and gene expression. Similar results are observed in the mouse, which indicates that replication timing is a main factor affecting nucleotide substitution dynamics at non-CpG sites and constitutes a major neutral process driving mammalian genome evolution.
Mutations are known to occur heterogeneously along genomes, but the causes of these fluctuations are unclear. Numerous works have revealed an increasing complexity of neutral mutation patterns in mammalian genomes. Substitution rates depend on the nucleotides flanking the mutated site. The rate of C → T transitions is much higher for CpG than for other dinucleotides due to the spontaneous deamination of methylcytosine that is mostly found at these sites (Ehrlich and Wang 1981). Other substitution rates also depend, but to a lesser extent, on the two flanking nucleotides (Zhao and Boerwinkle 2002; Hwang and Green 2004). Mutation rates in mammals also depend on the local G+C content as shown by studies of sequence divergence and by genome-wide studies of neutral substitution rates (Wolfe et al. 1989; Matassi et al. 1999; Hurst and Williams 2000). The correlation between mutation rate and G+C content is negative in low G+C content regions but positive in high G+C content regions (Waterston et al. 2002). Several studies reported positive correlations of the local rate of meiotic recombination with nucleotide diversity and substitution rates (Nachman 2001; Lercher and Hurst 2002; Waterston et al. 2002; Hellmann et al. 2003) and with the ratio of W (A or T) → S (G or C) to S → W substitution rates (Meunier and Duret 2004). This effect of recombination most likely results from the neutral process of biased gene conversion (Eyre-Walker and Hurst 2001; Galtier et al. 2001; Duret and Arndt 2008). Non-CpG mutation rates are lowest in regions with open chromatin structure. This was proposed to reflect lower rates of DNA damage or enhanced DNA repair in open chromatin (Prendergast et al. 2007).
Pioneering studies suggested that changes in nucleotide pools during replication could be responsible for mutation rate fluctuations (Wolfe et al. 1989; Gu and Li 1994), because different genomic regions replicate at different times during the S phase (MacAlpine et al. 2004; Woodfine et al. 2004; Karnani et al. 2007; Hiratani et al. 2008). Studies of limited portions of the human genome confirmed that mutation rates increase in late-replicating regions (Watanabe et al. 2002; Stamatoyannopoulos et al. 2009). It was proposed that the increase in mutation rates in non-CpG and CpG sites results from a single mechanism, namely, an increase of DNA damage during replication (Stamatoyannopoulos et al. 2009). However, this is in conflict with the notion that most non-CpG substitutions result from replication errors, whereas CpG transitions occur independently of replication (Li et al. 2002; Hwang and Green 2004; Taylor et al. 2006).
In this study, we have analyzed the links between nucleotide substitutions and replication timing. We have determined a high-resolution replication timing profile of the whole human genome and have correlated these data with human nucleotide substitution rates computed by aligning the human genome with three other primate genomes. These new genome-wide data, together with published rodent data, have allowed us to investigate non-CpG and CpG substitutions separately. The data show that both types of substitution rates increase monotonously with replication timing but by different mechanisms, and allow us to elucidate the contribution of DNA methylation and chromatin compaction. Overall, the results establish that replication timing is a major neutral process driving mammalian genome evolution.
Results
Determination of a human genome replication timing profile
To determine a human replication timing profile, HeLa cells were briefly pulsed with bromodeoxyuridine (BrdU) and sorted into four compartments of the S phase (Si) according to their DNA content; nascent DNA was labeled with Br-dU and the DNA corresponding to each Si compartment was immunoprecipitated with anti-BrdU antibodies and sequenced using the massively parallel sequencing Illumina technology (formerly Solexa sequencing technology; Methods). The enrichment of sequence reads along the genome was computed in each S-phase compartment (Fig. 1) (Methods). Replication timing of a defined genome region was estimated from the fraction of the S phase (S50) at which 50% of the sequence reads that map in this region were obtained (Methods). Small S50 values correspond to early replicating regions, and large S50 values correspond to late replicating regions. Significant correlations were observed between these S50 values and the timing data from HeLa cells (Karnani et al. 2007) measured in ENCODE regions (Pearson, R = 0.77, P < 10−15) or the low-resolution timing data from human lymphocytes (R = 0.72, P < 10−15) (Supplemental Fig. S1A,B; Woodfine et al. 2004). The fact that the correlation between the data of this study and the ENCODE data did not present higher values might result from differences between the experimental procedures (hybridization vs. massively parallel sequencing) and, in particular, from the cell synchronization by drug treatment (Karnani et al. 2007), a method that may alter the replication kinetics. Notably, the regions identified by Karnani et al. as replicating with a pan-S pattern were found in our study as mostly replicating at specific times in the S phase (Methods). The similarity of timing data between different cell types could be extended genome-wide to human/mouse homologous regions (R = 0.68, P < 10−15) (Supplemental Fig. S1C) in agreement with previous observations (Farkash-Amar et al. 2008; Hiratani et al. 2008).
Figure 1.

Analysis of the replication timing profiles. (A) Profile along human chromosome 15q of the enrichment of sequence reads E computed in 100-kb windows, in four periods of the S phase, _S_1, _S_2, _S_3, _S_4; S50, profile of the replication timing values (Methods). Small S50 values correspond to early replicating regions; large S50 values correspond to late replicating regions. (B) Enlarged view of E and S50 profiles along a fragment of chromosome 15. (C) Pairwise correlations (Pearson) between the enrichment E determined in the Si periods of the S phase and the S50 values of Experiments 1 and 2. Colors indicate the range of correlation coefficient values; positive correlations are observed only between neighboring Si fractions; S50 values are negatively correlated with _S_1 and positively correlated with _S_4. This confirmed that different alleles of the same region were usually replicated at similar periods of the S phase (Farkash-Amar et al. 2008). (D) Histogram of S50 values in the whole genome.
Although the timing profile is expected to present cell-type-specific variations, the similarity between different timing profiles strongly suggests that a large part of the replication program remains identical in the different cell types, including the germline. Since nucleotide substitutions specifically propagate in the germline, their rate profiles could thus be compared to replication timing data obtained in HeLa cells. This working hypothesis was supported by inspection of the substitution rates and of the timing profiles along the genome that clearly indicate a covariation between substitution rates and replication timing (Fig. 2A; Supplemental Fig. S2).
Figure 2.

Increase in non-CpG substitution rates during the S phase. (A) Substitution rate and replication timing profiles along human chromosome 17. (B) Global substitution rate (all panels). GC fixed: (black) GC ≤ 37%; (blue) 37% < GC ≤ 42%; (orange) 42% < GC ≤ 52%; (red) GC > 52%. CO fixed: (black) CO ≤ 1 cM/Mb; (blue) 1 < CO ≤ 2 cM/Mb; (orange) 2 < CO ≤ 4 cM/Mb; (red) CO > 4 cM/Mb; DT fixed: (black) DT > 50 Mb; (blue) 30 < DT ≤ 50 Mb; (orange) 10 < DT ≤ 30 Mb; (red) DT ≤ 10 Mb. In the abscissa, S50 determined in 100-kb windows (Methods) is shown. In the ordinate, the mean value of the substitution rate ± SEM in percent is shown. The distance between the lines shows dependency on the controlling factor. (C) Individual substitution rates when controlling for CO. Colors are as in B. S → W and W → W rates show moderate dependency on CO. In contrast, W → S rates, and to a lesser extent S → S, depend strongly on CO.
Non-CpG and CpG substitutions were investigated separately. They were tabulated in the human lineage since its divergence from chimpanzee using the macaque and pongo as outgroups (Methods). A reliable estimate of neutral substitution rates was obtained by eliminating coding regions, splice sites, and CpG islands (except when indicated), since these cannot be considered to evolve neutrally.
Neutral non-CpG substitutions and diversity increase monotonously with replication timing
The global non-CpG substitution rate increases monotonously with replication timing and displays a 28% increase from the earliest to latest replicating regions (Fig. 2B, left panel). The substitution rate is known to correlate with the G+C content (GC), meiotic crossover rate (CO), or distance to telomere (DT) (Kong et al. 2002; Lercher and Hurst 2002; Waterston et al. 2002; Hellmann et al. 2005; Duret and Arndt 2008; Berglund et al. 2009). When these variables are fixed in discrete bins, the global rate displays rather parallel lines that still increase with timing (Fig. 2B). The spacing between these lines is small for GC but larger for CO and DT, which is consistent with the high dependency of the substitution rate on CO and DT (Duret and Arndt 2008; Berglund et al. 2009). This dependency results from the neutral process of biased gene conversion (BGC) associated with meiotic recombination, which favors W → S substitutions.
Next, we showed that all individual non-CpG substitution rates increase with timing; the relative increase is G:C → A:T (35%), C:G → A:T (83%), T:A → A:T (55%), A:T → G:C (20%), T:A → G:C (23%), C:G → G:C (30%) (Supplemental Fig. S3). However, when examining the effect of CO (Fig. 2C), GC, and DT (Supplemental Fig. S4A,B), two types of dependence emerged, with N (all nucleotides) → W rates showing less dependency (line spacing) than N → S rates. The dependence of individual rates on CO and DT further supports that BGC influences substitution rates (Duret and Arndt 2008; Berglund et al. 2009). To disentangle the contributions of GC, CO, DT, and of replication timing (S50), we performed a multivariate regression analysis. Timing alone explains 38% of the global non-CpG substitution rate variability predicted by the full model, and from 14% to 73% of the individual rate variability (Table 1). Timing is the best predictor of N → W substitutions, and, as expected from the BGC model, CO and DT are the best predictors of N → S substitutions. These results were further confirmed by partial correlation analysis showing that all rates displayed strongly significant positive correlations with timing when controlling for other variables (R = 0.16 to 0.29, P < 10−115) (Supplemental Table S1). Similar results were obtained with different window sizes (100 kb, 200 kb, 500 kb, 1 Mb, 2 Mb, 5 Mb) (Table 1; Supplemental Tables S1–S3; data not shown).
Table 1.
Multivariate regression analysis of human substitution rates and diversity

We checked that replication affects substitution rates similarly in all regions, whether transcribed or not, and whether they consist of repeated elements or not (Supplemental Fig. S5). Since the substitution rate at a given site depends on the identity of the two flanking nucleotides (Hwang and Green 2004), we also checked that the observed increase of rate with timing does not simply result from differing trinucleotide compositions in the early- and late-replicating regions (Supplemental Fig. S6).
To compare the effect of timing on individual substitution rates, we examined the ratio of each rate to the global rate during the S phase. This ratio is constant for all substitutions except for C:G → A:T and to a lesser extent T:A → A:T, both of which increase significantly more than the global rate (Supplemental Fig. S7). This is confirmed by partial correlation analysis (Supplemental Table S4), indicating that if the same mechanism causes increase with timing in all non-CpG substitution rates, this mechanism has to produce a stronger increase of C:G → A:T substitutions.
Using single nucleotide polymorphism (SNP) data from several fully sequenced human genomes, we observed that human diversity (at non-CpG sites) is correlated with timing and shows the same relative increase as the global non-CpG substitution rate (29%) (Fig. 3A). Diversity displays correlation with timing that is similar to that observed with global substitution rate when controlling for GC, CO, or DT. Similar results were obtained with SNP data (Table 1) from the International HapMap Project (either homozygous or heterozygous SNP) (Supplemental Fig. S8) or from several individual genomes (Supplemental Fig. S9). In contrast, a recent study showed that human non-CpG diversity displays a relative increase with timing that is much greater than that found for human–chimpanzee divergence (Stamatoyannopoulos et al. 2009); this difference can be attributed to the limited fraction of human genome (ENCODE data) used in this latter study. Our data thus establish that the mechanism underlying the replication time dependence of non-CpG divergence and diversity has been acting in a stable mode since the common ancestor of human and chimpanzee.
Figure 3.

Increase in human and mouse non-CpG divergence and diversity during the S phase. (A) The human global substitution rate (blue) and diversity (Levy et al. 2007) (red) as a function of replication timing. The relative increase in the human global rate as a function of timing (28%) is the same as the relative increase in diversity (29%). (B) (Blue) Mouse–rat divergence; (red) mouse diversity; the relative increase of mouse divergence (16%) is smaller than that of diversity (30%). This likely results from substitution saturation due to long evolutionary time since the mouse/rat divergence. Correlation coefficients (Pearson): human diversity and timing (R = 0.23, P < 10−16); mouse diversity and timing (R = 0.21, P < 10−16). All rates are determined in 100-kb windows.
Substitution rates increase independently of chromatin compaction
Recently, the global non-CpG substitution rate has been shown to correlate with chromatin compaction as measured from hydrodynamic properties of chromatin segments (Gilbert et al. 2004; Prendergast et al. 2007). It has been suggested that this results from lower rates of DNA damage or enhanced DNA repair in open chromatin. Euchromatin has an open structure and replicates early, whereas heterochromatin has a more compact structure and replicates late in the S phase. We verified that chromatin compaction is negatively correlated with replication timing (R = −0.42, P < 10−15) (Fig. 4A). However, after controlling for chromatin compaction, the global non-CpG and CpG (see below) substitution rates still increase with timing and show no dependence on chromatin compaction (Fig. 4B,C). Partial correlation analysis shows that chromatin compaction does not contribute significantly to the non-CpG global substitution rate (_R_ = 0.01, _P_ > 0.7) and to the CpG transition rate (R = −0.02, P > 0.3). The reported correlation between compaction and substitution rate is therefore entirely accounted for by the correlation with replication time. This finding implies that the efficiency of mutation/repair processes is not influenced by chromatin compaction.
Figure 4.

(A) Dependence of chromatin compaction with replication timing. Compaction data were from Gilbert et al. (2004). Variation of global non-CpG substitution rate (B) and CpG substitution rate (C) with replication timing after controlling for chromatin compaction. Timing values and substitution rates are determined within all genomic regions for which chromatin compaction was determined (Gilbert et al. 2004). (Black) Log2(Open:Input) ≤ −1; (blue) −1 < log2(Open:Input) ≤ 0; (orange) 0 < log2(Open:Input) ≤ 1; (red) log2(Open:Input) > 1. The window size is as defined in Gilbert et al. (2004) (mean size: 146 kb).
Mouse diversity increases with replication timing
We next extended our study to rodents and computed mouse non-CpG diversity using recent SNP data. We observed an increase of SNP density with timing (30%) that was similar to that observed in human (29%) (Fig. 3B). Mouse diversity increased similarly with timing after controlling for GC, CO, or DT, although these variables had little effect on rate variation (Supplemental Fig. S10) as compared to their major impact on human diversity (Supplemental Figs. S8, S9). Overall, the data show that the mechanism responsible for the observed increase of non-CpG substitution rates with replication timing has been operating similarly for primates and rodents, a conclusion that can likely be extended to most mammals.
CpG transition rate increases in later-replicating regions
We next investigated the variation of the CpG transition rate (excluding CpG islands) and observed that it increases with timing similarly to non-CpG substitutions (49% relative increase) (Fig. 5A). This increase was also observed when controlling for GC, CO, or DT or chromatin compaction; the CpG transition rate showed little dependence on these variables (Fig. 4C; Supplemental Fig. S11D–F). Most CpG transitions do not result from replication errors, but from spontaneous deamination of methylcytosine into thymine (Ehrlich and Wang 1981). They are known to occur in a clock-like fashion, that is, independently of replication (Hwang and Green 2004; Taylor et al. 2006), which apparently contradicts their increase during the S phase. Repeated sequences display higher CpG transition rates than nonrepeated sequences, but both rates increase with timing (Supplemental Figs. S2C, S11A–C). This difference likely results from higher methylation levels in repeated than in nonrepeated sequences (Goll and Bestor 2005).
Figure 5.

Increase of human and mouse CpG substitution rates in later-replicating regions explained by the increase in methylation level. Replication timing and CpG substitution rates are determined in noncoding regions excluding CpG islands (Methods). (A, left) Human CpG substitution rate as function of S50; (center) methylation level determined in human sperm cells (Eckhardt et al. 2006) plotted as a function of the replication timing S50; (right) human CpG substitution rate when controlling for methylation level (ML). (Black) ML ≤ 20%; (blue) 20% < ML ≤ 60%; (red) ML > 60%. (B) Analyses performed with mouse replication timing data TR50 (Farkash-Amar et al. 2008). (Left) Mouse CpG diversity was computed with SNP data (The International HapMap Consortium 2007); (center) methylation level determined in mouse embryonic stem cells (Meissner et al. 2008) plotted as a function of replication timing; (right) Mouse CpG SNP density when controlling for ML. (Black) ML ≤ 45%; (blue) 45% < ML ≤ 60%; (orange) 60% < ML ≤ 70%; (red) ML > 70%. DNA methylation levels, substitution rates, divergence, and replication timing were computed as indicated in Methods. The window size is as indicated in Methods (DNA Methylation section).
To find out whether the increase in CpG transitions with timing could result from enhanced methylation of CpG cytosines in later-replicating regions, we used genome-wide methylation data determined in human sperm cells (Eckhardt et al. 2006) to search for a possible correlation between the methylation level and timing. Actually, the methylation level increases with replication timing (Fig. 5A). The transition rate at the fully methylated CpG sites (excluding CpG islands) can be estimated from the mean values of the methylation level (ML) in early- and late-replicating regions (see Methods, Determination of the Transition Rate at Methylated CpG Sites): it is almost constant, slightly decreasing from 8.98% to 8.86%. This is in full agreement with the observation that the transition rate is rather constant from early to late regions when controlling for ML values: it increases with ML values from ∼2% (for ML < 20%) to ∼5% (for ML > 60%) (Fig. 5A, right). The value a ≈ 8.9% can be considered as an upper limit of the CpG transition rate corresponding to a methylation level of 100%. Multiple regression analysis of the CpG transition rate indicated that the methylation level is the best predictor, explaining most (78%; P = 10−6) of the rate variability explained by the model, while timing explains no significant fraction (Supplemental Table S5). As a control, timing is the best predictor for the non-CpG global rate model, explaining most (61%; P < 10−7) of the rate variability compared to the methylation level (29%; P = 10−3); this high contribution likely results from the replication timing of HeLa cells that can differ from germline replication timing in ∼20% of the genome (Hiratani et al. 2008). Similar results were obtained in the mouse, using embryonic stem cell methylation data (Fig. 5B; Supplemental Table S5). This demonstrates that in the germline, the apparent increase in transitions at CpG sites (excluding CpG islands) during the S phase mostly results from a higher methylation level in later-replicating regions. Since the variation in replication timing between HeLa cells and the various cell types of the germline lineage may be quite large (Hiratani et al. 2010), it cannot be excluded that the increase in CpG transition rate might be also partly due to some other mechanism like the increase in DNA damage as proposed by Stamatoyannopoulos et al. (2009). We measured the G:C → A:T transition rate at CpG sites in CpG islands (0.55% ± 0.02%), where cytosines present a low methylation level (Cross and Bird 1995); as expected, the transition rate is similar to that of the other cytosines in non-CpG sites (0.36% ± 0.001%). In the mouse, we observed negative correlations between expression level and timing (R = −0.11, P < 10−13), and between expression level and methylation level (excluding CpG islands; R = −0.13, P < 10−18) (Supplemental Fig. S12C–F); we also observed strong negative correlations between expression breadth and timing (R = −0.20, P < 10−117) and between expression breadth and methylation level (R = −0.14, P < 10−51) (Supplemental Fig. S12A,B). These results indicate that the correlation between timing and methylation level results from negative correlations of transcriptional activity with both replication timing and methylation level.
Discussion
What mechanism(s) could be responsible for the correlations between non-CpG substitution rates and replication timing? (1) The fidelity of the replication machinery may decrease during the S phase. A pioneering study has proposed that changes in nucleotide pools during the S phase could alter mutation rates (Wolfe et al. 1989). To our knowledge, no study has reported changes in dNTP pools during the S phase that could account for the mutation spectrum observed here, and in particular for the increase in C:G → A:T substitutions relative to other substitution types (Supplemental Fig. S7). (2) Head-on collisions between the replication and transcription machineries can generate mutations (Mirkin and Mirkin 2007). It is possible that the number of such collisions increases in later-replicating regions, thus generating a corresponding increase in mutation rates, as observed here. However, to our knowledge, such increase has not yet been observed; in addition, transcription is inversely correlated with replication timing. (3) It is theoretically possible that the contribution of different translesion DNA polymerases changes during the S phase in a manner that would account for the observed patterns of substitution rates by their specific mutation signatures. (4) DNA lesions may increase during the S phase, for example, due to changes in general metabolism (Yu et al. 2009) or to generation of single-stranded DNA (Stamatoyannopoulos et al. 2009). (5) DNA repair activities may decrease during the S phase. The correction of replication errors requires the mismatch repair mechanism (MMR) (Kunkel and Erie 2005). In vivo studies indicate that MMR activity and fidelity are greater during the S phase than during the G1 and G2 phases (Edelbrock et al. 2009). In addition, A:G mismatches are repaired less efficiently than T:G mismatches (repair is nick-directed to the underlined nucleotide), resulting in a larger proportion of C:G → A:T than G:C → A:T substitutions, as a signature of MMR activity and fidelity (Edelbrock et al. 2009). Our observation that C:G → A:T increases at a rate higher than that of the other substitutions (Supplemental Fig. S7) is consistent with a model in which MMR activity and fidelity would be highest at the onset of the S phase and decrease progressively with replication timing. Although no available human data support such a model, it would explain the pattern of replication-dependent substitution rates, suggesting a decrease of MMR activity during the S phase as a possible cause of the observed correlation pattern between non-CpG substitutions and replication timing. (6) It is also possible that a decrease in other DNA repair activities (e.g., base and nucleotide excision repair) during the S phase could contribute to the observed patterns of non-CpG substitution rates. In particular, decreasing repair of 8-oxo-guanine, the most prevalent DNA lesion, could thus contribute to the greater increase of the C:G → A:T mutation rate (Barnes and Lindahl 2004).
Our data establish that the increase in the CpG transition rate, although strikingly similar to the increase in the non-CpG substitution rate, is only indirectly associated with replication, but results from increasing methylation from early- to late-replicating regions in the germline. These results can be understood in the light of previous studies showing a negative correlation between replication timing and transcriptional potential estimated by the expression level (Goldman et al. 1984), or, more significantly, by the probability of expression (Woodfine et al. 2004), expression breadth, or mean expression level over many tissues (Farkash-Amar et al. 2008). Along this line, we propose that the positive correlation between replication timing and methylation level results from negative correlations of gene transcriptional potential with both replication timing and methylation level. This implies that the correlation between the CpG transition rate and timing results from an increase in methylation level from early-replicating transcription-prone regions, to late-replicating transcription-silent regions.
Recent studies have identified biased gene conversion (BGC) as the cause of strong variations along genomes of A or T toward G or C (W → S) mutation rates in regions that undergo recombination (Duret and Arndt 2008). Here, we have identified large mutation rate variations, which are similar in amplitude to those induced by BGC, but that affect all types of substitution rates. These variations result from mechanisms that are independent from BGC; their effects are superimposed over those of BGC (see the parallel curves for W → S rates in Fig. 2C). Notably, these mechanisms induce an increase of substitution rates in later-replicating regions that is greater for C:G → A:T than for other mutations; in these regions this should counteract the increase in G+C content due to the effect of BGC.
Although we have observed significant correlations between S50 values and replication timing data from various cell types (Supplemental Fig. S1), timing can differ among cell types in particular for genes expressed in small tissue numbers, resulting in differences in the timing profiles of HeLa and germline cells (Hiratani et al. 2010). Thus, it can be reasonably anticipated that the actual correlation between substitution rate and replication timing existing in germline cells is larger than that observed in the present study.
In conclusion, genome-wide analyses revealed distinct mechanisms responsible for the correlations between non-CpG and CpG mutation rates with replication timing. The data demonstrate that replication timing has been modulating mutation rates along genomes in a stable fashion since the human/mouse divergence. Replication timing is thus the major factor affecting all non-CpG substitution rates. These results open new avenues toward understanding the evolutionary mechanisms that shape the mutational landscape of mammalian genomes.
Methods
Massively parallel sequencing of BrdU-labeled nascent replicated DNA
Asynchronous HeLa cells were pulse-labeled with 50 μM BrdU for 40 min. Equal numbers of replicating cells (3 × 105) were collected according to their DNA content by using a fluorescence activated cell sorter (FACS), namely, _S_1, _S_2, _S_3, and _S_4. Similarly labeled unsorted cells were used as control (_S_0). The isolation of BrdU-labeled nascent strands was adapted from Azuara (2006) with the following modifications. Total DNA was extracted without salmon sperm carrier DNA yielding 1.9 μg of S1, 2.3 μg of S2, 2.8 μg of S3, and 3.4 μg of S4. BrdU-labeled DNA was immunoprecipitated without addition of Drosophila BrdU-labeled DNA yielding 100–120 ng of DNA for each Si. Double-stranded DNA was produced from immunoprecipitated DNA by brief (10 min) random priming (resulting in a fivefold DNA amplification) using the Bioprime labeling system (Invitrogen). The resulting DNA was sequenced using an Illumina sequencing device. The libraries were prepared following the Illumina protocol for chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) library construction. The DNA sample overhangs were first converted into phosphorylated blunt-ends using T4 DNA polymerase, Klenow DNA polymerase, and T4 polynucleotide kinase. An adenine residue was then added in 3′ position by the Klenow (exo-) polymerase. After ligation of the Illumina adapter (diluted 1/10), the mix was gel-purified to select 200–400-bp fragments; a final PCR amplification step (14–18 cycles) was performed using specific primers to complete the flanking sequences. Cloning a fraction of the library and sequencing a few transformants verified the quality of the constructs. The libraries, at a final concentration of 2–6 pM, were sequenced on the Illumina Genome Analyzer with 36 cycle runs.
Two replicate experiments (Experiments 1 and 2) were performed that produced seven to 15 million sequence reads for each fraction of the S phase (Supplemental Table S6). The sequence reads were identified using the standard Illumina base-calling software ELAND (GAPipeline 1.0) and then aligned to the human genome (assembly NCBI build 36.1, hg18) allowing up to two mismatches. Only reads that aligned at a single locus in the genome were considered. The 32-mer read-associated GC biases were corrected as described in Hillier et al. (2008). The data can be accessed at http://www.cgm.cnrs-gif.fr/thermes/donnees_sequencage/index.html.
Data processing
For each of the Experiments 1 and 2, the density of sequence reads (density Di,w) was computed along the human genome in 100-kb nonoverlapping windows (w) for each sample of the different fractions Si of the S phase (i = 1–4) and for the control sample _S_0 (density Dwc). We then searched for background regions within each Si fraction: a background window in an Si fraction was defined as a window that is not enriched compared to the control window (P > 10−2, binomial test) in the adjacent fraction(s) and significantly enriched (P < 10−3) in the nonadjacent fraction(s). For example, a window w of _S_1 is a background window if this window is not enriched in _S_2 and enriched in _S_3 or _S_4. To perform the binomial test for the windows of each Si fraction, we used the total number of sequence reads of this fraction divided by the total number of sequence reads of the control fraction _S_0. A new background ratio Ri was then calculated as:
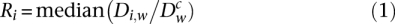
for all background windows w. These Ri values were used to recalculate the probabilities using a binomial test to redefine new background regions and obtain new Ri values. This process was reiterated several times in order to obtain stable ratios defined as the final Ri (stability was reached after at most 11 iterations). The enrichment value E for a given window n was then computed as:
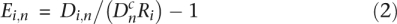
All negative values were reset to 0. The distributions of the numbers of sequence reads in the background regions selected for each Si fraction after stabilization of the iterative process showed that they are strongly correlated with the numbers of sequence reads in the corresponding regions of the control sample _S_0 (Supplemental Fig. S13). These correlations illustrate the variations of the number of reads either due to experimental factors (e.g., sequence-associated variations of the efficiency of the immunoprecipitation step) or to copy number variations (frequently observed in the HeLa genome) (Macville et al. 1999) illustrated by the bimodality of the distribution of the read number (Supplemental Fig. S13A′). The ratios between the density of sequence reads in the Si fractions and in the control sample are similar for the low-copy number and the high-copy number regions (Supplemental Fig. S13A–D). This procedure of detection of background regions requires that the regions presenting significant enrichment values in nonadjacent fractions of the S phase (e.g., in _S_1 and _S_3) and nonsignificant values in the intermediate fraction (e.g., _S_2) represent a small proportion of the genome; we checked that such regions represented a small genome fraction (5.46%) (Supplemental Table S7).
Calculation of the replication time estimator S50
The ratio S50, defined as the fraction of the S phase (0 < S50 < 1) at which 50% of DNA is replicated in a defined genome region (50% of the cumulative enrichment) was computed by linear interpolation of enrichment values in the four compartments of the S phase as described in Jeon et al. (2005). When a region was not significantly enriched in all four Si periods, no S50 value was attributed (5.01% of genome regions). The S50 values of Experiments 1 and 2 were strongly correlated to each other (R = 0.97, P < 10−15) (Fig. 1C; Supplemental Fig. S2A). For each window, the mean S50 value of the two experiments was used as the final value. Since the FACS profile of the sorted cells showed an approximately equal abundance of all Si periods of the S phase (data not shown), we have therefore assumed an approximately linear mapping between the DNA content (S50) and the S-phase progression (replication time). S50 data are available in the Supplemental material.
Synchronously replicated regions
A defined window can be significantly enriched in one, two, three, or four Si periods (the corresponding amounts are given in Supplemental Table S7). Each window was classified as replicating either in a temporally specific or nonspecific manner as described in Karnani et al. (2007). A window is temporally specific (synchronously replicated) (1) if the enrichment in any Si period was at least twice the enrichment of each of the three other periods; or (2) if the sum of any two adjacent periods was at least three times the enrichment of each of the two other periods. Windows that do not satisfy these criteria are designated as temporally nonspecific or asynchronously replicated (Supplemental Table S7). We observed that a small amount of regions were asynchronously replicated (7.41%). As expected, asynchronously replicated regions mostly presented mid-replication timing values as shown by the corresponding S50 distributions (Supplemental Fig. S14B). By comparison, Karnani et al. (2007) observed that 20% of the ENCODE regions had a pan-S replication profile. Conversely, when analyzing our enrichment values in these (pan-S) regions, we found that 83% of them were synchronously replicated, with S50 timing values evenly distributed along the S phase (Supplemental Fig. S14C).
Determination of the substitution rates
The four-way catarrhini-specific alignments of Homo sapiens (assembly hg18), Pan troglodytes (panTro2), Pongo pygmaeus abelii (ponAbe2), and Macaca mulatta (rheMac2), that were generated using the Enredo Pecan Ortheus (EPO) pipeline (Paten et al. 2008a,b) were retrieved from the Ensembl Genome Browser (http://www.ensembl.org). Annotations of the human genome were retrieved from the UCSC Genome Browser (http://genome.ucsc.edu). To delineate the most reliable intergenic regions, transcribed regions were retrieved from “all_mrna,” one of the largest sets of annotated transcripts. To obtain gene sequences, we used the RefSeq annotation. Coding regions and CpG islands were not considered in the analyses (except when indicated). The first and last 50 bp of each intronic sequence were also excluded, since those regions likely contain control elements and evolve in a nonneutral fashion (Touchon et al. 2004). Nucleotide substitutions were tabulated in the human lineage since its divergence from chimpanzee using both the orangutan and macaque as outgroups. The analysis has been only performed with the autosomes. To minimize the effects of alignment artifacts, only isolated substitutions defined as those flanked by sites that are identical in the four species were tabulated. Sequences were divided into CpG and non-CpG sites. CpG sites were defined as the sites having the following human/chimpanzee/orangutan/macaque pattern: NG/CG/CG/CG or CG/NG/CG/CG or CN/CG/CG/CG or CG/CN/CG/CG, where N is any nucleotide. Substitution rates were calculated within nonoverlapping windows by dividing the number of substitution events of the appropriate type by the number of potentially mutable sites that meet the same criteria. Since the divergence between these four catarrhini species is small, possible multiple substitutions were ignored (using two outgroups instead of one lowers the amount of multiple mutations, in particular at CpG sites, and preferentially eliminates sites that are not ancestral to human and chimpanzee).
Determination of the transition rate at methylated CpG sites
The observed overall CpG transition rate, β, results from two types of substitutions: substitutions at methylated CpG sites and substitutions at unmethylated CpG sites.
The rate β is computed as follows. a is the transition rate at methylated CpG sites; b, the transition rate at unmethylated CpG sites; _m_1, the number of methylated CpG sites among the CpG sites; and _m_2, the number of unmethylated CpG sites among the CpG sites (_m_1 + _m_2 is the total number of CpG sites). By using two outgroup genomes (macaque and orangutan), we observe only a fraction, _k_1, of the ancestral methylated CpG sites and a fraction _k_2 of the ancestral unmethylated CpG sites. Thus, we have:
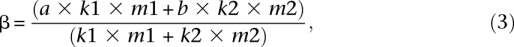
where _k_1 × _m_1 is the number of methylated CpG sites among the observed ancestral CpG sites and _k_2 × _m_2 is the number of unmethylated CpG sites among the observed ancestral CpG sites (_k_1 × _m_1 + _k_2 × _m_2 is the total number of observed ancestral CpG sites).
Equation 3 leads to:
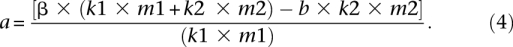
To compute the values of a in the early (_a_E) and late (_a_L) replicating regions, we first compute the early and late values of β, b, _m_1, and _m_2. βE = 3.97% and βL = 5.75% (Fig. 5A, left); _b_E = 0.30% and _b_L = 0.40% (the transition rate is similar at unmethylated CpG sites and at non-CpG cytosines) (Supplemental Fig. S3, G:C → A:T); the proportion of methylated sites (mean methylation level) increases from _m_1E/(_m_1E + _m_2E) = 0.63 in early regions, to _m_1L/(_m_1L + _m_2L) = 0.80 in late regions (Fig. 5A, center). To compute _k_1 and _k_2 (which does not depend on replication timing) (data not shown), we first compute the proportion of CpG sites that have been retained in the analysis (in the 1.5-Gb human sequences aligned with the chimpanzee, macaque, and orangutan sequences), that is, the ratio between the number of observed ancestral CpG sites (7.7 × 106) and the total number of CpGs (1.4 × 107). Thus, we obtain:
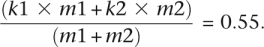
We compute _k_2 (using the property that the transition rate is similar at unmethylated CpG sites and non-CpG cytosines) as the ratio between the observed ancestral cytosines (2.9 × 108 retained for the analysis of transitions) and the total number of non-CpG cytosines (3.1 × 108), that is, _k_2 = 0.93. Since the mean methylation level of the whole analyzed sequences is _m_1/(_m_1 + _m_2) = 0.70 (Eckhardt et al. 2006), we obtain:
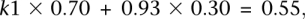
which leads to _k_1 = 0.39. We finally deduce the transition rates at the fully methylated CpG sites, _a_E = 8.98% and _a_L = 8.86%.
Determination of human diversity
The SNP data (rel27) of The International HapMap Project (Frazer et al. 2007) for each of four human populations—Yoruba in Ibadan (YRI), Japanese in Tokyo (JPT), Han Chinese in Beijing (CHB), and Utah residents with ancestry from northern and western Europe (CEU)—retrieved from the UCSC Genome Browser (http://genome.ucsc.edu) were used to calculate the human diversity. SNP data from four fully sequenced individual genomes of C. Venter (Levy et al. 2007), J. Watson (Wheeler et al. 2008), an anonymous Asian male (YH) (Wang et al. 2008), and a male Yoruba from Ibadan (Bentley et al. 2008) were also used to evaluate the human diversity.
Mouse data
Pairwise sequence alignments of Mus musculus (mm9) and Rattus norvegicus (rn4) and mouse genome annotations were retrieved from the UCSC Genome Browser (http://genome.ucsc.edu). The mouse–rat divergence was calculated in noncoding regions as described previously. Mouse diversity was calculated by using SNP data from the Perlegen Mouse SNP project (http://mouse.perlegen.com/mouse/) (The International HapMap Consortium 2007). Replication timing data of the mouse genome were obtained from Farkash-Amar et al. (2008).
Recombination rates
Crossover rate data of the human genome were retrieved from the International HapMap Project (http://www.hapmap.org) (Frazer et al. 2007). The crossover rate for a given window was computed as a weighted average of crossover rates in chromosomal regions overlapping with the corresponding window. The single nucleotide polymorphism genetic maps (Shifman et al. 2006), with a resolution of 160 ± 140 kb, were used to calculate crossover rates within the mouse genome. The recombination rate between all pairs of adjacent markers was calculated by dividing the distance between the markers in the genetic map (centimorgans, CM) by the distance between the markers in the sequence map in megabases (Mb).
DNA methylation
CpG methylation data of human chromosomes 6, 20, and 22 were retrieved from the Human Epigenome Project (HEP; http://www.epigenome.org/) (Eckhardt et al. 2006), and the mean methylation level was computed for each amplicon, excluding CpG islands. CpG and non-CpG substitution rates were computed in 10-kb windows centered on each amplicon; S50 values were computed in 50-kb windows centered on amplicons. Mouse genome CpG methylation data were retrieved from the Broad Institute (http://www.broad.mit.edu/) (Meissner et al. 2008). Mean methylation level and TR50 values were computed in 1-Mb non-overlapping windows. CpG and non-CpG SNP densities were computed in the restriction fragments (MspI) selected in Meissner et al. (2008), in 1-Mb nonoverlapping windows. Only the CpG sites located outside CpG islands were used in the analyses. For human sperm cells, the mean density of CpG sites (excluding CpG islands) was 42/Mb. For the mouse, the mean density of CpG sites was 235/Mb.
Statistical analysis
Statistical analyses were performed using R (http://www.r-project.org). For multivariate linear regression analysis, all parameters were standardized (zero mean value, variance equal to 1) so that the given slopes (i.e., the standardized regression coefficient, _r_std) of the various parameters are directly comparable measurements of the strength of the relationship between explanatory variables, xj, and the response variable, y. To determine the contribution of each parameter to the variance of the response variable, we used the notation defined in Scherrer (1984). The multiple correlation coefficient (_R_2) of the full model was computed as:

where _r_stdj is the standardized regression coefficient of the _j_th explanatory variable and ryxj is the simple correlation coefficient (Pearson r) between the response variable (y) and the _j_th explanatory variable (xj). The contribution of the _j_th explanatory variable to the variability explained by the full model was computed as:
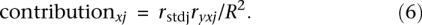
Acknowledgments
We thank the cell sorting facility (Influx 500) of the Institut Jacques Monod, UMR CNRS 7592, Universities Paris VII and VI (supported by the Région Ile-de-France, contract no. E539); and D. Casane, M. Ghosh, and M. Kwapisz for critical reading of this manuscript. This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Agence Nationale de la Recherche (NT05-3_41825), and grants from the Association pour la Recherche sur le Cancer, the Ligue Contre le Cancer (Comité de Paris), and the Fondation pour la Recherche Médicale (équipe labellisée) to O.H.
Footnotes
References
- Azuara V. Profiling of DNA replication timing in unsynchronized cell populations. Nat Protoc. 2006;1:2171–2177. doi: 10.1038/nprot.2006.353. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e26. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev. 1995;5:309–314. doi: 10.1016/0959-437x(95)80044-1. [DOI] [PubMed] [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelbrock MA, Kaliyaperumal S, Williams KJ. DNA mismatch repair efficiency and fidelity are elevated during DNA synthesis in human cells. Mutat Res. 2009;662:59–66. doi: 10.1016/j.mrfmmm.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science. 1981;212:1350–1357. doi: 10.1126/science.6262918. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Hurst LD. The evolution of isochores. Nat Rev Genet. 2001;2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L. GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics. 2001;159:907–911. doi: 10.1093/genetics/159.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Goldman MA, Holmquist GP, Gray MC, Caston LA, Nag A. Replication timing of genes and middle repetitive sequences. Science. 1984;224:686–692. doi: 10.1126/science.6719109. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH. A model for the correlation of mutation rate with GC content and the origin of GC-rich isochores. J Mol Evol. 1994;38:468–475. doi: 10.1007/BF00178846. [DOI] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet. 2003;72:1527–1535. doi: 10.1086/375657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE. Why do human diversity levels vary at a megabase scale? Genome Res. 2005;15:1222–1231. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010;20:155–169. doi: 10.1101/gr.099796.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Williams EJ. Covariation of GC content and the silent site substitution rate in rodents: Implications for methodology and for the evolution of isochores. Gene. 2000;261:107–114. doi: 10.1016/s0378-1119(00)00489-3. [DOI] [PubMed] [Google Scholar]
- Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Bekiranov S, Karnani N, Kapranov P, Ghosh S, Macalpine D, Lee C, Hwang DS, Gingeras TR, Dutta A. Temporal profile of replication of human chromosomes. Proc Natl Acad Sci. 2005;102:6419–6424. doi: 10.1073/pnas.0405088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor C, Malhotra A, Dutta A. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res. 2007;17:865–876. doi: 10.1101/gr.5427007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002;18:337–340. doi: 10.1016/s0168-9525(02)02669-0. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Yi S, Makova K. Male-driven evolution. Curr Opin Genet Dev. 2002;12:650–656. doi: 10.1016/s0959-437x(02)00354-4. [DOI] [PubMed] [Google Scholar]
- MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes & Dev. 2004;18:3094–3105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macville M, Schrock E, Padilla-Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res. 1999;59:141–150. [PubMed] [Google Scholar]
- Matassi G, Sharp PM, Gautier C. Chromosomal location effects on gene sequence evolution in mammals. Curr Biol. 1999;9:786–791. doi: 10.1016/s0960-9822(99)80361-3. [DOI] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Duret L. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 2004;21:984–990. doi: 10.1093/molbev/msh070. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 2001;17:481–485. doi: 10.1016/s0168-9525(01)02409-x. [DOI] [PubMed] [Google Scholar]
- Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: Genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008a;18:1814–1828. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, Holmes I, Birney E. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008b;18:1829–1843. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast JG, Campbell H, Gilbert N, Dunlop MG, Bickmore WA, Semple CA. Chromatin structure and evolution in the human genome. BMC Evol Biol. 2007;7:72. doi: 10.1186/1471-2148-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer B. Biostatistique, Gaëtan Morin edition. Boucherville; Québec, Canada: 1984. [Google Scholar]
- Shifman S, Bell JT, Copley RR, Taylor MS, Williams RW, Mott R, Flint J. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. Human mutation rate associated with DNA replication timing. Nat Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J, Tyekucheva S, Zody M, Chiaromonte F, Makova KD. Strong and weak male mutation bias at different sites in the primate genomes: Insights from the human–chimpanzee comparison. Mol Biol Evol. 2006;23:565–573. doi: 10.1093/molbev/msj060. [DOI] [PubMed] [Google Scholar]
- Touchon M, Arneodo A, d'Aubenton-Carafa Y, Thermes C. Transcription-coupled and splicing-coupled strand asymmetries in eukaryotic genomes. Nucleic Acids Res. 2004;32:4969–4978. doi: 10.1093/nar/gkh823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Guo Y, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Fujiyama A, Ichiba Y, Hattori M, Yada T, Sakaki Y, Ikemura T. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum Mol Genet. 2002;11:13–21. doi: 10.1093/hmg/11.1.13. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM, Li WH. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337:283–285. doi: 10.1038/337283a0. [DOI] [PubMed] [Google Scholar]
- Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. Replication timing of the human genome. Hum Mol Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- Yu FX, Dai RP, Goh SR, Zheng L, Luo Y. Logic of a mammalian metabolic cycle: An oscillated NAD+/NADH redox signaling regulates coordinated histone expression and S-phase progression. Cell Cycle. 2009;8:773–779. doi: 10.4161/cc.8.5.7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Boerwinkle E. Neighboring-nucleotide effects on single nucleotide polymorphisms: A study of 2.6 million polymorphisms across the human genome. Genome Res. 2002;12:1679–1686. doi: 10.1101/gr.287302. [DOI] [PMC free article] [PubMed] [Google Scholar]