Exome sequencing identifies the cause of a Mendelian disorder (original) (raw)
. Author manuscript; available in PMC: 2010 Jul 1.
Published in final edited form as: Nat Genet. 2009 Nov 13;42(1):30–35. doi: 10.1038/ng.499
Abstract
We demonstrate the first successful application of exome sequencing to discover the gene for a rare, Mendelian disorder of unknown cause, Miller syndrome (OMIM %263750). For four affected individuals in three independent kindreds, we captured and sequenced coding regions to a mean coverage of 40X, and sufficient depth to call variants at ~97% of each targeted exome. Filtering against public SNP databases and a small number of HapMap exomes for genes with two novel variants in each of the four cases identified a single candidate gene, DHODH, which encodes a key enzyme in the pyrimidine de novo biosynthesis pathway. Sanger sequencing confirmed the presence of DHODH mutations in three additional families with Miller syndrome. Exome sequencing of a small number of unrelated, affected individuals is a powerful, efficient strategy for identifying the genes underlying rare Mendelian disorders and will likely transform the genetic analysis of monogenic traits.
Rare monogenic diseases are of substantial interest because identification of their genetic basis provides important knowledge about disease mechanisms, biological pathways, and potential therapeutic targets. However, to date, allelic variants underlying fewer than half of all monogenic disorders have been discovered. This is because the identification of allelic variants for many rare disorders is fundamentally limited by factors such as the availability of only a small number of cases/families, locus heterogeneity, or substantially reduced reproductive fitness, each of which lessens the power of traditional positional cloning strategies and often restricts the analysis to a priori identified candidate genes. In contrast, deep resequencing of all human genes for discovery of allelic variants could potentially identify the gene underlying any given rare monogenic disease. Massively parallel DNA sequencing technologies1 have rendered the whole genome resequencing of individual humans increasingly practical, but cost remains a key consideration. An alternative approach involves the targeted resequencing of all protein-coding subsequences (i.e., the “exome”)2-4, which requires ~5% as much sequencing as a whole human genome2.
Sequencing of the exome, rather than the entire human genome, is well justified as an efficient strategy to search for alleles underlying rare Mendelian disorders. First, positional cloning studies focused on protein-coding sequences have, when adequately powered, proven highly successful at identification of variants for monogenic diseases. Second, the clear majority of allelic variants known to underlie Mendelian disorders disrupt protein-coding sequences5. Splice acceptor and donor sites represent an additional class of sequences that are enriched for highly functional variation, and are therefore targeted here as well. Third, a large fraction of rare non-synonymous variants in the human genome are predicted to be deleterious6. This contrasts with non-coding sequences, where variants are more likely to have neutral or weak effects on phenotypes, even in well conserved non-coding sequences7,8. The exome therefore represents a highly enriched subset of the genome in which to search for variants with large effect sizes.
We recently showed how exome sequencing of a small number of affected, unrelated individuals could potentially be applied to identify a causal gene underlying a monogenic disorder2. Specifically, we performed targeted enrichment of the exome by hybridization to programmable microarrays and then sequenced each enriched shotgun genomic library on an Illumina Genome Analyzer II. The exome was conservatively defined using the National Center for Biotechnology Information (NCBI) Consensus Coding Sequence (CCDS) database9 (version 20080902), which covers approximately 164,000 discontiguous regions over 27.9 Mb, of which 26.6 Mb were “mappable” with 76 bp single-end reads. Approximately 96% of targeted, mappable bases comprising the exomes of 8 HapMap individuals and 4 individuals with Freeman-Sheldon syndrome (FSS (OMIM #193700)) were successfully sequenced to high quality2. Using both dbSNP and HapMap exomes as filters to remove common variants, we showed that we could accurately identify the causal gene for FSS by exome sequencing alone. This effort demonstrated that low-cost, high throughput technologies for deep resequencing have the potential to rapidly accelerate the discovery of allelic variants for rare diseases. However, it provided only a proof-of-concept as the causal gene for FSS had previously been identified10. A more recent report by Choi et al. describes the generation of exome sequences of similar quality with a different capture platform, and the application of exome sequencing to make an unanticipated genetic diagnosis of congenital chloride diarrhea3.
To evaluate the effectiveness of this strategy on a Mendelian condition of unknown cause, we sought to find the gene for a rare multiple malformation disorder named Miller syndrome11, the cause of which has been intractable to standard approaches of discovery12. The clinical characteristics of Miller syndrome include severe micrognathia, cleft lip/palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids, and supernumerary nipples (Figure 1a,b). Miller syndrome has been hypothesized to be an autosomal recessive disorder. However, only three multiplex families, each of which consists of two affected sibs born to unaffected, nonconsanguineous parents, have been described among a total of ~30 reported cases for which substantial clinical information is available11,13-17. Accordingly, there has been speculation that Miller syndrome is an autosomal dominant disorder18 and the rare occurrence of affected siblings the result of germline mosaicism. While we thought it likely that Miller is recessive, we also considered a dominant model of inheritance.
Figure 1. Clinical characteristics of an individual with Miller syndrome (A,B) and an individual with methotrexate embryopathy (C,D).
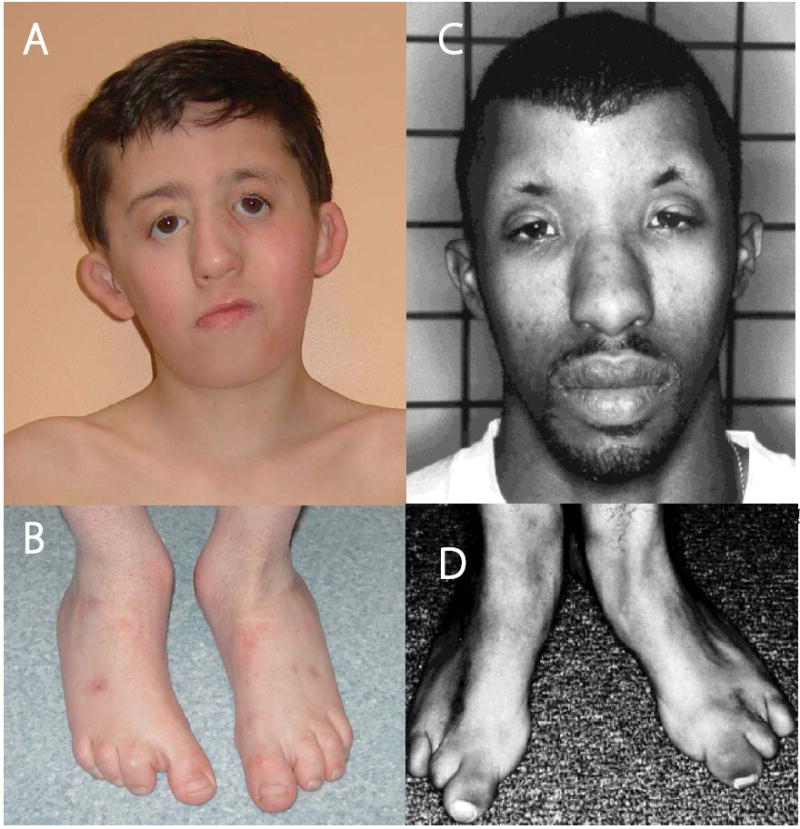
A 9 year-old boy with Miller syndrome (A and B) caused by mutations in DHODH. Facial anomalies (A) include cupped ears, coloboma of the lower eyelids, prominent nose, micrognathia and absence of the 5th digits of the feet (B). A 26 year-old man with methotrexate embryopathy (C and D). Note the cupped ears, hypertelorism, sparse eyebrows, and prominent nose (C) accompanied by absence of the 4th and 5th digits of the feet (D). C and D are reprinted with permission from Bawle et al. Teratology 57:51-55 (1978).
Results
Exome sequencing identifies a candidate gene for Miller syndrome
Exomes were sequenced in a total of two siblings with Miller syndrome (kindred 1 in Table 1) and two additional unrelated affected individuals (kindreds 2 and 3 in Table 1), i.e. a total of 4 affected individuals in three independent kindreds. An average of 5.1 gigabases (Gb) of sequence was generated per affected individual as single-end, 76 bp reads. After discarding reads that had duplicated start sites, we achieved ~40-fold coverage of the 26.6 Mb mappable, targeted exome defined by Ng et al. (2009)2 (Table 2). About 97% of targeted bases were sufficiently covered to pass our thresholds for variant calling. To distinguish potentially pathogenic mutations from other variants, we focused only on nonsynonymous (NS) variants, splice acceptor and donor site mutations (SS) and coding indels (I), anticipating that synonymous variants were far less likely to be pathogenic. We also predicted that the variants responsible for Miller syndrome would be rare, and therefore likely to be novel. A novel variant was defined as one that did not exist in the datasets used for comparison, including dbSNP129, exome data from 8 HapMap individuals previously sequenced by us2, and both (Table 1).
Table 1. Direct identification of the gene for a Mendelian disorder by exome resequencing.
Each cell indicates the number of genes with nonsynonymous (NS) variants, splice acceptor and donor site mutations (SS), and coding indels (I). Filtering by either requiring the presence of NS/SS/I variants in siblings (kindred 1 (A+B)), multiple unrelated individuals (columns), or by excluding annotated variants (rows) identifies 26 and 8 candidate genes under a dominant model and only a single candidate gene, DHODH, under a recessive model (light grey cells). Exclusion of mutations predicted to be benign using PolyPhen (row 5) increases sensitivity under a dominant model but excludes DHODH under a recessive model because a variant in kindred 1 is predicted to be benign. A single candidate gene is identified in kindred 1 under a recessive model (dark grey cell) but this candidate is excluded in comparisons with unrelated cases of Miller syndrome. Mutations in this candidate, DNAH5, were found to cause a primary ciliary dyskinesia in kindred 1. The asterisk indicates that a second gene, CDC27, was also identified as a candidate gene but on manual review it appears to be multiple copies of a processed pseudogene that cause a false positive.
| Filter | Kindred 1-A | Kindred 1-B | Kindred1 (A + B) | Kindreds 1+2 | Kindred 1+2+3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dominant | Recessive | Dominant | Recessive | Dominant | Recessive | Dominant | Recessive | Dominant | Recessive | |
| NS/SS/I | 4670 | 2863 | 4687 | 2859 | 3940 | 2362 | 3099 | 1810 | 2654 | 1525 |
| … not in dbSNP129 | 641 | 102 | 647 | 114 | 369 | 53 | 105 | 25 | 63 | 21 |
| … notin HapMap 8 | 898 | 123 | 923 | 128 | 506 | 46 | 117 | 7 | 38 | 4 |
| … not in either | 456 | 31 | 464 | 33 | 228 | 9 | 26 | 1* | 8 | 1* |
| … AND predicted damaging | 204 | 6 | 204 | 12 | 83 | 1 | 5 | 0 | 2 | 0 |
Table 2. Summary statistics for exome sequencing of four individuals with Miller Syndrome.
The total number of unpaired 76-bp sequencing reads per individual is reported (“Total”), along with the number that map uniquely to the human genome (“Uniquely Mapping”, Maq map score > 0), the number that overlap at least one base of the target space (“Overlapping Target”), and the number left after removing reads with duplicate start sites (“Non-duplicated”). Mean coverage over the whole of CCDS2008 is also given. Called bases refer to bases passing quality and coverage thresholds (Maq consensus quality >= 20 and read depth >= 8X). % of CCDS refers to the fraction of the mappable 26.6 megabases of CCDS2008 (i.e. masked for poorly mappable coordinates, as described in Ng et al. 2009) that is called in each exome.
| Sequencing reads | Called coverage | ||||||
|---|---|---|---|---|---|---|---|
| Kindred-Sibling | Total | Uniquely Mapping | Overlapping Target | Non-duplicated | Mean Coverage | Called Bases | % of CCDS |
| 1-A | 62,974,440 | 52,854,115 | 25,267,592 | 17,872,660 | 36.85 | 25,720,216 | 97 |
| 1-B | 72,539,306 | 61,940,123 | 40,335,280 | 21,971,509 | 44.24 | 25,825,104 | 97 |
| 2 | 63,839,828 | 55,022,098 | 29,987,198 | 19,686,779 | 40.31 | 25,790,427 | 97 |
| 3 | 68,690,600 | 57,970,901 | 36,180,596 | 19,649,281 | 39.81 | 25,617,361 | 96 |
Each sibling (i.e., A and B) in kindred 1 was found to have at least a single NS/SS/I variant in ~4,600 genes and two or more NS/SS/I variants in ~2,800 genes. For a dominant model, in which each sib was required to have at least one novel NS/SS/I variant in the same gene, filtering these variants against dbSNP129 and 8 HapMap exomes reduced the candidate gene pool ~40-fold. For a recessive model in which each sib was required to have at least two novel NS/SS/I variants in the same gene, the candidate pool was reduced >500-fold compared to all known human genes. Both siblings were predicted to share the causal variant for Miller syndrome so we next considered novel NS/SS/I variants shared between them. Under our dominant model, this reduced the pool of candidate genes to 228, and under our recessive model, the number of candidate genes was reduced to 9.
To further exclude candidate genes containing non-pathogenic variants, we next compared the NS/SS/I variants from both siblings in kindred 1 to the novel variants in two unrelated individuals with Miller syndrome (kindreds 2 and 3). Using both dbSNP129 and the 8 HapMap exomes as filters, comparison between the siblings in kindred 1 and the unrelated simplex case in kindred 2 reduced the number of candidate genes to 26 under our dominant model. Under the autosomal recessive model, this comparison revealed that only a single gene, DHODH, which encodes the enzyme, dihydroorotate dehydrogenase, was a shared candidate. Thus, comparison of exome data from two affected sibs and one unrelated, affected individual was sufficient to identify DHODH as the sole candidate gene for Miller syndrome. Comparison between the siblings in kindred 1 and the unrelated simplex cases in kindreds 2 and 3 reduced the number of candidate genes to 8 under a dominant model, while retaining DHODH as the sole candidate under the recessive model.
We calculated a Bonferroni corrected p-value for the null hypothesis of no deviation from the expected frequency of two variants in the same gene in three out of three unrelated, affected individuals over the ~17,000 genes in CCDS2008. Assuming all genes are of the same length and have the same mutation rate, the rate of novel NS/SS/I variants per gene was 0.0309 (i.e., ~526 novel NS/SS/I variants per 17,000 genes). If the variants occur independently of one another, two variants occur in the same gene at a rate of (0.0309)2 or 9.57 × 10-4 so the p-value is ((9.57 × 10-4)3 × 17,000) or ~0.000015. Hence, even after correcting for searching across all genes, the result remains highly significant.
We also examined the effect on the size of the candidate gene list when analyzing the exomes of affected individuals in different pairwise or three-way combinations, and the potential consequences of genetic heterogeneity by relaxing selection criteria such that only a subset of the exomes of affected individuals were required to contain novel variants in a given gene for it to be considered as a candidate gene (Table 3). Heterogeneity clearly increases the number of candidate genes that must be considered under any fixed number of exomes analyzed. However, this likely can be overcome by the inclusion of a greater number of cases with mutations in the same gene.
Table 3. Number of candidate genes identified based on different filtering strategies.
Under the dominant model, at least one non-synonymous variant, splice acceptor or donor site variant or coding indel (NS/SS/I) in a gene was required in the gene. Under the recessive model, at least two novel variants were required, and these could be either at the same position (i.e. a homozygous variant) or at two different positions in the same gene (i.e. a potential compound heterozygote, though we were unable to ascertain phase at this stage). In each column is the range for the number of candidate genes for exomes considered individually (column 1) and all combinations of 2 to 4 exomes (columns 2-4). Note that the upper bound on the ranges may be inflated relative to what would be the case if four unrelated, affected individuals had been used because the comparisons in which the two siblings were included provided reduced power compared to unrelated individuals. Columns 5-9 show the number of candidate genes when at least 1, 2, or 3 individuals is required to have one variant in a gene (dominant model) or two or more variants in a gene (recessive model). This is a simple model of genetic heterogeneity or incomplete data. For example, the total number of candidate genes common to any 3 of all 4 exomes is shown in column 9. For columns 5-6, one of the siblings (Kindred 1-B) was not included in the analysis as sibs share 50% of variants.
| Dominant Model | Number of Affected Exomes | Subsets of 3 Exomes | Subsets of all 4 Exomes | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | Any 1 | Any 2 | Any 1 | Any 2 | Any 3 | |
| NS/SS/I | 4645-4687 | 3358-3940 | 2850-3099 | 6658 | 4489 | 6943 | 5167 | 3920 |
| … not in dbSNP129 | 634-695 | 136-369 | 72-105 | 1617 | 274 | 1829 | 553 | 172 |
| … not in HapMap 8 | 898-979 | 161-506 | 55-117 | 2336 | 409 | 2628 | 835 | 222 |
| … not in either | 453-528 | 40-228 | 10-26 | 1317 | 109 | 1516 | 333 | 44 |
| … AND predicted damaging | 204-284 | 10-83 | 3-6 | 682 | 37 | 787 | 126 | 11 |
| Recessive Model | Number of Affected Exomes | Subsets of 3 Exomes | Subsets of all 4 Exomes | |||||
| 1 | 2 | 3 | Any 1 | Any 2 | Any 1 | Any 2 | Any 3 | |
| NS/SS/I | 2780-2863 | 1993-2362 | 1646-1810 | 4097 | 2713 | 4293 | 3172 | 2329 |
| … not in dbSNP129 | 92-115 | 30-53 | 22-31 | 226 | 61 | 270 | 90 | 42 |
| … not in HapMap 8 | 111-133 | 13-46 | 5-13 | 329 | 32 | 397 | 75 | 19 |
| … not in either | 31-45 | 2-9 | 2-3 | 100 | 6 | 121 | 14 | 4 |
| … AND predicted damaging | 6-16 | 0-2 | 0-1 | 35 | 2 | 44 | 4 | 1 |
Most variants underlying rare Mendelian diseases either affect highly conserved sequence and/or are predicted to be deleterious. Accordingly, we also sought to investigate to what extent the pool of candidate genes could be reduced by combining variant filtering with predictions of whether NS/SS/I variants were “damaging.” This strategy further reduced the pool of candidate genes for each of the comparison made previously (Table 1). However, DHODH was not identified as a candidate under a recessive model in any of these comparisons. Review of predicted biophysical consequences of DHODH variants revealed that the effect of one variant, c.G605A, found in both siblings in kindred 1, was benign. As a result, DHODH was eliminated from further consideration as a candidate under a recessive model in kindred 1 and all subsequent comparisons. However, because the other variant found in kindred 1, c.G454A, was predicted to be damaging, as was every other novel DHODH variant identified, DHODH was the only candidate gene for Miller syndrome under a dominant model in a comparison of kindreds 1, 2, and 3.
Combinatorial filtering supplemented by PolyPhen predictions initially identified a second candidate gene, DNAH5, in kindred 1 under a recessive model (Table 1). However, this candidate was excluded in subsequent comparisons to kindreds 2 and 3. DNAH5 encodes a dynein heavy chain found in cilia, and mutations in DNAH5 are a well-known cause of primary ciliary dyskinesia (PCD; OMIM #608644), a disorder characterized by recurrent sinopulmonary infections, bronchiectasis, and chronic lung disease. This was of particular interest because some of the clinical findings in the siblings in kindred 1 are unique among reported cases of Miller syndrome. Specifically, both siblings have recurrent lung infections, bronchiectasis, and chronic obstructive pulmonary disease for which they have received medical management in a specialty clinic for individuals with cystic fibrosis. Accordingly, exome analysis revealed that both siblings in kindred 1 have, in addition to Miller syndrome, PCD due to mutations in DNAH5.
Sanger sequencing of implicated gene
To confirm that mutations in DHODH were responsible for Miller syndrome, we screened three additional unrelated kindreds (one pair of affected siblings and two simplex cases) by directed Sanger sequencing. All four individuals were found to be compound heterozygotes for missense mutations in DHODH that are predicted to be deleterious. Collectively, 11 different mutations in 6 kindreds were identified in DHODH by a combination of exome and targeted resequencing (Table 4 and Figure 2). Each parent of an affected individual who was tested was found to be heterozygous carriers and none of the mutations appeared to have arisen de novo. In the kindred with affected siblings, none of the unaffected siblings were compound heterozygotes. None of these mutations were found in 200 control chromosomes from individuals of matched geographical ancestry that were genotyped. Ten of these mutations were missense mutations, two of which affected the same amino acid codon, and one was a 1-bp indel that is predicted to cause a frameshift and a termination codon seven amino acids downstream. One mutation, c1036T, was shared between two unrelated individuals with Miller syndrome who are of different self-identified geographical ancestry. Each of the amino acid residues affected by a DHODH mutation is highly conserved among homologues studied to date (Supplementary Figure 1). A single validated nonsynonymous polymorphism in human DHODH has been studied by Grabar et al.19 This polymorphism causes a lysine to glutamine substitution in the relatively diverse N-terminal extension of dihydroorotate dehydrogenase that is responsible for the association of the enzyme with the inner mitochondrial membrane.
Table 4. Summary of DHODH mutations in kindreds with Miller syndrome.
| Kindred | Mutation | Exon | Amino acid change | Location |
|---|---|---|---|---|
| 1* | c.454G>A | 4 | p.G152R | chr16:70608443 |
| c.605G>C | 5 | p.G202A | chr16: 70612611 | |
| 2* | c.403C>T | 3 | p.R135C | chr16: 70606041 |
| c.1036C>T | 8 | p.R346W | chr16: 70614936 | |
| 3* | c.595C>T | 5 | p.R199C | chr16: 70612601 |
| c.611delT | 5 | p.L204PfsX8 | chr16: 70612617 | |
| 4 | c.605G>A | 5 | p.G202D | chr16: 70612611 |
| c.730C>T | 6 | p.R244W | chr16: 70613786 | |
| 5 | c.56G>A | 2 | p.G19E | chr16: 70603484 |
| c.1036C>T | 8 | p.R346W | chr16: 70614936 | |
| 6 | c.851C>T | 7 | p.T284I | chr16: 70614596 |
| c.1175A>G | 9 | p.D392G | chr16: 70615586 |
Figure 2. Genomic structure of the exons encoding the open reading frame of DHODH.
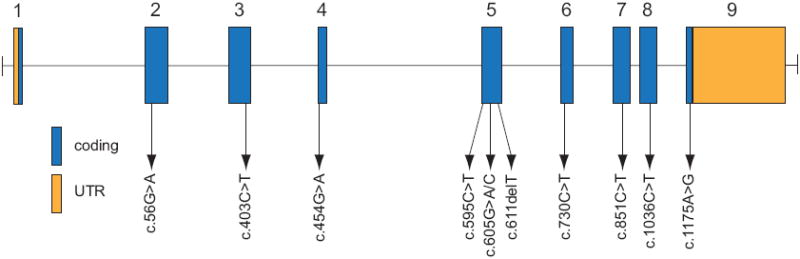
DHODH is composed of 9 exons that encode untranslated regions (orange) and protein coding sequence (blue). Arrows indicate the locations of 11 different mutations found in 6 families with Miller syndrome.
Discussion
We show that the sequencing of the exomes of affected individuals from a few unrelated kindreds, with appropriate filtering against public SNP databases and a small number of HapMap exomes, is sufficient to identify a single candidate gene for a previously unsolved monogenic disorder, Miller syndrome. Several factors were important to the success of this study. First, Miller syndrome is a very rare disorder that is inherited in an autosomal recessive pattern. Therefore, the causal variants were unlikely to be found in public SNP databases or control exomes. Second, genes for recessive diseases will, in general, be easier to find than genes for dominant disorders because fewer genes in any single individual have 2 or more novel or rare nonsynonymous variants. Third, we were fortunate that there was no genetic heterogeneity in our sample of Miller cases. In the presence of heterogeneity, it is possible to relax stringency by allowing for genes common to subsets of all affected individuals to be considered candidates, although this will reduce power (Table 3). Third, all of the individuals with Miller syndrome for whom exomes were sequenced were of European ancestry. Sequencing exomes of affected individuals sampled from populations with a different geographical ancestry who have a higher number of novel and/or rare variants (e.g., sub-Saharan African, East Asian) will make the identification of candidate genes more difficult. This will become less of an issue as databases of human polymorphisms become increasingly comprehensive.
Additional factors could facilitate the future application of this strategy. Mapping information, such as blocks of homozygosity, could focus the search to a smaller pool of candidates. The number of candidate variants can also be reduced further by comparison between variants in a case to those found in each parent. For autosomal dominant disorders, this strategy can discover de novo coding variants as neither parent is predicted to have a mutation that causes a fully penetrant dominant disorder, whereas for recessive disorders, parents are predicted to be carriers of the disease-causing variants.
There are at least three aspects of this approach where we see significant scope for improvement. The first relates to missed variant calls, either due to low coverage or because some variants are not identified easily with current sequencing platforms (e.g. within repeat tracts in coding sequences). The second is that our filtering relied on a public SNP database (dbSNP) that is a highly uneven ascertainment of variation across the genome. It would be better to rely on catalogues of common variation that are ascertained in a single study either exome-wide (as with the 8 HapMap exomes2) or genome-wide (e.g. as with the 1000 Genomes project), and where estimates of allele frequency are available. Increasing the number of control exomes progressively reduces the relevance of dbSNP to this analysis (Supplementary Figure 2). Furthermore, as increasingly deep catalogs of polymorphism become available, it may be necessary to establish frequency-based thresholds for defining “common” variation that is unlikely to be causal. A third concern is that the specificity of this approach is currently reduced by a subset of genes that recurrently appear enriched for novel variants. These include long genes, but also genes that are subject to systematic technical artifacts (e.g. mis-mapped reads due to duplicated or highly similar sequence in the genome). For sequences that are known to be duplicated or have paralogues (e.g. genes from large gene families, or pseudogenes), these artifacts are mostly removed during read alignment (as reads with non-unique placements are removed from consideration). However, duplicated sequences not represented in the reference genome are not removed and spuriously appear as enriched for novel variants (e.g. CDC27).
The mechanism by which mutations in DHODH cause Miller syndrome is unclear. The primary known function of dihydroorotate dehydrogenase is to catalyze the conversion of dihydroorotate to orotic acid, an intermediate in the pyrimidine de novo biosynthesis pathway (Supplementary Figure 3)20. Orotic acid is subsequently converted to uridine monophosphate (UMP) by UMP synthase. Pyrimidine biosynthesis might be particularly sensitive to the step mediated by dihydroorotate dehydrogenase21 and the classical rudimentary phenotype in D. melanogaster, reported by T.H. Morgan in 1910 and characterized by wing anomalies, defective oogenesis, and malformed posterior legs, is caused by mutations in the same pathway22-24. However, the clinical characteristics of the other inborn errors of pyrimidine biosynthesis such as orotic aciduria, caused by mutations in UMP synthase, do not include malformations. Indeed, inborn errors of metabolism are, in general, a rare cause of birth defects so DHODH would be given little weight a priori as a candidate for a multiple malformation disorder. Thus, the discovery that mutations in DHODH cause Miller syndrome reveals both a new role for pyrimidine metabolism in craniofacial and limb development as well as a novel function of dihydroorotate dehydrogenase that remains to be explored.
Selective inhibition of pyrimidine or purine biosynthesis has long been used as a therapeutic option to treat various cancers and autoimmune disorders. Leflunomide, a prodrug that is converted in the gastrointestinal tract to the active metabolite, A771726, reduces de novo pyrimidine biosynthesis by selectively inhibiting dihydroorotate dehydrogenase21. In mice, use of leflunomide during pregnancy causes a wide range of limb and craniofacial defects, the most common of which are exencephaly, cleft palate, and “open eye” or failure of eyelid to close25. These phenotypic characteristics recapitulate some of the malformations observed in individuals with Miller syndrome providing further evidence that it is caused by mutations in DHODH .
The developmental pathways disrupted by leflunomide are unknown but their elucidation could help understand the mechanism by which DHODH mutations cause malformations. In the liver of mice treated with leflunomide, TNF-α production is repressed by the direct inhibition of NF-κB activity26. Interruption of NF-κB signaling during development can result in disrupted cell migration, diminished cellular proliferation, and increased apoptosis27. Indeed, open eye is a defect observed in mice with targeted disruption of TNF-α28C. Furthermore, NF-κB plays an important role in limb morphogenesis, specifically as a transducer of signals that regulate Sonic hedgehog (Shh) expression. Shh controls, in part, anterior-posterior patterning of the digits and Shh-/- knockout mice fail to form digits 2-529. These observations suggest that the malformations observed in individuals with Miller syndrome could be caused by perturbed NF-κB signaling due to loss of DHODH function.
The pattern of malformations observed in individuals with Miller syndrome is similar to those of individuals with fetal exposure to methotrexate (Figure 1c,d). Methotrexate is a well-established inhibitor of de novo purine biosynthesis, and its anti-proliferative actions are thought to be due to its inhibition of dihydrofolate reductase and folate-dependent transmethylations. Accordingly, defects of both purine and pyrimidine biosynthesis appear to be capable of causing a similar pattern of birth defects. However, at low doses methotrexate also decreases plasma levels of pyrimidines as well as purines. This observation raises the possibility that methotrexate embryopathy might indeed be caused by its effects on pyrimidine rather than purine metabolism. Given that not all embryos exposed to methotrexate manifest birth defects, functional polymorphisms in DHODH or other genes in the de novo pyrimidine biosynthesis pathway could influence susceptibility to methotrexate embryopathy.
Individuals with Miller syndrome have similar phenotypic characteristics to those with Nager syndrome, another rare monogenic disorder that primarily affects the craniofacial skeleton. In contrast to Miller syndrome, the limb defects observed in individuals with Nager syndrome affect the anterior elements of the upper limb. Nevertheless, it has been hypothesized that Miller and Nager syndromes were caused by different mutations in the same gene. We resequenced DHODH in twelve unrelated individuals diagnosed with Nager syndrome but found no pathogenic mutations (data not shown). Accordingly, Nager syndrome and Miller syndrome are either not allelic or Nager syndrome is caused exclusively by mutations in regulatory elements that alter the expression of DHODH.
Rare diseases are arbitrarily defined as those that affect fewer than 200,000 individuals in the U.S. Per this definition, more than 7,000 rare diseases have been delineated, and in aggregate these affect more than 25 million people [Rare Diseases Act of 2002, Section 2, Findings]. The majority of these diseases are “genetic disorders” and many of them are thought to be monogenic. The bulk of genes underlying these rare monogenic diseases remain unknown. Lack of information about the genes and pathways that underlie rare monogenic diseases is a major gap in our scientific knowledge. Discovery of the genetic basis of a large collection of rare disorders that have, to date, been unyielding to analysis will substantially expand our understanding of biology of rare diseases, facilitate accurate diagnosis and improved management, and provide initiative for further investigation of novel therapeutics.
We have demonstrated that exome sequencing of a small number of affected family members or affected unrelated individuals is a powerful, efficient, and cost-effective strategy for markedly reducing the pool of candidate genes for rare monogenic disorders, and may even identify the responsible gene(s) specifically. This approach will likely become a standard tool for the discovery of genes underlying rare monogenic diseases and provide important guidance for developing an analytical framework for finding rare variants influencing risk of common disease.
Methods
Patients and Samples
For exome resequencing, we selected four individuals of self-reported European ancestry with Miller syndrome from three unrelated families. In one family, two siblings were affected (i.e., kindred 1 in Table 1), and in two families a single individual had been diagnosed with Miller syndrome (i.e., kindreds 2 and 3 in Table 1). For validation, we selected samples from four individuals in three additional families including a family with two affected siblings and two simplex cases. All participants provided written consent and the Institutional Review Boards of Seattle Children’s Hospital and the University of Washington approved all studies.
A referral diagnosis of Miller syndrome made by a clinical geneticist was required for inclusion. The clinical characteristics of several of the individuals who had been diagnosed with Miller syndrome have been reported previously11,13,14. Phenotypic data were collected from review of medical records, phone interviews, and photographs.
Targeted capture and massive parallel sequencing
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols, and ten micrograms of DNA from each of four individuals with Miller syndrome in kindred’s 1, 2, and 3 was used for construction of a shotgun sequencing library as described previously2 using adaptors for single-end sequencing on an Illumina Genome Analyzer II (GAII). Each shotgun library was hybridized to two Agilent 244K microarrays for target enrichment, followed by washing, elution and additional amplification2. The first array targeted CCDS (2007), while the second was designed against targets poorly captured by the first array plus updates to CCDS in 2008. Enriched libraries were then sequenced on a GAII.
Read mapping and variant analysis
Reads were mapped to the reference human genome (UCSC hg18), initially with ELAND (Illumina) for quality recalibration, and then again with Maq30. Sequence calls were also performed by Maq, and filtered to coordinates with >= 8x coverage and a _phred_-like30 consensus quality >= 20. Indels affecting coding sequence were identified as described previously2. Sequence calls were compared against 8 HapMap individuals for whom we had previously reported exome data2. Annotations of variants were based on NCBI and UCSC databases, supplemented with PolyPhen Grid Gateway predictions generated for nearly all nonsynonymous SNPs. Any non-synonymous variant that was not assigned a “benign” PolyPhen prediction was considered to be damaging, as were all splice acceptor and donor site mutations and all coding indels.
Mutation validation
Sanger sequencing of PCR amplicons from genomic DNA was used to confirm the presence and identity of variants in the candidate gene identified via exome sequencing and to screen the candidate gene in additional cases of Miller syndrome.
Supplementary Material
1
Acknowledgments
We thank the families for their participation and the Foundation of Nager and Miller Syndrome for their support. We thank M. McMillin for assistance with project coordination. We thank R. Scott, T. Cox, L. Cox, R. Jack, E. Eichler, G. Cooper, J. Kidd, and R. Waterston for discussions. Our work was supported in part by grants from the National Institutes of Health/National Heart Lung and Blood Institute, the National Institutes of Health/National Human Genome Research Institute, and the National Institutes of Health/National Institute of Child Health and Human Development. S.B.N. is supported by the Agency for Science Technology and Research, Singapore. A.W.B. is supported by a training fellowship from the National Institutes of Health/National Human Genome Research Institute.
Footnotes
Author contributions The project was conceived and experiments planned by M.B., D.A.N., and J.S. Review of phenotypes and sample collection were performed by E.W.J. and M.B. Experiments were performed by S.B.N., K.J.B., and C.L. Genetic counseling and ethical consultation were provided by K.M.D and H.K.T. Data analysis were performed by S.B.N., K.J.B., A.W.B., C.D.H., P.T.S., and J.S. The manuscript was written by M.B., J.S., S.B.N., and K.J.B. All aspects of the study were supervised by M.B., D.A.N., and J.S.
References
- 1.Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 2.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009 doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi M, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodges E, et al. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–7. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 5.Stenson PD, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–39. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CT, Wang JC, Cohen BA. The strength of selection on ultraconserved elements in the human genome. Am J Hum Genet. 2007;80:692–704. doi: 10.1086/513149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahituv N, et al. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5:e234. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pruitt KD, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–23. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toydemir RM, et al. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet. 2006;38:561–5. doi: 10.1038/ng1775. [DOI] [PubMed] [Google Scholar]
- 11.MIller M, Fineman R, Smith DW. Postaxial acrofacial dysostosis syndrome. J Pediat. 1979;95:970–975. doi: 10.1016/s0022-3476(79)80285-1. [DOI] [PubMed] [Google Scholar]
- 12.Splendore A, Passos-Bueno MR, Jabs EW, Van Maldergem L, Wulfsberg EA. TCOF1 mutations excluded from a role in other first and second branchial arch-related disorders. Am J Med Genet. 2002;111:324–7. doi: 10.1002/ajmg.10567. [DOI] [PubMed] [Google Scholar]
- 13.Fineman RM. Recurrence of the postaxial acrofacial dysostosis syndrome in a sibship: implications for genetic counseling. J Pediat. 1981;98:87–88. doi: 10.1016/s0022-3476(81)80546-x. [DOI] [PubMed] [Google Scholar]
- 14.Oglivy-Stuart AL, Parsons AC. Miller syndrome (postaxial acrofactial dysostosis): further evidence for autosomal recessive inheritance and expansion of the phenotype. J Med Genet. 1991;28 doi: 10.1136/jmg.28.10.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnai D, Hughes HE, Winter RM. Postaxial acrofacial dysostosis (Miller) syndrome. J Med Genet. 1987;24:422–425. doi: 10.1136/jmg.24.7.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genee E. Une forme extensive de dysostose mandibulo-faciale. J Genet Hum. 1969;17:45–52. [PubMed] [Google Scholar]
- 17.Pereira SCS, Rocha CMG, Guion-Almeida ML, Richieri-Costa A. Postaxial acrofacial dysostosis: report on two patients. Am J Med Genet. 1992;44:274–279. doi: 10.1002/ajmg.1320440303. [DOI] [PubMed] [Google Scholar]
- 18.Robinow M, Johnson GF, Apesos J. Robin sequence and oligodactyly in mother and son. Am J Med Genet. 1986;25:293–7. doi: 10.1002/ajmg.1320250214. [DOI] [PubMed] [Google Scholar]
- 19.Grabar PB, Rozman B, Logar D, Praprotnik S, Dolzan V. Dihydroorotate dehydrogenase polymorphism influences the toxicity of leflunomide treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2009;68:1367–8. doi: 10.1136/ard.2008.099093. [DOI] [PubMed] [Google Scholar]
- 20.Brosnan ME, Brosnan JT. Orotic Acid Excretion and Arginine Metabolism. The Journal of Nutrition. 2007;137:1656–1660. doi: 10.1093/jn/137.6.1656S. [DOI] [PubMed] [Google Scholar]
- 21.Breedveld FC, Dayer J-M. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Annals of the Rheumatic Diseases. 2000;59:841–849. doi: 10.1136/ard.59.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgan TH. Sex Limited Inheritance in Drosophila. Science. 1910;32:120–122. doi: 10.1126/science.32.812.120. [DOI] [PubMed] [Google Scholar]
- 23.Jarry B, Falk D. Functional diversity within the rudimentary locus of Drosophila melanogaster. Mol Gen Genet. 1974;135:113–22. doi: 10.1007/BF00264779. [DOI] [PubMed] [Google Scholar]
- 24.Conner TW, Rawls JM., Jr Analysis of the phenotypes exhibited by rudimentary-like mutants of Drosophila melanogaster. Biochem Genet. 1982;20:607–19. doi: 10.1007/BF00483960. [DOI] [PubMed] [Google Scholar]
- 25.Fukushima R, et al. Teratogenacity study of the dihydroorotate-dehydrogenase inihibitor and protein tyrosine kinase inhibitor Lefunomide in mice. ScienceDirect. 2007;24:310–316. doi: 10.1016/j.reprotox.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Imose M, et al. Lefunomide Protects From T-Cell--Mediated Liver Injury in Mice Through Inhibition of Nuclear Factor κB. Hepatology. 2004;40:1160–1169. doi: 10.1002/hep.20438. [DOI] [PubMed] [Google Scholar]
- 27.Bushid PB, Brantley DM, Yull FE. Inhibition of NF-κB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature. 1998;392:615–618. doi: 10.1038/33435. [DOI] [PubMed] [Google Scholar]
- 28.Luetteke NC, et al. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–78. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- 29.Chiang C, et al. Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev Biol. 2001;236:421–35. doi: 10.1006/dbio.2001.0346. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1