Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies (original) (raw)
Abstract
Chromosomal microarray (CMA) is increasingly utilized for genetic testing of individuals with unexplained developmental delay/intellectual disability (DD/ID), autism spectrum disorders (ASD), or multiple congenital anomalies (MCA). Performing CMA and G-banded karyotyping on every patient substantially increases the total cost of genetic testing. The International Standard Cytogenomic Array (ISCA) Consortium held two international workshops and conducted a literature review of 33 studies, including 21,698 patients tested by CMA. We provide an evidence-based summary of clinical cytogenetic testing comparing CMA to G-banded karyotyping with respect to technical advantages and limitations, diagnostic yield for various types of chromosomal aberrations, and issues that affect test interpretation. CMA offers a much higher diagnostic yield (15%–20%) for genetic testing of individuals with unexplained DD/ID, ASD, or MCA than a G-banded karyotype (∼3%, excluding Down syndrome and other recognizable chromosomal syndromes), primarily because of its higher sensitivity for submicroscopic deletions and duplications. Truly balanced rearrangements and low-level mosaicism are generally not detectable by arrays, but these are relatively infrequent causes of abnormal phenotypes in this population (<1%). Available evidence strongly supports the use of CMA in place of G-banded karyotyping as the first-tier cytogenetic diagnostic test for patients with DD/ID, ASD, or MCA. G-banded karyotype analysis should be reserved for patients with obvious chromosomal syndromes (e.g., Down syndrome), a family history of chromosomal rearrangement, or a history of multiple miscarriages.
Introduction
Scope and Purpose
Clinical genetic testing, including chromosome analysis, is a standard practice for patients with diagnoses including unexplained developmental delay/intellectual disability (DD/ID), autism spectrum disorders (ASD), and multiple congenital anomalies (MCA). These categories of disorders account for the largest proportion of cytogenetic testing because of their high prevalence in the population. The incidence of DD/ID in the general population approaches 3%,1 and ASD affects ∼1:150 individuals.2,3 Most patients lack sufficient specific history or features from physical examination to suggest a specific genetic (or non-genetic) cause. Published guidelines for testing such patients have emphasized (1) testing for chromosomal abnormalities by G-banded karyotyping and (2) testing for common single-gene disorders, such as fragile X syndrome.4
Microarray-based genomic copy-number analysis is now a commonly ordered clinical genetic test for this patient population and is offered under various names, such as “chromosomal microarray” (CMA) and “molecular karyotyping.”5–10 CMA, as used here, encompasses all types of array-based genomic copy number analyses, including array-based comparative genomic hybridization (aCGH) and single nucleotide polymorphism (SNP) arrays. G-banded karyotyping allows a cytogeneticist to visualize and analyze chromosomes for chromosomal rearrangements, including genomic gains and losses. CMA performs a similar function, but at a much higher resolution for genomic imbalances. G-banded karyotyping has been the standard first-tier test for detection of genetic imbalance in this population for more than 35 years, whereas CMA is not yet standard in all clinical settings.
Clinical Interpretation of CMA Results
Although clinical genetic laboratories are familiar with recurrent copy-number changes mediated by segmental duplication architecture, population studies suggest that the vast majority of copy-number variation is not recurrent.11 Determining the clinical significance of variants identified by CMA can be challenging. Although CMA offers the sensitivity of high-resolution genome-wide detection of clinically significant copy-number variants (CNVs), the additional challenge of interpreting _v_ariants of _u_ncertain _c_linical _s_ignificance (VOUS), the preferred terminology based on a recent study of variant terminology, can impose a burden on clinicians and laboratories.12 Furthermore, recent efforts to evaluate reporting of CNVs among clinical laboratories indicates variability of interpretation.13
Lack of uniform guidelines for the expected clinical yield of CMA, in terms of resolution and coverage (i.e., whole genome or locus specific), impedes standardization of clinical practice for CMA testing. CMA is currently performed in many different laboratories with different technology platforms and different array design and content. Uniform interpretation of results is an equally formidable challenge to standardization. Most clinical laboratories maintain internal databases of pathogenic and benign CNVs, but they might not agree on interpretations.13 Although College of American Pathology (CAP) proficiency testing is now available for CMA, there is not yet an openly accessible centralized resource for comparing clinical interpretations for thousands of possible variants among dozens of laboratories performing this testing. These problems can be addressed through more uniform array content, a rational approach to variant interpretation, and increased data sharing among laboratories and clinicians. Sharing of information across laboratories will provide the most benefit to laboratories, clinicians, and—ultimately and most importantly—patients.
We evaluated the benefits and limitations of CMA, as compared to G-banded karyotyping, for detecting pathogenic genomic imbalances in patients with DD/ID, ASD, and/or MCA. Recommendations for CMA use and standardization as a first-tier genetic test in this patient population are provided.
Subjects and Methods
Assembly and Focus of the ISCA Consortium
The International Standard Cytogenomic Array (ISCA) Consortium is an independent group assembled, through voluntary participation of an international group of experts in this field, to address mutual concerns about standardization and collaboration for clinical CMA testing. The ISCA held two international workshops sponsored by a grant from the American College of Medical Genetics (ACMG) Foundation and Luminex to explore and implement improvements in CMA testing. The workshops included ten clinicians (clinical geneticists or genetic counselors (D.T.M., M.P.A., L.G.B., C.J.E., W.A.F., J.M.F., A.H., L.J., I.D.K., and D.J.W.); 17 clinical laboratory geneticists (D.T.M., S.A., A.R.B., J.A.C., J.M.F., L.J., K.K., C.L., C.R., N.B.S., D.J.S., J.H.T., E.C.T., J.R.V., M.S.W., C.L.M, and D.H.L.); and nine genome scientists and bioinformaticians (N.P.C., D.M.C., E.E.E., L.F., E.B.K., R.M.K., C.L., J.M.O., and S.W.S.).
The goals of the ISCA Consortium are analogous to the goals of the Newborn Screening (NBS) Expert Group convened by the ACMG in 2006 to address the lack of uniformity in state-based NBS programs.14,15 For many years, each state made independent decisions regarding which diseases to include in NBS, leading to a wide range of diseases tested by different states. An expert panel developed an evidence-based recommendation for a uniform, core panel of 29 well-defined disorders. Testing for the vast majority of these core conditions has now been implemented or mandated (but not yet implemented) in all 50 states.16
The ISCA Consortium is focused on the clinical application of CMA as opposed to laboratory technical guidelines. This report is intended as a review of available data to determine whether CMA should be adopted as a first-tier test before routine G-banded karyotyping in this patient population (defined below). We reviewed previously published clinical-practice guidelines9,17 and laboratory guidelines18 for clinical CMA testing. CMA is a high-complexity assay with many technical considerations beyond the scope of this discussion. Suggested guidelines about specimen requirements, DNA preparation, labeling, analysis algorithms, and assay validation are beyond the scope of this discussion but have been outlined elsewhere.9,18
Patient Population
The analysis and recommendations in this manuscript are focused solely on CMA for constitutional abnormalities in postnatal testing of patients with DD/ID, ASD, or MCA. These analyses and conclusions are not necessarily applicable to other CMA applications, such as prenatal testing, hematological malignancies, or other forms of cancer. Although the ISCA Consortium recognizes the potential for CMA to play a role in prenatal diagnosis, current evidence is not sufficient to allow recommendations regarding prenatal CMA, and traditional cytogenetic methods, such as G-banded karyotyping and fluorescence in situ hybridization (FISH), are still the standard for prenatal diagnosis, as supported in a recent American College of Obstetrics and Gynecology (ACOG) Statement.19 Multicenter studies making a direct comparison of the performance of CMA to conventional cytogenetic analysis in a prenatal setting are currently underway.
Systematic Literature Review
We conducted a systematic literature review focused on clinical CMA by searching the PubMed database of the National Center for Biotechnology Information (NCBI) and using the following controlled vocabulary MeSH terms: (Microarray Analysis) AND ((chromosomal disorders) OR (chromosomal aberration)) AND ((mental retardation) OR (developmental disabilities) OR (Autism) OR (congenital abnormalities)). We included case series or cohort studies that were posted to PubMed prior to April 15, 2009 and that included either bacterial artificial chromosome (BAC) or oligonucleotide arrays. We considered studies with (1) a clear description of the patient population, including patient selection criteria; (2) a description of the CMA platform and resolution; and (3) a description of the process for interpretation of CMA results. We also included published studies that were not necessarily identified by this combination of search terms if they met the aforementioned criteria. We excluded studies that presented only validation samples for new techniques or platforms or were focused on a particular medical condition or syndrome (e.g., only one type of congenital anomaly). We identified 33 original reports (not reviews), including 21,698 patients for expert review and discussion by ISCA Consortium workshop participants.
Classification of CMA Variants
Typical strategies for interpreting pathogenic or benign status for CNVs (Table 1) have been outlined elsewhere,9,20,21 In general, copy-number variants are assigned the following interpretations (1) abnormal (e.g., well-established syndromes, de novo variants, and large changes); (2) VOUS; (3) likely benign (e.g., not previously reported but inherited from a healthy parent). Diagnostic yield was defined as the number of patients with abnormal variants divided by the total number of patients tested and was derived directly from each original study. Data were not systematically collected on the number of VOUS in each study.
Table 1.
Assessment of Pathogenicity of a CNVa
| Primary Criteria | Indicates CNV Is Probably | |
|---|---|---|
| Pathogenic | Benign | |
| 1. | a. Identical CNV inherited from a healthy parentb | ✓ |
| b. Expanded or altered CNV inherited from a parent | ✓ | |
| c. Identical CNV inherited from an affected parent | ✓ | |
| 2. | a. Similar to a CNV in a healthy relative | ✓ |
| b. Similar to a CNV in an affected relative | ✓ | |
| 3. | CNV is completely contained within genomic imbalance defined by a high-resolution technology in a CNV database of healthy individuals | ✓ |
| 4. | CNV overlaps a genomic imbalance defined by a high-resolution technology in a CNV database for patients with ID/DD, ASD, or MCA | ✓ |
| 5. | CNV overlaps genomic coordinates for a known genomic-imbalance syndrome (i.e., previously published or well-recognized deletion or duplication syndrome) | ✓ |
| 6. | CNV contains morbid OMIM genesc | ✓ |
| 7. | a. CNV is gene rich | ✓ |
| b. CNV is gene poor | ✓ | |
| General Findingsd | ||
| 1. | a. CNV is a deletion | ✓ |
| b. CNV is a homozygous deletion | ✓ | |
| 2. | a. CNV is a duplication (no known dosage-sensitive genes) | ✓ |
| b. CNV is an amplification (greater than 1 copy gain) | ✓ | |
| 3. | CNV is devoid of known regulatory elements | ✓ |
Development of a CNV Database through the National Institutes of Health
The ISCA Consortium has initiated a new database for CNV and phenotype data generated from clinical CMA laboratories as a project within dbGaP (Database of Genotype and Phenotype at NCBI). This database is capable of receiving and managing raw data and normalized data files from all current CMA platforms, including both copy-number and SNP arrays. The common denominator for displaying and comparing data from different labs and platforms will be the use of genome sequence coordinates, based on the UCSC Genome Browser, for defining the start and stop points for any copy-number imbalance (loss or gain). To minimize barriers to data contribution by clinical laboratories and maximize the empirical data available on CNVs from this patient population, an opt-out mechanism to notify patients that their deidentified data will be contributed to the database has been developed and approved by the central institutional review board (IRB) at the National Institutes of Health and the IRBs at individual contributing centers. All participating centers will also be strongly encouraged to obtain a full informed consent and submit qualified cases to DECIPHER (Databas_e_ of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources) as well.
The opt-out mechanism is a relatively new concept that allows deidentified data collected as part of clinical testing to be used for multiple reasons, including improving the accuracy and quality of test results, test validation, educational purposes, or research. Patients are notified of the opt-out mechanism through various avenues, such as test requisition forms, clinical laboratory reports, and laboratory websites. It is also the responsibility of the ordering physician or other downstream providers who review requisitions and reports containing the opt-out language to inform patients how samples are managed and data are used and that they can opt out of inclusion at any time. Because this mechanism is a growing area that will be of great use to the clinical and research communities and will ultimately improve clinical care, educational materials are also being developed for patients and healthcare providers. Our current NIH-funded grant supports an education and ethics working group comprised of experienced genetic counselors and clinicians who will continually address these important issues.
Working groups have been established to determine standard data formats and vocabularies for genotypic and phenotypic data, as well as to make recommendations for standard interpretation guidelines for CMA results. An expert curation process is being established so that discrepancies within the data can be identified and resolved, and public data releases are planned on a quarterly basis. These will be available to major genomics resources (e.g., UCSC Genome Browser, ENSEMBL, DECIPHER, Database of Genomic Variants (DGV), etc.) via the Database of Structural Variation (dbVar), but also to individual laboratories and commercial vendors for CMA software and database software products.
Results
Diagnostic Yield of CMA Is Greater than that of G-Banded Karyotyping
We reviewed 33 studies, together including 21,698 patients tested by CMA (Table 2). CMA detected pathogenic genomic imbalances with an average diagnostic yield of 12.2% across all studies in this patient population, about 10% more than G-banded karyotyping alone. We also reviewed studies related to potential limitations of CMA as a clinical test, especially with regard to detection of VOUS, balanced translocations, and low-level mosaicism. Our results also include an overview of the CNV Database developed through the ISCA Consortium.
Table 2.
Summary of Clinical CMA Studies on 21,698 Subjects
| Author | Location | Patients | Phenotype | Controls | Array Type | Resolution | Diagnostic Yield |
|---|---|---|---|---|---|---|---|
| Vissers et al. (2003)22 | Netherlands and U.S. | 20 | ID, DF | 4 (2M, 2F) | BAC | Whole genome (1.0 Mb) | 10.0% |
| Shaw-Smith et al. (2004)23 | U.K., France | 50 | ID (moderate to severe) | Pooled DNA (20M, 20F) | BAC | Whole genome (1.0 Mb) | 14.0% |
| de Vries et al. (2005)6 | Netherlands | 100 | ID | 72 parents of probands | BAC | Whole genome (50 kb) | 10.0% |
| Schoumans et al. (2005)24 | Sweden | 41 | ID (mild to severe) | Pooled DNA (10 subjects) | BAC | Whole genome (1.3 Mb) | 9.8% |
| Tyson et al. (2005)25 | Canada | 22 | ID, DF | Pooled normal of four to six males or females (Promega) | BAC | Whole genome (1.3 Mb) | 13.7% |
| Wong et al. (2005)26 | U.S. | 102 | ID | Normal controls | BAC | Telomere | 18.6% |
| Ballif et al. (2006)27 | U.S. | 3600 | ID, DD | BAC | Targeted | 5.1% | |
| Friedman et al. (2006)28 | Canada | 100 | ID | 8 unaffected siblings | SNP Oligo (Affy 100k) | Whole genome (30 kb) | 11.0% |
| Krepischi-Santos et al. (2006)29 | Brazil | 95 | ID, DF, MCA | 100 control observations for each chromosome pair | BAC | Whole genome (1.0 Mb) | 16.8% |
| Menten et al. (2006)30 | Belgium | 140 | ID, MCA | Other patient samples in cohort | BAC | Whole genome (1.0 Mb) | 13.6% |
| Ming et al. (2006)31 | U.S. | 10 | MCA | 128 (42 Caucasian, 42 African-American, 20 Asian, 20 NIGMS) | SNP Oligo (Affy 100k) | Whole genome (30 kb) | 20.0% |
| Miyake et al. (2006)32 | Japan | 30 | ID, MCA | 2 negative (1M, 1F), 1 positive | BAC | Targeted (1.4 Mb) | 16.7% |
| Rosenberg et al. (2006)33 | Netherlands, Brazil, U.K. | 81 | ID (Mild to severe), DF | 100 control observations for each chromosome pair | BAC | Whole genome (1.0 Mb) | 16.0% |
| Sharp et al. (2006)34 | U.K., U.S. | 290 | ID +/− DF; +/− MCA | 316 controls from various ethnicities | BAC | Targeted | 5.5% |
| Shaffer et al. (2006)35 | U.S. | 1500 | Various indications | Opposite gender control | BAC | Targeted | 5.6% |
| Aradhya et al. I(2007)36 | U.S. | 20 (12M, 8F) | ID or DD +/− DF; +/− MCA, +/− GR | Normal male or female (Promega) | Oligo (Agilent 44k) | Whole genome (70 kb) | 35.0% |
| Baris et al. (2007)37 | U.S. | 234 | ID or DD, DF, MCA | 50 normal controls (25M, 25F) and 36 patients with abnormal chromosome testing | BAC | Targeted | 5.6% |
| Engels et al. (2007)38 | Germany | 60 | ID | Pooled DNA (10M, 10F) | BAC | Whole genome and Targeted (0.5 Mb) | 10.0% |
| Fan et al. (2007)39 | U.S. | 100 | ID or DD | 7M, 7F gender matched | Oligo (Agilent 44k) | Whole genome (35 kb) | 15.0% |
| Hoyer et al. (2007)10 | Germany | 104 | ID | Not specified | SNP Oligo (Affy 100k) | Whole genome (30 kb) | 9.6% |
| Lu et al. (2007)40 | U.S. | 2513 | ID or DD, DF, MCA, ASD | Normal (1M, 1F) | BAC | Targeted | 7.0% |
| Newman et al. (2007)41 | U.K. | 36 | DD, LD, DF | Not specified | BAC | Whole genome (1.0 Mb) | 13.8% |
| Shaffer et al. (2007)42 | U.S. and “abroad“ | 8789 | ID or DD, MCA | Normal (1M) | BAC | Targeted | 6.9% |
| Shen et al. (2007)43 | U.S. | 211 | ID or DD, MCA, MCA | Normal (1M, 1F) | Oligo (Agilent) | Targeted (35 kb in target regions) | 7.6% |
| Thuresson et al. (2007)44 | Sweden | 48 | ID, DF, MCA | 8M, 8F gender matched | BAC | 1 Whole genome (1.0 Mb) | 6.3% |
| Wagenstaller et al. (2007)45 | Germany | 67 | ID (mild to severe) | 44 unaffected parents and 4 children with known translocations | SNP Oligo (Affy 100k) | Whole genome (30 kb) | 16.4% |
| Aston et al. (2008)46 | U.S. | 1075 | ID, DD, DF, MCA | Normal control of the opposite gender (Promega) | BAC | Whole genome (1.3 Mb) | 5.3% |
| Baldwin et al. (2008)47 | U.S. | 211 | ID, DD, DF, MCA, ASD | Normal control of the opposite gender (Promega) | Oligo (custom Agilent 44k) | Whole genome and Targeted (75 kb) | 15.6% |
| Pickering et al. (2008)48 | U.S. | 1176 | ID or DD | Normal (1M, 1F) | BAC | Targeted (n = 822) and whole genome (1 Mb; n = 354) | 7.8% |
| Shevell et al. (2008)49 | Canada | 94 | ID, DD | Not specified | BAC | Targeted | 6.4% |
| Xiang et al. (2008)50 | U.S. | 50 | ID, DD | Gender mismatched | Oligo (Agilent 44k) | Whole genome (35 kb) | 18.0% |
| Nowakowska et al. (2008)51 | U.S. | 91 | ID, DF | Normal (1M, 1F) | BAC | Targeted | 11.8% |
| Lu et al. (2008)52 | U.S. | 638 | MCA | Normal (1M, 1F) | BAC (n = 372) and Oligo (n = 266) | Targeted with backbone | 17.1% |
Diagnostic yield has improved as the resolution of cytogenetic testing for patients with developmental disabilities has evolved. G-banded karyotyping can sometimes detect genomic imbalances as small as 3 Mb, but it can often miss genomic imbalances in the 5–10 Mb range, depending on the genomic region involved and/or conditions of the assay (Figure 1). If patients with Down syndrome (MIM 190685) are excluded, G-banded karyotyping of patients with ID has typically identified abnormalities in fewer than 3% of individuals.1,53–56 Karyotyping is also limited because it is based on the subjective assessment of gains and losses and is prone to considerable interpersonal and interlaboratory variation in detection rates.
Figure 1.
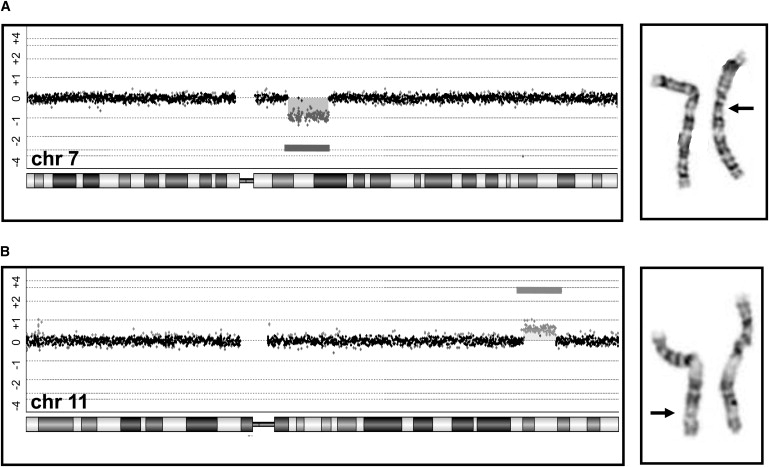
Examples of Genomic Imbalances Detected by a CMA but Not by G-Banded Karyotyping
(A) A 10.9 Mb deletion, including more than 60 genes. The deletion includes the Williams-Beuren syndrome region at chromosome region 7q11 but extends beyond the typical breakpoints for this syndrome. The arrow is pointing to the deleted chromosome that was observed by retrospective analysis of G-banded slides.
(B) A 7.2 Mb duplication on the long arm of chromosome 11. Again, the arrow is pointing to the chromosome that has the duplication shown by the darker G-positive band.
The addition of subtelomeric fluorescence in situ hybridization (FISH) for identifying submicroscopic deletions and duplications of genomic regions proximal to the telomeres has been shown to almost double the diagnostic yield. In the largest analysis of subtelomeric FISH testing, presumed pathogenic changes were found in 2.6% of 11,688 unselected cases,57 and a recent review of another 7,000 cases found subtelomeric rearrangements in 2.4% of the patients studied.58 Most FISH assays used in a clinical cytogenetic setting detect submicroscopic changes in the range of 100 kb or larger. BAC-based arrays can detect genomic imbalances 100 kb and larger, depending on probe coverage.
The increased yield of clinical genetic diagnoses with CMA parallels the evolution of array designs (Figure 2). Early clinical CMA tests included BAC clones and targeted subtelomeric and pericentromeric regions, “known” recurrent microdeletion and microduplication syndromes, or whole-genome coverage with clones at ∼1 Mb genomic intervals (Figure 2),59,60 and diagnostic yields were 7%–11% for children with normal G-banded cytogenetic analysis.22–24,33,35,61–63 Subsequent versions of clinical CMA tests included additional probes, so-called genomic “backbone” coverage, allowing for the detection of pathogenic genomic imbalances outside of the well-described critical regions for microdeletion and microduplication syndromes. Rapid increases in the availability of genome-wide arrays with density sufficient to allow detection of copy-number changes of ∼100 kb throughout the genome increased diagnostic yields to between 11% and 15%28,10,39 based on studies of DD/ID patients who had normal G-banded cytogenetic analysis; these yields are far higher than what was obtained via traditional cytogenetic methods.
Figure 2.
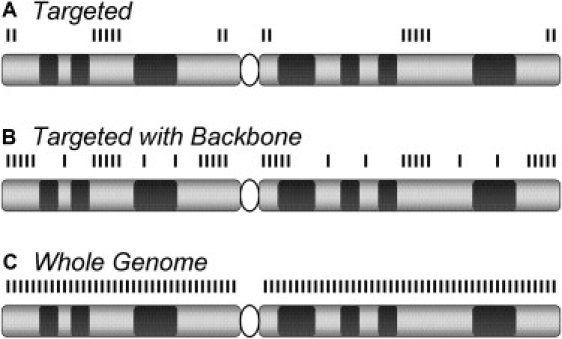
Evolution of a Constitutional CMA Design
(A) Early versions of array-based Comparative Genomic Hybridization (aCGH) platforms used for constitutional cytogenetic testing targeted the subtelomeric and pericentromeric regions and defined microdeletion and microduplication syndromes.61,62
(B) Later, more extensive coverage was added at the subtelomeric and pericentromeric regions and included additional probes outside the targeted regions; this is so-called “backbone” coverage.
(C) Higher-density backbone coverage or high-density genome-wide arrays provide essentially whole-genome coverage, yielding even higher detection rates.47
CMA Platform Comparison
Most commercial and academic laboratories have now converted to oligonucleotide- or SNP-based arrays. Some clinical laboratories use oligonucleotide-based arrays that emulate the coverage of earlier versions of BAC-based arrays. Advantages of oligonucleotide-based arrays include more flexibility in terms of probe selection, thus facilitating higher probe density and customization of array content. Arrays designed specifically for copy-number analysis use longer oligonucleotide probes (∼60-mer), which have been shown to provide better signal-to-noise ratios (SNRs) than the shorter oligonucleotide probes (∼22-mer) used in some SNP-detecting array platforms.64 Longer oligonucleotides (50-mer) on some SNP-detecting arrays result in a better signal-to-noise ratio than short oligonucleotide SNP arrays. Also, SNP-detecting arrays now typically include a mixture of SNP and copy-number probes to address this issue.
In general, high specificity can be achieved by current BAC or oligonucleotide arrays, including SNP arrays, depending on coverage. BAC-based arrays employ large-insert genomic clones of 100–150 kb, and accurate copy-number determination can be made for individual clones. Single oligonucleotide probes do not provide accurate determination of copy number, and multiple consecutive probes indicating the same copy-number change are required for accurately determining a gain or loss. The number of consecutive probes required may vary between arrays of long-oligonucleotide probes and SNP probes, and SNP arrays typically require more consecutive probes to achieve comparable specificity.
SNP arrays have the advantage of also detecting long stretches of homozygosity, which might represent uniparental disomy (UPD) or consanguinity not suspected on the basis of clinical history. However, routine CMA analysis with SNP-based platforms will detect heterodisomic UPD only in that portion of cases where there are blocks of isodisomy. UPD occurring by trisomy rescue usually contains blocks of heterodisomy and isodisomy but can result in complete heterodisomy.
Arrays based entirely on probes for SNPs are biased to certain genomic segments containing common SNPs. One earlier SNP-detecting array could detect only ∼26% of the CNVs that were detected by fosmid end sequence mapping strategies.65 For this reason, newer SNP-detecting arrays also contain nonpolymorphic oligonucleotide probes exclusively designed for copy-number detection to provide more robust and uniform coverage. Overall, with sufficiently dense probe coverage, all current array platforms are able to provide sufficient sensitivity for clinical CMA testing.
Recommendations for CMA Coverage and Resolution
The clinically effective resolution of CMA represents a balance between sensitivity and specificity. Analytical sensitivity is primarily influenced by probe coverage, resolution, and genomic spacing of probes selected for the array. For CMA to identify genomic imbalances at a higher resolution than a conventional G-banded karyotype, the array must consistently detect clinically significant genomic imbalances smaller than 5 Mb. Clinical sensitivity of CMA should be considered in relation to G-banded karyotyping and not in comparison to gene-specific molecular-based assays for Mendelian disorders.
Available oligonucleotide platforms can now detect genomic imbalances as small as 500 bp,66 permitting the detection of genomic copy-number changes as small as 10–20 kb in many regions of the genome. Clinical arrays are typically designed to detect imbalances of 20–50 kb in targeted regions (e.g., within known Mendelian genes) and imbalances of 100–250 kb in nontargeted (backbone) regions of the genome. The ability to identify smaller deletions allows diagnosis of diseases caused by as few as one haploinsufficient gene, which increases the diagnostic capability of this testing. However, this level of resolution is not necessary as a genome-wide clinical test of genomic imbalance.
Most current clinical CMA platforms can detect copy-number changes with a lower limit of resolution of ∼400 kb throughout the genome, representing a ≥10-fold improvement in resolution in comparison to G-banded karyotyping. This level of resolution will provide a broad genomic survey and reliably identify all known recurrent microdeletion and microduplication syndromes mediated by segmental duplication architecture and most nonrecurrent pathogenic imbalances that are unequivocally pathogenic. The smallest known recurrent microdeletion syndromes, such as deletions at 17q21.31 (MIM 610443)34,67–69 and 16p11.2 (MIM 611913),70–72 are ∼500 kb or larger.
Although copy-number alterations smaller than ∼400 kb may certainly be pathogenic and smaller disease-causing CNVs are likely to be identified on an ongoing basis, higher-resolution CMA detects an increasing proportion of benign CNVs. Decisions about clinically interpretable, effective resolution for CMA should therefore also consider empiric data about size distribution of CNVs in the human genome. Research-based tiling arrays used on HapMap samples (considered healthy individuals) revealed a median size for presumably benign CNVs of ∼200 kb and showed that 90%–95% of these CNVs are less than 500 kb.11,73 More recent data with higher-density arrays suggest that many more “small” CNVs exist, that there is an overall median CNV length of ∼2.9 kb, and that 95% of CNVs are less than 100 kb.66 These smaller CNVs were previously undetectable because of technological limitations, and CNV sizes from previous studies have often been overestimated for the majority of CNVs.11,74 These recent studies demonstrate that only 1%–2% of all CNVs in normal individuals are larger than 1 Mb.11,66 A summary of recommendations appears in Table 3 and in the Supplemental Data.
Table 3.
Recommendations for CMA Testing of Individuals with Unexplained Developmental Delay/Intellectual Disability, Autism Spectrum Disorders, or Multiple Congenital Anomalies
| Recommendation for CMA Platform: |
|---|
| 1. CMA standards should not be specific to a particular array platform. Arrays based on BAC clones, long oligonucleotides or SNP-detecting shorter oligonucleotides can achieve the recommended coverage and level of resolution. |
| Recommendations for CMA Coverage and Probe Density: |
| 1. In order to perform the same intended purpose as a karyotype, CMA must have uniform coverage to detect all areas of imbalance at a resolution exceeding that of a karyotype (∼5 Mb). Currently, to detect CNV we recommend a resolution of ≥400 kb throughout the genome as a balance of analytical and clinical sensitivity. |
| 2. For oligonucleotide and SNP arrays, multiple consecutive probes are needed to permit a call to be made, so the array must be designed to include sufficient probe density for each targeted region. Note that SNP arrays may require a greater number of consecutive probes to permit a reliable call to be made. |
| 3. Laboratories that choose to add probes to cover Mendelian disease loci should explicitly state the minimum detectable imbalance and clinical sensitivity of the assay for each disease at each locus and point out the availability of sequence-based technology for detecting mutations that are not detectable by CMA. |
| Recommendations Related to Balanced Rearrangements, Low-Level Mosaicism, and Positive Family History of Known Chromosomal Abnormalities or Reproductive Loss: |
| 1. CMA can detect many more submicroscopic pathogenic genomic imbalances than the number of balanced rearrangements it would miss. Cytogenetically balanced rearrangements and low-level mosaicism, which would not be detected by CMA, cause only a small proportion of all cases of unexplained DD/ID, ASD, and/or MCA. |
| 2. G-banded karyotyping should be offered to patients with a family history of a balanced chromosomal rearrangement, a history of multiple miscarriages, or certain other conditions, as discussed. |
| Recommendations for a CMA Database: |
| 1. CMA Database should be “platform neutral” and able to incorporate information based on chromosomal position according to the human genome (hg) build. |
| 2. All raw data should be freely accessible to all qualified researchers who register with dbGaP at NCBI. |
| 3. Curated data should be publicly released on a quarterly basis and made available to major genomics resources and commercial vendors, as well as individuals and clinical laboratories. |
Impact of Balanced Rearrangements and Low-level Mosaicism on Clinical Sensitivity of CMA
G-banded karyotyping is still important in the detection of balanced rearrangements and low-level mosaicism, both of which are not uniformly detectable by CMA. The clinical sensitivity of CMA depends on the proportion of potentially pathogenic balanced rearrangements, and mosaic chromosomal material, in this patient population. The available evidence suggests that (1) truly balanced rearrangements represent only a small proportion of clinically significant genomic events in this patient population and (2) many “apparently balanced” rearrangements detected by G-banding are not balanced at the DNA level.
Balanced rearrangements account for a minority of cytogenetically detectable events in patients with ID. G-banded karyotyping of large cohorts of individuals with ID shows apparently balanced structural abnormalities in 1/500 patients, and in 1/2,000 patients, these occur de novo.75 Balanced rearrangements make up only about 10% of cytogenetically visible abnormalities in patients with ID, meaning that only about 0.3% of patients with ID who are tested by karyotype analysis would have such changes.1,54,76 In the general population, balanced rearrangements are also quite common. A study of 377,357 amniocenteses showed a de novo reciprocal translocation in approximately 1/2,000 and a balanced Robertsonian translocation in 1/9,000.77
Moreover, although cytogenetic events might appear balanced at the microscopic level of resolution, many have a submicroscopic imbalance, especially among individuals with an abnormal phenotype.78,79 In one study of ten patients with abnormal phenotypes and chromosomal rearrangements that appeared balanced by conventional cytogenetic methods, six had submicroscopic imbalance according to CMA with 1 Mb resolution.78 In a larger study of 59 patients with apparently balanced rearrangements (41 de novo reciprocal translocations and 18 de novo complex chromosomal rearrangements), 27 patients were found to have an abnormal phenotype attributed to the chromosomal rearrangement itself. Analysis of these 27 patients with higher-resolution oligonucleotide arrays (Agilent 44k and 244k) revealed a submicroscopic imbalance in 11/27 patients, or 41%.79 A more recent study of 14 patients with an apparently balanced chromosomal rearrangement revealed a submicroscopic imbalance in 4/14 patients, or 29%.80
Another approach to this question is to consider the impact of balanced translocations on phenotypically normal individuals. Most apparently balanced reciprocal translocations in phenotypically normal individuals do not contain genomic imbalance.81 Among balanced rearrangements that interrupt a gene, approximately half (16/31) are found in healthy individuals.80 In the setting of multiple miscarriages, a balanced translocation in one of the parents could be the explanation for unbalanced offspring, and G-banded karyotyping should still be the standard of care for this indication. In some cases, family history can be a clue to segregation of a balanced translocation. In clinical settings, probands are more likely to carry an imbalance, and parents and other family members would subsequently be tested by traditional cytogenetic methods.
The incidence of low-level mosaicism is relatively low compared to the incidence of other detectable chromosomal imbalances in this patient population. Conlin et al. (2010) observed mosaic aneuploidy in 1% of 2,019 patients with a variety of diagnoses (the most common diagnosis was DD),82 and G-banded karyotyping of a standard 20 cells is estimated to detect such mosaicism at a level of 14%.83 Detection of mosaicism as low as 10% is possible with BAC-based arrays,27 but the trend among laboratories is for increasing use of oligonucleotide arrays. Although similar validation studies are needed for long-oligo-based arrays, clinical experience indicates that mosaicism for segmental aneuploidy is detectable to a level of about 20%–30% mosaic cells with most current platforms.84 SNP arrays can detect lower levels of mosaicism (below 5% in some cases) because of the increased resolution afforded by genotyping in conjunction with probe intensity.82 Many types of mosaicism are missed by karyotyping either because they are not present in the relevant lymphocytes or because the cells with the imbalance are unable to respond to the mitogen.27,85,86 Regardless of the method, ease of detection will be proportional to the size of mosaic chromosome fragments. At this time, CMA performs comparably to G-banded karyotyping for detection of mosaicism.
Development of an Open-Access CMA Database
Difficulties interpreting VOUS can be overcome with the cooperation of researchers, clinicians, clinical laboratory directors, and bioinformaticians.
The most comprehensive existing online database for presumed benign CNVs is DGV, but this valuable resource is based on testing of presumably healthy individuals and does not provide clinical information. A recently launched database of structural variation from both normal control populations and disease populations is dbVAR, developed and housed at NCBI within the NIH. Correlation of clinical phenotypes with specific genomic rearrangements requires databases that include data from individuals with DD/ID or MCA and is exemplified by the interactive web-based database DECIPHER, which incorporates a suite of tools designed to aid in the interpretation of submicroscopic chromosomal imbalances, inversions, and translocations.87 DECIPHER enables free public access to fully consented molecular and phenotypic data as well as password-protected protocols for individual labs to securely upload and manage patient information.
With more than 100 clinical CMA laboratories currently participating in the ISCA Consortium, it can be anticipated that this database will exceed several hundred thousand CMA cases within the next 2–3 years. Data at the individual level will be stored in a controlled-access environment in dbGaP, and curated summary data, including CNV region calls and phenotypic associations, will be available and visualized through various resources, including public databases and individual vendor software. This large dataset, in combination with DECIPHER, DGV, and dbVAR, should substantially reduce the current frequency of VOUS results. However, CNVs with incomplete penetrance and variable expression will remain significant challenges, particularly in the prenatal setting, unless the molecular basis of the phenotypic variation can be identified.
Discussion
Clinical geneticists, pediatric neurologists, and developmental pediatricians are increasingly ordering CMA to obtain a genetic diagnosis for their patients with unexplained DD/ID, ASD, and MCA. A specific genetic diagnosis facilitates comprehensive medical care and accurate recurrence risk counseling for the family. Similar to our results, a recent meta-analysis of CMA on 13,926 subjects with ID and/or MCA, most of whom had normal conventional cytogenetic studies, reported an overall diagnostic rate of 10% for pathogenic genomic imbalances.88 Another retrospective analysis of 36,325 patients with DD estimated that a pathogenic abnormality could be detected in ∼19% of unselected DD/ID patients via genome-wide array-based assays with a 30–70 kb median probe spacing.89
Our recommendation based on current evidence is to offer CMA as the first-tier genetic test, in place of G-banded karyotype, for patients with unexplained DD/ID, ASD, or MCA. Although others have proposed that CMA should be considered a first-tier test,63,89 this point of view is still not widely accepted. Except in special cases, such as those involving family history of multiple miscarriages, a karyotype is not cost effective in a child with DD/ID, ASD, or MCA and a negative array study. CMA testing is not inexpensive, but the cost is less than the cost of a G-banded karyotype plus a customized FISH test such as subtelomeric FISH, and the yield is greater.
G-banding has been available for more than 38 years and has the advantage of being a widely accepted and uniform technique with an international system of cytogenetic nomenclature (ISCN). By contrast, CMA is new and more diverse in terms of techniques used, coverage, and approach to data interpretation. Overcoming this lack of uniformity will allow clinicians to leverage the increased sensitivity and specificity of CMA with genome-wide coverage to detect genomic imbalances at higher resolution and with less subjectivity in clinical settings. In addition, the more precise delineation of deletions and duplications provides direct links to the genome content in these areas, allowing recognition of disease-causing genes.
CMA offers additional advantages beyond the ability to detect submicroscopic genomic imbalances. Although G-banding is better at detecting a small marker chromosome that contains exclusively pericentromeric repeat sequences, the clinical significance of such an event is negligible, and CMA is better than traditional cytogenetic techniques for identifying the composition of small marker chromosomes when they contain sufficient euchromatic material.90 CMA is also superior to FISH for detecting submicroscopic duplications because of its higher resolution (multiple small oligonucleotide probes can recapitulate the coverage of a single BAC probe) and because of the technical difficulty of visualizing tandem duplications by metaphase FISH analysis.26 Clinically significant submicroscopic duplications, including the reciprocal duplication of known microdeletion syndromes such as the 7q11 Williams-Beuren syndrome region91 or the 17p11.2 Potocki-Lupski syndrome region,92 are more easily identified by CMA.
The ISCA Consortium proposes the clinical algorithm in Figure 3 to guide postnatal testing in this patient population. For clinical testing, traditional cytogenetic methods, such as FISH, might offer the best confirmation for certain abnormal findings. For example, terminal deletions or duplications are more likely than interstitial events to be involved in a rearrangement, especially when more than one deletion or duplication is identified in a single individual. Some labs might use other methods, such as quantitative PCR (qPCR) and multiplex ligation and probe amplification (MLPA). The need for confirmatory testing purely for copy-number determination is debatable in cases such as those involving very large deletions or duplications (typically involving dozens of consecutive probes).
Figure 3.
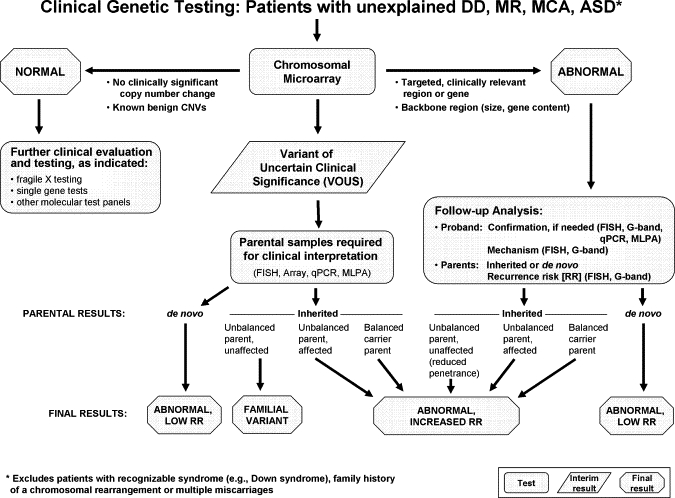
Algorithm for CMA Testing in Patients with Unexplained DD, MR, MCA, and ASD
This algorithm assumes that the patient does not present with features of a recognizable syndrome or metabolic disorder or that tests have been negative for a suspected disorder. The first-tier test is a chromosomal copy-number array or CMA. If no copy-number changes are identified, or if only known CNVs that are known to be benign are identified, this testing is considered “normal” (left side of figure), and further clinical evaluation is warranted to determine whether other testing should be pursued on the basis of the clinical presentation. If a CNV is detected within a known, clinically relevant region or gene, or if the CNV is in the genomic backbone and meets recommended size and gene content guidelines, then the result is considered a pathogenic CNV and “abnormal” (right side of figure). For these cases, follow-up analyses include confirmation studies and determination of the mechanism of imbalance in the proband and parental analysis to determine recurrence risk. All other results are considered VOUS until parental analysis is performed to aid in the final clinical interpretation. After the parental analyses of “abnormal” and “VOUS” results, final results may be classified into three major categories: familial variant, abnormal with a low recurrence risk (RR), or abnormal with an increased RR. In addition, the final interpretation may remain VOUS in some instances, even after parental testing.
In general, traditional cytogenetic methods are still needed for single-cell analysis. Other circumstances in which traditional cytogenetic methods are indicated instead of (or at least before) CMA include when the patient has a recognizable chromosomal syndrome such as trisomy 21, trisomy 13, Turner syndrome, or Klinefelter syndrome. For these circumstances, conventional cytogenetic analysis or interphase FISH analysis might provide a more rapid turn-around time, allow more sensitive detection of low-level mosaicism, and provide information regarding position to distinguish free trisomy from translocation-associated trisomy. For couples with a history of recurrent miscarriage or other significant family history of reproductive loss, conventional chromosome analysis to identify potential balanced translocations would still be the most appropriate first-tier test.
Some might consider FISH follow-up testing to be important for detection of possible insertional translocations (IT). These events are rare at the karyotypic level; they occur in approximately 1:80,000 live births.93 IT might be more common at the submicroscopic level, but specific incidence rates have not been published. The potential implications of IT for genetic counseling about recurrence risk are one argument in favor of FISH testing after identification of a copy-number gain by CMA. Other evidence suggests that parental testing is best done by CMA because breakpoints may be unstable on transmission.94 However, on the basis of limited published studies95 and clinical experience, most CNVs have similar, if not identical, breakpoints between parent and child.
CMA testing of this patient population must provide whole-genome coverage in order to replace a karyotype, as outlined above. Clinical labs might increasingly utilize custom arrays to detect intragenic deletions and duplications in individual genes associated with Mendelian disorders, and we agree that first-tier clinical testing for most Mendelian disorders should be based on sequencing and/or platforms, such as custom arrays, with high resolution for small intragenic deletions. For CMA with resolution of ∼400 kb, as proposed here, information about “targeted” Mendelian genes should clearly state that CMA is intended to detect large deletions or duplications that include part, or all, of the targeted gene but that it is not intended to replace complete gene sequencing or high-resolution array.
We acknowledge that concerns about identification of VOUS might also cause hesitation about adoption of CMA as a first-tier test, but we argue that this issue can be adequately addressed through (1) choosing a level of resolution that balances sensitivity and specificity; (2) increased data sharing through the established database; and (3) parental studies to determine whether CNVs are de novo or inherited (Figure 3). The fact that no single CMA platform has been found to be clearly superior to all of the others for clinical purposes and the absence of published clinical standards for coverage and resolution have led to a lack of uniformity in arrays offered in different laboratories. The variety of available tests with nonoverlapping lists of covered genomic regions is a potential barrier to the widespread adoption of CMA as the first-tier cytogenetic test, but our recommendations (summarized in Table 3 and the Supplemental Data) address this issue.
Even for arrays that contain comparable genomic content, accurate interpretation of test results requires increased sharing of primary data among clinical laboratories so that benign copy-number variants can be distinguished from pathogenic ones. New recommendations for CMA nomenclature outlined in ISCN 2009 should facilitate interlaboratory comparisons of copy-number changes, but they do not address the interpretation of clinical significance.96 Clinical utility will be improved through a standard approach to interpretation of copy-number variation applicable to all technology platforms, and especially through efforts such as the widely accessible ISCA database of pathogenic and benign copy-number changes for comparison of clinical interpretations.
Performing large numbers of parental samples is costly by any method (CMA, FISH,.MLPA, or qPCR), but the need for parental testing will diminish with accumulating data about benign CNVs. Population studies suggest that >99% of all benign CNVs are inherited, and the vast majority of inherited CNVs are much smaller than 500 kb.95 Most pathogenic copy-number alterations are larger than 1 Mb, and most occur de novo. Inherited CNVs, such as those seen in 1q21.1 (MIM 612474 and 612475),97 1q41q42,98 3q29 (MIM 609425 and 611936),99 15q11.2,100 15q13.2q13.3 (MIM 612001),101–103 16p11.2 (MIM 611913),70–72 16p13.11,100,103 and 22q11.2 (MIM 188400 and 608363),104 can also be pathogenic, but they have incomplete penetrance and variable expressivity. Variable clinical implications also result from mechanisms of inheritance that influence the expression of a trait, for example when an inherited deletion uncovers an imprinted region or a pathogenic recessive allele on a homologous chromosome.105–107
Most genomic copy-number changes associated with DD/ID are sporadic, but others can be inherited with a recurrence risk as high as 50%. More highly penetrant syndromes may be more easily recognized through history and clinical exam, but such syndromes are more likely to occur because of de novo changes that confer minimal recurrence risk. Thus, syndromes exhibiting nonpenetrance in some individuals can be more challenging to diagnose, and the cost to the family for failure to diagnose might be higher because there might be more inherited variants for these types of syndromes and thus a significant recurrence risk. In general, VOUS within the resolution of CMA should be reported so that they can be interpreted clinically as the field advances.
An extensive discussion of ethical and legal issues associated with advances in CMA technology is beyond the scope of the current manuscript. Brief examples include the ability of SNP arrays to identify unsuspected consanguinity that might point to an autosomal-recessive disease. In some cases, the degree of consanguinity revealed may be indicative of parent-child incest, and there are potential ethical and legal implications of reporting of such results. Whole-genome analysis also raises important questions regarding intellectual-property issues around gene patents, leading some institutions to instruct their CMA laboratories to omit or mask coverage of such patented genes, to the detriment of patient care. Current legal challenges to gene patenting in the United States might alleviate this particular concern.
As a community of clinical and laboratory geneticists, we have an opportunity to increase the uniformity of CMA and its clinical interpretation in laboratories around the world. Clinical geneticists have always worked closely with genetics laboratories to interpret results in the context of the patient's clinical findings. In this regard, clinical geneticists must have or develop a basic understanding of CMA, including indications for testing, interpretation of results, and counseling of families. As more medical specialists utilize this technology, clinical and laboratory geneticists will play an important role in helping these specialists interpret the tests and communicate results to the families. Educational tools should be developed and utilized in the setting of ongoing continuing medical education for individuals in practice as well as for those in laboratory and clinical training programs.
Acknowledgments
Workshops and conference calls related to the ISCA Consortium were funded through a grant from the ACMG Foundation and Luminex to D.H.L. and C.L.M., and ongoing standards and database development are funded in part by grants from the National Institutes of Health (RC2HD064525 and MH074090 to D.H.L. and C.L.M.). This work was also supported by Simons Foundation grant 137578 (to E.E.E.). E.E.E. is an investigator of the Howard Hughes Medical Institute. The opinions expressed here are those of the authors and the ISCA Consortium and do not necessarily reflect the views of the institutions to which the authors are affiliated, the ACMG, or the American Society of Human Genetics (ASHG). The authors would like to thank Dr. Omer Gokcumen for his assistance with summary of population CNV data. Regarding conflict of interest, the majority of authors are involved in CMA testing and/or cytogenetic testing, either as clinicians who order the testing, laboratory professionals who perform the testing, or researchers in the genomics field. S.A. is a director of clinical cytogenetics at GeneDx, a subsidiary of BioReference Laboratories; A.R.B. is a director of clinical cytogenetics and molecular cytogenetics at ARUP Laboratories; E.E.E. is a member of the Pacific Biosciences Scientific Advisory Board. S.W.S. is on the Scientific Advisory Board of Combimatrix Diagnostics and the Scientific Advisory Committee of Autism Speaks. J.T. is a director of clinical cytogenetics at Laboratory Corporation of America. J.V. is on the board of directors of Cartagenia. M.W. raises funds from industry to support the educational activities of the ACMG Foundation.
Contributor Information
David T. Miller, Email: david.miller2@childrens.harvard.edu.
David H. Ledbetter, Email: david.ledbetter@emory.edu.
Supplemental Data
Document S1. General Recommendations of the ISCA Consortium
Web Resources
The URLs for data presented herein are as follows:
- DatabasE of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER), https://decipher.sanger.ac.uk/application/
- Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
- Database of Genotype and Phenotype, dbGaP; http://www.ncbi.nlm.nih.gov/gap
- Database of Structural Variation, dbVAR ;http://www.ncbi.nlm.nih.gov/dbvar/
- International Standard Cytogenomic Array Consortium, https://isca.genetics.emory.edu
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
- PubMed at National Center for Biotechnology Information, www.ncbi.nlm.nih.gov/PubMed
- UCSC Genome Bioinformatics Site, http://genome.ucsc.edu/
References
- 1.Shevell M., Ashwal S., Donley D., Flint J., Gingold M., Hirtz D., Majnemer A., Noetzel M., Sheth R.D. Practice parameter: evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–380. doi: 10.1212/01.wnl.0000031431.81555.16. [DOI] [PubMed] [Google Scholar]
- 2.Autism and Developmental Monitoring Network Surveillance Year 2000 Principal Investigators Prevalence of autism spectrum disorders–autism and developmental disabilities monitoring network, six sites, United States, 2000. MMWR Surveill. Summ. 2007;56:1–11. [PubMed] [Google Scholar]
- 3.Newschaffer C.J., Croen L.A., Daniels J., Giarelli E., Grether J.K., Levy S.E., Mandell D.S., Miller L.A., Pinto-Martin J., Reaven J. The epidemiology of autism spectrum disorders. Annu. Rev. Public Health. 2007;28:235–258. doi: 10.1146/annurev.publhealth.28.021406.144007. [DOI] [PubMed] [Google Scholar]
- 4.Moeschler J.B., Shevell M. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117:2304–2316. doi: 10.1542/peds.2006-1006. [DOI] [PubMed] [Google Scholar]
- 5.Rauch A., Ruschendorf F., Huang J., Trautmann U., Becker C., Thiel C., Jones K.W., Reis A., Nurnberg P. Molecular karyotyping using an SNP array for genomewide genotyping. J. Med. Genet. 2004;41:916–922. doi: 10.1136/jmg.2004.022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries B.B., Pfundt R., Leisink M., Koolen D.A., Vissers L.E., Janssen I.M., Reijmersdal S., Nillesen W.M., Huys E.H., Leeuw N. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hochstenbach R., Ploos van Amstel H.K., Poot M. Microarray-based genome investigation: Molecular karyotyping or segmental aneuploidy profiling? Eur. J. Hum. Genet. 2006;14:262–265. doi: 10.1038/sj.ejhg.5201553. [DOI] [PubMed] [Google Scholar]
- 8.Vermeesch J.R., Rauch A. Reply to Hochstenbach et al. ‘Molecular karyotyping’. Eur. J. Hum. Genet. 2006;14:1063–1064. doi: 10.1038/sj.ejhg.5201663. [DOI] [PubMed] [Google Scholar]
- 9.Hoyer J., Dreweke A., Becker C., Gohring I., Thiel C.T., Peippo M.M., Rauch R., Hofbeck M., Trautmann U., Zweier C. Molecular karyotyping in patients with mental retardation using 100K single-nucleotide polymorphism arrays. J. Med. Genet. 2007;44:629–636. doi: 10.1136/jmg.2007.050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vermeesch J.R., Fiegler H., de Leeuw N., Szuhai K., Schoumans J., Ciccone R., Speleman F., Rauch A., Clayton-Smith J., Van Ravenswaaij C. Guidelines for molecular karyotyping in constitutional genetic diagnosis. Eur. J. Hum. Genet. 2007;15:1105–1114. doi: 10.1038/sj.ejhg.5201896. [DOI] [PubMed] [Google Scholar]
- 11.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vos J., van Asperen C.J., Wijnen J.T., Stiggelbout A.M., Tibben A. Disentangling the Babylonian speech confusion in genetic counseling: an analysis of the reliability and validity of the nomenclature for BRCA1/2 DNA-test results other than pathogenic. Genet. Med. 2009;11:742–749. doi: 10.1097/GIM.0b013e3181b2e608. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchiya K.D., Shaffer L.G., Aradhya S., Gastier-Foster J.M., Patel A., Rudd M.K., Biggerstaff J.S., Sanger W.G., Schwartz S., Tepperberg J.H. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet. Med. 2009;11:866–873. doi: 10.1097/GIM.0b013e3181c0c3b0. [DOI] [PubMed] [Google Scholar]
- 14.American College of Medical Genetics Newborn screening: toward a uniform screening panel and system. Genet. Med. 2006;8(Suppl 1):1S–252S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green N.S., Rinaldo P., Brower A., Boyle C., Dougherty D., Lloyd-Puryear M., Mann M.Y., Howell R.R. Committee Report: advancing the current recommended panel of conditions for newborn screening. Genet. Med. 2007;9:792–796. doi: 10.1097/gim.0b013e318159a38e. [DOI] [PubMed] [Google Scholar]
- 16.National Newborn Screening and Genetics Resource Center (2009). National Newborn Screening Status Report. http://genes-r-us.uthscsa.edu/nbsdisorders.pdf.
- 17.Manning M., Hudgins L. Use of array-based technology in the practice of medical genetics. Genet. Med. 2007;9:650–653. doi: 10.1097/gim.0b013e31814cec3a. [DOI] [PubMed] [Google Scholar]
- 18.Shaffer L.G., Beaudet A.L., Brothman A.R., Hirsch B., Levy B., Martin C.L., Mascarello J.T., Rao K.W. Microarray analysis for constitutional cytogenetic abnormalities. Genet. Med. 2007;9:654–662. doi: 10.1097/gim.0b013e31814ce3d9. [DOI] [PubMed] [Google Scholar]
- 19.American College of Obstetrics and Gynecology ACOG Committee Opinion No. 446: Array comparative genomic hybridization in prenatal diagnosis. Obstet. Gynecol. 2009;114:1161–1163. doi: 10.1097/AOG.0b013e3181c33cad. [DOI] [PubMed] [Google Scholar]
- 20.Lee C., Iafrate A.J., Brothman A.R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet. 2007;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Revenga L., Mila M., Rosenberg C., Lamb A., Lee C. Structural variation in the human genome: The impact of copy number variants on clinical diagnosis. Genet. Med. 2007;9:600–606. doi: 10.1097/gim.0b013e318149e1e3. [DOI] [PubMed] [Google Scholar]
- 22.Vissers L.E., de Vries B.B., Osoegawa K., Janssen I.M., Feuth T., Choy C.O., Straatman H., van der Vliet W., Huys E.H., van Rijk A. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw-Smith C., Redon R., Rickman L., Rio M., Willatt L., Fiegler H., Firth H., Sanlaville D., Winter R., Colleaux L. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoumans J., Ruivenkamp C., Holmberg E., Kyllerman M., Anderlid B.M., Nordenskjold M. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J. Med. Genet. 2005;42:699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyson C., Harvard C., Locker R., Friedman J.M., Langlois S., Lewis M.E., Van Allen M., Somerville M., Arbour L., Clarke L. Submicroscopic deletions and duplications in individuals with intellectual disability detected by array-CGH. Am. J. Med. Genet. A. 2005;139:173–185. doi: 10.1002/ajmg.a.31015. [DOI] [PubMed] [Google Scholar]
- 26.Wong A., Lese Martin C., Heretis K., Ruffalo T., Wilber K., King W., Ledbetter D.H. Detection and calibration of microdeletions and microduplications by array-based comparative genomic hybridization and its applicability to clinical genetic testing. Genet. Med. 2005;7:264–271. doi: 10.1097/01.gim.0000160076.14102.ec. [DOI] [PubMed] [Google Scholar]
- 27.Ballif B.C., Rorem E.A., Sundin K., Lincicum M., Gaskin S., Coppinger J., Kashork C.D., Shaffer L.G., Bejjani B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- 28.Friedman J.M., Baross A., Delaney A.D., Ally A., Arbour L., Armstrong L., Asano J., Bailey D.K., Barber S., Birch P. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am. J. Hum. Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krepischi-Santos A.C., Vianna-Morgante A.M., Jehee F.S., Passos-Bueno M.R., Knijnenburg J., Szuhai K., Sloos W., Mazzeu J.F., Kok F., Cheroki C. Whole-genome array-CGH screening in undiagnosed syndromic patients: Old syndromes revisited and new alterations. Cytogenet. Genome Res. 2006;115:254–261. doi: 10.1159/000095922. [DOI] [PubMed] [Google Scholar]
- 30.Menten B., Maas N., Thienpont B., Buysse K., Vandesompele J., Melotte C., de Ravel T., Van Vooren S., Balikova I., Backx L. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: A new series of 140 patients and review of published reports. J. Med. Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ming J.E., Geiger E., James A.C., Ciprero K.L., Nimmakayalu M., Zhang Y., Huang A., Vaddi M., Rappaport E., Zackai E.H. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum. Mutat. 2006;27:467–473. [Google Scholar]
- 32.Miyake N., Shimokawa O., Harada N., Sosonkina N., Okubo A., Kawara H., Okamoto N., Kurosawa K., Kawame H., Iwakoshi M. BAC array CGH reveals genomic aberrations in idiopathic mental retardation. Am. J. Med. Genet. A. 2006;140:205–211. doi: 10.1002/ajmg.a.31098. [DOI] [PubMed] [Google Scholar]
- 33.Rosenberg C., Knijnenburg J., Bakker E., Vianna-Morgante A.M., Sloos W., Otto P.A., Kriek M., Hansson K., Krepischi-Santos A.C., Fiegler H. Array-CGH detection of micro rearrangements in mentally retarded individuals: Clinical significance of imbalances present both in affected children and normal parents. J. Med. Genet. 2006;43:180–186. doi: 10.1136/jmg.2005.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 35.Shaffer L.G., Kashork C.D., Saleki R., Rorem E., Sundin K., Ballif B.C., Bejjani B.A. Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J. Pediatr. 2006;149:98–102. doi: 10.1016/j.jpeds.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Aradhya S., Manning M.A., Splendore A., Cherry A.M. Whole-genome array-CGH identifies novel contiguous gene deletions and duplications associated with developmental delay, mental retardation, and dysmorphic features. Am. J. Med. Genet. A. 2007;143A:1431–1441. doi: 10.1002/ajmg.a.31773. [DOI] [PubMed] [Google Scholar]
- 37.Baris H.N., Tan W.H., Kimonis V.E., Irons M.B. Diagnostic utility of array-based comparative genomic hybridization in a clinical setting. Am. J. Med. Genet. A. 2007;143A:2523–2533. doi: 10.1002/ajmg.a.31988. [DOI] [PubMed] [Google Scholar]
- 38.Engels H., Brockschmidt A., Hoischen A., Landwehr C., Bosse K., Walldorf C., Toedt G., Radlwimmer B., Propping P., Lichter P. DNA microarray analysis identifies candidate regions and genes in unexplained mental retardation. Neurology. 2007;68:743–750. doi: 10.1212/01.wnl.0000256367.70365.e0. [DOI] [PubMed] [Google Scholar]
- 39.Fan Y.S., Jayakar P., Zhu H., Barbouth D., Sacharow S., Morales A., Carver V., Benke P., Mundy P., Elsas L.J. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridization. Hum. Mutat. 2007;28:1124–1132. doi: 10.1002/humu.20581. [DOI] [PubMed] [Google Scholar]
- 40.Lu X., Shaw C.A., Patel A., Li J., Cooper M.L., Wells W.R., Sullivan C.M., Sahoo T., Yatsenko S.A., Bacino C.A. Clinical implementation of chromosomal microarray analysis: Summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman W.G., Hamilton S., Ayres J., Sanghera N., Smith A., Gaunt L., Davies L.M., Clayton-Smith J. Array comparative genomic hybridization for diagnosis of developmental delay: An exploratory cost-consequences analysis. Clin. Genet. 2007;71:254–259. doi: 10.1111/j.1399-0004.2007.00756.x. [DOI] [PubMed] [Google Scholar]
- 42.Shaffer L.G., Bejjani B.A., Torchia B., Kirkpatrick S., Coppinger J., Ballif B.C. The identification of microdeletion syndromes and other chromosome abnormalities: Cytogenetic methods of the past, new technologies for the future. Am. J. Med. Genet. C. Semin. Med. Genet. 2007;145C:335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- 43.Shen Y., Irons M., Miller D.T., Cheung S.W., Lip V., Sheng X., Tomaszewicz K., Shao H., Fang H., Tang H.S. Development of a focused oligonucleotide-array comparative genomic hybridization chip for clinical diagnosis of genomic imbalance. Clin. Chem. 2007;53:2051–2059. doi: 10.1373/clinchem.2007.090290. [DOI] [PubMed] [Google Scholar]
- 44.Thuresson A.C., Bondeson M.L., Edeby C., Ellis P., Langford C., Dumanski J.P., Anneren G. Whole-genome array-CGH for detection of submicroscopic chromosomal imbalances in children with mental retardation. Cytogenet. Genome Res. 2007;118:1–7. doi: 10.1159/000106434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagenstaller J., Spranger S., Lorenz-Depiereux B., Kazmierczak B., Nathrath M., Wahl D., Heye B., Glaser D., Liebscher V., Meitinger T. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aston E., Whitby H., Maxwell T., Glaus N., Cowley B., Lowry D., Zhu X.L., Issa B., South S.T., Brothman A.R. Comparison of targeted and whole genome analysis of postnatal specimens using a commercially available array based comparative genomic hybridisation (aCGH) microarray platform. J. Med. Genet. 2008;45:268–274. doi: 10.1136/jmg.2007.055319. [DOI] [PubMed] [Google Scholar]
- 47.Baldwin E.L., Lee J.Y., Blake D.M., Bunke B.P., Alexander C.R., Kogan A.L., Ledbetter D.H., Martin C.L. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet. Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- 48.Pickering D.L., Eudy J.D., Olney A.H., Dave B.J., Golden D., Stevens J., Sanger W.G. Array-based comparative genomic hybridization analysis of 1176 consecutive clinical genetics investigations. Genet. Med. 2008;10:262–266. doi: 10.1097/GIM.0b013e31816b64ad. [DOI] [PubMed] [Google Scholar]
- 49.Shevell M. Global developmental delay and mental retardation or intellectual disability: Conceptualization, evaluation, and etiology. Pediatr. Clin. North Am. 2008;55:1071–1084. doi: 10.1016/j.pcl.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Xiang B., Li A., Valentin D., Nowak N.J., Zhao H., Li P. Analytical and clinical validity of whole-genome oligonucleotide array comparative genomic hybridization for pediatric patients with mental retardation and developmental delay. Am. J. Med. Genet. A. 2008;146A:1942–1954. doi: 10.1002/ajmg.a.32411. [DOI] [PubMed] [Google Scholar]
- 51.Nowakowska B., Stankiewicz P., Obersztyn E., Ou Z., Li J., Chinault A.C., Smyk M., Borg K., Mazurczak T., Cheung S.W. Application of metaphase HR-CGH and targeted chromosomal microarray analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features. Am. J. Med. Genet. A. 2008;146A:2361–2369. doi: 10.1002/ajmg.a.32475. [DOI] [PubMed] [Google Scholar]
- 52.Lu X.Y., Phung M.T., Shaw C.A., Pham K., Neil S.E., Patel A., Sahoo T., Bacino C.A., Stankiewicz P., Kang S.H. Genomic imbalances in neonates with birth defects: High detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sutherland G.R., Murch A.R., Gardiner A.J., Carter R.F., Wiseman C. Cytogenetic survey of a hospital for the mentally retarded. Hum. Genet. 1976;34:231–245. doi: 10.1007/BF00295286. [DOI] [PubMed] [Google Scholar]
- 54.Jacobs P.A., Matsuura J.S., Mayer M., Newlands I.M. A cytogenetic survey of an institution for the mentally retarded: I. Chromosome abnormalities. Clin. Genet. 1978;13:37–60. doi: 10.1111/j.1399-0004.1978.tb04127.x. [DOI] [PubMed] [Google Scholar]
- 55.Faed M.J., Robertson J., Field M.A., Mellon J.P. A chromosome survey of a hospital for the mentally subnormal. Clin. Genet. 1979;16:191–204. doi: 10.1111/j.1399-0004.1979.tb00990.x. [DOI] [PubMed] [Google Scholar]
- 56.Kondo I., Hamaguchi H., Nakajima S., Haneda T. A cytogenetic survey of 449 patients in a Japanese institution for the mentally retarded. Clin. Genet. 1980;17:177–182. doi: 10.1111/j.1399-0004.1980.tb00130.x. [DOI] [PubMed] [Google Scholar]
- 57.Ravnan J.B., Tepperberg J.H., Papenhausen P., Lamb A.N., Hedrick J., Eash D., Ledbetter D.H., Martin C.L. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ballif B.C., Sulpizio S.G., Lloyd R.M., Minier S.L., Theisen A., Bejjani B.A., Shaffer L.G. The clinical utility of enhanced subtelomeric coverage in array CGH. Am. J. Med. Genet. A. 2007;143A:1850–1857. doi: 10.1002/ajmg.a.31842. [DOI] [PubMed] [Google Scholar]
- 59.Ylstra B., van den Ijssel P., Carvalho B., Brakenhoff R.H., Meijer G.A. BAC to the future! Or oligonucleotides: A perspective for micro array comparative genomic hybridization (array CGH) Nucleic Acids Res. 2006;34:445–450. doi: 10.1093/nar/gkj456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aradhya S., Cherry A.M. Array-based comparative genomic hybridization: Clinical contexts for targeted and whole-genome designs. Genet. Med. 2007;9:553–559. doi: 10.1097/gim.0b013e318149e354. [DOI] [PubMed] [Google Scholar]
- 61.Bejjani B.A., Saleki R., Ballif B.C., Rorem E.A., Sundin K., Theisen A., Kashork C.D., Shaffer L.G. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: Is less more? Am. J. Med. Genet. A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- 62.Cheung S.W., Shaw C.A., Yu W., Li J., Ou Z., Patel A., Yatsenko S.A., Cooper M.L., Furman P., Stankiewicz P. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet. Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 63.Shevell M.I., Bejjani B.A., Srour M., Rorem E.A., Hall N., Shaffer L.G. Array comparative genomic hybridization in global developmental delay. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008;147B:1101–1108. doi: 10.1002/ajmg.b.30730. [DOI] [PubMed] [Google Scholar]
- 64.Sharp A.J., Itsara A., Cheng Z., Alkan C., Schwartz S., Eichler E.E. Optimal design of oligonucleotide microarrays for measurement of DNA copy-number. Hum. Mol. Genet. 2007;16:2770–2779. doi: 10.1093/hmg/ddm234. [DOI] [PubMed] [Google Scholar]
- 65.Cooper G.M., Zerr T., Kidd J.M., Eichler E.E., Nickerson D.A. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat. Genet. 2008;40:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 68.Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 69.Koolen D.A., Sharp A.J., Hurst J.A., Firth H.V., Knight S.J., Goldenberg A., Saugier-Veber P., Pfundt R., Vissers L.E., Destree A. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J. Med. Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar R.A., KaraMohamed S., Sudi J., Conrad D.F., Brune C., Badner J.A., Gilliam T.C., Nowak N.J., Cook E.H., Jr., Dobyns W.B. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 71.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 73.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacobs P.A., Browne C., Gregson N., Joyce C., White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J. Med. Genet. 1992;29:103–108. doi: 10.1136/jmg.29.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Funderburk S.J., Spence M.A., Sparkes R.S. Mental retardation associated with “balanced” chromosome rearrangements. Am. J. Hum. Genet. 1977;29:136–141. [PMC free article] [PubMed] [Google Scholar]
- 77.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am. J. Hum. Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 78.Gribble S.M., Prigmore E., Burford D.C., Porter K.M., Ng B.L., Douglas E.J., Fiegler H., Carr P., Kalaitzopoulos D., Clegg S. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J. Med. Genet. 2005;42:8–16. doi: 10.1136/jmg.2004.024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Gregori M., Ciccone R., Magini P., Pramparo T., Gimelli S., Messa J., Novara F., Vetro A., Rossi E., Maraschio P. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: A study of 59 patients. J. Med. Genet. 2007;44:750–762. doi: 10.1136/jmg.2007.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baptista J., Mercer C., Prigmore E., Gribble S.M., Carter N.P., Maloney V., Thomas N.S., Jacobs P.A., Crolla J.A. Breakpoint mapping and array CGH in translocations: Comparison of a phenotypically normal and an abnormal cohort. Am. J. Hum. Genet. 2008;82:927–936. doi: 10.1016/j.ajhg.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baptista J., Prigmore E., Gribble S.M., Jacobs P.A., Carter N.P., Crolla J.A. Molecular cytogenetic analyses of breakpoints in apparently balanced reciprocal translocations carried by phenotypically normal individuals. Eur. J. Hum. Genet. 2005;13:1205–1212. doi: 10.1038/sj.ejhg.5201488. [DOI] [PubMed] [Google Scholar]
- 82.Conlin L.K., Thiel B.D., Bonnemann C.G., Medne L., Ernst L.M., Zackai E.H., Deardorff M.A., Krantz I.D., Hakonarson H., Spinner N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hook E.B. Exclusion of chromosomal mosaicism: tables of 90%, 95% and 99% confidence limits and comments on use. Am. J. Hum. Genet. 1977;29:94–97. [PMC free article] [PubMed] [Google Scholar]
- 84.Scott S.A., Cohen N., Brandt T., Toruner G., Desnick R.J., Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet. Med. 2010;12:85–92. doi: 10.1097/GIM.0b013e3181cc75d0. [DOI] [PubMed] [Google Scholar]
- 85.Cheung S.W., Shaw C.A., Scott D.A., Patel A., Sahoo T., Bacino C.A., Pursley A., Li J., Erickson R., Gropman A.L. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am. J. Med. Genet. A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- 86.Theisen A., Rosenfeld J.A., Farrell S.A., Harris C.J., Wetzel H.H., Torchia B.A., Bejjani B.A., Ballif B.C., Shaffer L.G. aCGH detects partial tetrasomy of 12p in blood from Pallister-Killian syndrome cases without invasive skin biopsy. Am. J. Med. Genet. A. 2009;149A:914–918. doi: 10.1002/ajmg.a.32767. [DOI] [PubMed] [Google Scholar]
- 87.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sagoo G.S., Butterworth A.S., Sanderson S., Shaw-Smith C., Higgins J.P., Burton H. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet. Med. 2009;11:139–146. doi: 10.1097/GIM.0b013e318194ee8f. [DOI] [PubMed] [Google Scholar]
- 89.Hochstenbach R., van Binsbergen E., Engelen J., Nieuwint A., Polstra A., Poddighe P., Ruivenkamp C., Sikkema-Raddatz B., Smeets D., Poot M. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur. J. Med. Genet. 2009;52:161–169. doi: 10.1016/j.ejmg.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 90.Ballif B.C., Hornor S.A., Sulpizio S.G., Lloyd R.M., Minier S.L., Rorem E.A., Theisen A., Bejjani B.A., Shaffer L.G. Development of a high-density pericentromeric region BAC clone set for the detection and characterization of small supernumerary marker chromosomes by array CGH. Genet. Med. 2007;9:150–162. doi: 10.1097/gim.0b013e3180312087. [DOI] [PubMed] [Google Scholar]
- 91.Berg J.S., Brunetti-Pierri N., Peters S.U., Kang S.H., Fong C.T., Salamone J., Freedenberg D., Hannig V.L., Prock L.A., Miller D.T. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet. Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- 92.Potocki L., Bi W., Treadwell-Deering D., Carvalho C.M., Eifert A., Friedman E.M., Glaze D., Krull K., Lee J.A., Lewis R.A. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grati F.R., Lesperance M.M., De Toffol S., Chinetti S., Selicorni A., Emery S., Grimi B., Dulcetti F., Malvestiti B., Taylor J. Pure monosomy and pure trisomy of 13q21.2-31.1 consequent to a familial insertional translocation: exclusion of PCDH9 as the responsible gene for autosomal dominant auditory neuropathy (AUNA1) Am. J. Med. Genet. A. 2009;149A:906–913. doi: 10.1002/ajmg.a.32754. [DOI] [PubMed] [Google Scholar]
- 94.South S.T., Rope A.F., Lamb A.N., Aston E., Glaus N., Whitby H., Maxwell T., Zhu X.L., Brothman A.R. Expansion in size of a terminal deletion: A paradigm shift for parental follow-up studies. J. Med. Genet. 2008;45:391–395. doi: 10.1136/jmg.2008.057315. [DOI] [PubMed] [Google Scholar]
- 95.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 96.Shaffer L.G., Slovak M.L., Campbell L.J. Karger; Basel, Switzerland: 2009. ISCN: An International System for Human Cytogenetic Nomenclature. [Google Scholar]
- 97.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent Rearrangements of Chromosome 1q21.1 and Variable Pediatric Phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shaffer L.G., Theisen A., Bejjani B.A., Ballif B.C., Aylsworth A.S., Lim C., McDonald M., Ellison J.W., Kostiner D., Saitta S. The discovery of microdeletion syndromes in the post-genomic era: Review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet. Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- 99.Willatt L., Cox J., Barber J., Cabanas E.D., Collins A., Donnai D., FitzPatrick D.R., Maher E., Martin H., Parnau J. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Kovel C.G., Trucks H., Helbig I., Mefford H.C., Baker C., Leu C., Kluck C., Muhle H., von Spiczak S., Ostertag P. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sharp A.J., Mefford H.C., Li K., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., De Gregori M., Ciccone R. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miller D.T., Shen Y., Weiss L.A., Korn J., Anselm I., Bridgemohan C., Cox G.F., Dickinson H., Gentile J., Harris D.J. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatirc disorders. J. Med. Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Derbent M., Bikmaz Y.E., Yilmaz Z., Tokel K. Variable phenotype and associations in chromosome 22q11.2 microdeletion. Am. J. Med. Genet. A. 2006;140:659–660. doi: 10.1002/ajmg.a.31120. [DOI] [PubMed] [Google Scholar]
- 105.Feuk L., Carson A.R., Scherer S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 106.Feuk L., Marshall C.R., Wintle R.F., Scherer S.W. Structural variants: Changing the landscape of chromosomes and design of disease studies. Hum. Mol. Genet. 2006;15(Spec No 1):R57–R66. doi: 10.1093/hmg/ddl057. [DOI] [PubMed] [Google Scholar]
- 107.Curry C.J., Mao R., Aston E., Mongia S.K., Treisman T., Procter M., Chou B., Whitby H., South S.T., Brothman A.R. Homozygous deletions of a copy number change detected by array CGH: A new cause for mental retardation? Am. J. Med. Genet. A. 2008;146A:1903–1910. doi: 10.1002/ajmg.a.32450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. General Recommendations of the ISCA Consortium