Mechanisms of Hepatic Fibrogenesis (original) (raw)
. Author manuscript; available in PMC: 2010 Jun 21.
Published in final edited form as: Gastroenterology. 2008 May;134(6):1655–1669. doi: 10.1053/j.gastro.2008.03.003
Abstract
Substantial improvements in the treatment of chronic liver disease have accelerated interest in uncovering the mechanisms underlying hepatic fibrosis and its resolution. Activation of resident hepatic stellate cells into proliferative, contractile, and fibrogenic cells in liver injury remains a dominant theme driving the field. However, several new areas of rapid progress in the past 5–10 years also have taken root, including: (1) identification of different fibrogenic populations apart from resident stellate cells, for example, portal fibroblasts, fibrocytes, and bone-marrow– derived cells, as well as cells derived from epithelial mesenchymal transition; (2) emergence of stellate cells as finely regulated determinants of hepatic inflammation and immunity; (3) elucidation of multiple pathways controlling gene expression during stellate cell activation including transcriptional, post-transcriptional, and epigenetic mechanisms; (4) recognition of disease-specific pathways of fibrogenesis; (5) re-emergence of hepatic macrophages as determinants of matrix degradation in fibrosis resolution and the importance of matrix cross-linking and scar maturation in determining reversibility; and (6) hints that hepatic stellate cells may contribute to hepatic stem cell behavior, cancer, and regeneration. Clinical and translational implications of these advances have become clear, and have begun to impact significantly on the management and outlook of patients with chronic liver disease.
The field of hepatic fibrosis is flourishing thanks to continued experimental advances complemented by exciting progress in the treatment of chronic liver disease.1 Control of chronic hepatitis B and C by antiviral therapies has established that advanced fibrosis can regress in association with improved clinical outcomes,2-4 thereby intensifying enthusiasm to uncover the mechanistic basis for hepatic fibrogenesis and its attenuation. At the same time, simple paradigms defining the cellular sources of extracellular matrix (ECM), and the roles of cytokines and paracrine interactions among resident liver cells and inflammatory cells have yielded a more nuanced understanding of how the liver responds to injury. These advances have had a collateral benefit towards understanding fibrosis in other organs, particularly the pancreas.5 Thus, a review of the mechanisms underlying hepatic fibrosis is not only timely, but is more clinically relevant than ever. This article focuses on recent advances in the field, building on established principles from earlier reviews6,7 while weaving in their clinical relevance, but also emphasizing the molecular subtleties that have emerged through continued progress in the field.
General Principles
Fibrosis, or scarring of the liver, is a wound-healing response that engages a range of cell types and mediators to encapsulate injury. Although even acute injury will activate mechanisms of fibrogenesis, the sustained signals associated with chronic liver disease caused by infection, drugs, metabolic disorders, or immune attack are required for significant fibrosis to accumulate. Occasionally, fibrosis may be rapidly progressive over weeks to months, for example, as a result of drug injury, hepatitis C virus (HCV) after liver transplantation,8 or human immunodeficiency virus (HIV)/HCV co-infection,9 but for the most part this is a response that evolves over decades. The protracted nature of this response, in contrast to the more rapid progression of fibrosis in kidney or lung, has been classically ascribed to the liver’s unique regenerative capacity, but the molecular underpinnings of this capacity remain mysterious.
In many ways, the liver’s response to injury is an angiogenic one, with evidence of new blood vessel formation, sinusoidal remodeling, and pericyte (ie, stellate cell) expansion.10 Thus, mediators familiar to the angiogenesis field are equally relevant in understanding hepatic fibrosis, including platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and their cognate receptors, as well as vasoactive mediators that include nitric oxide and carbon monoxide. For example, increased VEGF concentrations may contribute to accelerated progression of fibrosis in smokers who have hepatitis C.11
Cirrhosis, the most advanced stage of fibrosis, connotes not only more scar than fibrosis alone, but also distortion of the liver parenchyma associated with septae and nodule formation, altered blood flow, and risk of liver failure. However, cirrhosis still remains a dynamic and evolving state, as discussed later (see Clinical and Translational Implications section), such that interventions even at these advanced stages could regress scar and improve clinical outcomes.
Continued progress in the field also has exploited steady refinements in both cell culture and animal models of fibrosis.12 Although there are no rodent models that closely mimic hepatitis B virus (HBV), HCV, or nonalcoholic steatohepatitis (NASH), the development of genetic mouse models has continued to accelerate progress by enabling reductionist approaches that focus on the role of individual gene products in fibrogenesis, and by permitting genetic lineage tracing to define cellular phenotypes and their evolution.13 Animal and culture models also have benefited from improving technology, in particular the use of gene array and proteomics. For example, gene expression patterns from stellate cells (the key resident fibrogenic cell type) isolated from rats with fibrosis from either CCl4 or bile duct ligation are remarkably similar, but differ substantially from ultrapurified (ie, using flow cytometry) culture-activated stellate cells.14 However, stellate cells isolated using standard gradient methods more closely resembled activated cells from fibrotic liver, indicating that standard cell isolation methods yield gene expression data that are relevant to the biology of stellate cells during fibrosis in vivo.14
Liver Injury and Inflammation: Established Mediators and New Players
Fibrosis requires some element of liver injury, albeit not necessarily defined by the presence of inflammatory cells. For example, although the most prevalent diseases in clinical practice (viral hepatitis, alcoholic steatohepatitis [ASH], and NASH) are characterized by leukocyte infiltration, metabolic diseases such as hemachromatosis are notable for their lack of inflammatory cells, yet they too lead to cirrhosis and risk of hepatocellular carcinoma. Thus, any chronic perturbation of hepatic homeostasis, whether visible by light microscopy or not, may elicit the signals necessary to stimulate fibrogenesis.
One family of such established mediators are reactive oxygen species. These unstable compounds include superoxide and hydroxyl radicals, hydrogen peroxide, and aldehydic end products including 4-hydroxy-2,3-nonenal and 4-hydroxy-2,3-alkenals. These mediators are generated through lipid peroxidation, and can derive from hepatocytes, macrophages, stellate cells, and inflammatory cells.15,16 Several substrates may enhance reactive oxygen species production including ethanol, polyunsaturated fatty acids, and iron. The classic pathway of reactive oxygen species generation in hepatocytes results from induction of cytochrome P450 2E1, especially in either ASH or NASH,17,18 leading to pericentral (zone 3) injury. More recently, however, reduced nicotinamide adenine dinucleotide phosphate oxidase has emerged as another source of oxidant stress that mediates pathways of fibrogenic activation in hepatic stellate cells, as well as in Kupffer cells, the resident liver macrophages.19 Reduced nicotinamide adenine dinucleotide phosphate oxidase also may mediate liver injury and fibrosis generated by angiotensin signaling because animals genetically lacking the p47 subunit of reduced nicotinamide adenine dinucleotide phosphate oxidase have reduced superoxide production and attenuated hepatic fibrosis after injury.20
More recently, increasing attention also has been directed to nitrosative stress generated by hepatocyte mitochondrial injury and induction of nitric oxide synthase 2,21,22 although links from this pathway to fibrogenesis are not as well defined.
Apoptosis of parenchymal cells is no longer viewed as a silent consequence of liver injury, but rather as an important inflammatory stimulus23 that activates stellate cells, which display a surprising capacity to phagocytose apoptotic bodies,24 leading to induction of reduced nicotinamide adenine dinucleotide phosphate oxidase.25 This response to apoptotic hepatocytes in part reflects the interaction of hepatocyte DNA with Toll-like receptor 9 (TLR9) expressed on stellate cells.26 A profibrogenic response also can be elicited by hepatocyte apoptosis after disruption of the anti-apoptotic mediator Bcl-xL,27 and by Fas.28 Thus, efforts to block hepatocyte apoptosis therapeutically are being developed as a potential antifibrotic strategy.29 On the other hand, selective stimulation of apoptosis in stellate cells rather than hepatocytes would be antifibrotic, mediated by either tumor necrosis factor-related apoptosis-inducing ligand,30 gliotoxin,31 or proteasome inhibitors.32
Although necrosis of cells is a classic morphologic feature of liver injury, its pathogenic contribution to hepatic fibrosis has been overlooked, largely because there are no classic biochemical or molecular hallmarks of necrosis similar to those that have been uncovered for apoptosis.33 Yet, in human disease both necrosis and apoptosis are evident in liver sections, although specific inflammatory pathways of necrosis have not been identified. Necrosis may simply represent a more severe cellular response than apoptosis when concentrations of injurious stimuli are higher,15,16 but the relative potencies of necrosis compared with apoptosis in stimulating fibrogenesis are unknown.
Among the most compelling pathways of injury are those recently uncovered for innate immune signaling in liver. Specifically, the discovery of TLRs has led to major advances in understanding how the human organism responds to pathogens.34 The identification of TLR4, the receptor for bacterial lipopolysaccharide, on Kupffer cells, was therefore not a surprise, but its expression on stellate cells was unexpected.35 Moreover, although TLR4 signaling in macrophages may be essential for inflammatory responses,36 recent studies have indicated that signaling by stellate cells in response to lipopolysaccharide and possibly endogenous ligands of TLR4 (eg, high-mobility group box 1, biglycan, and heparan sulfate) may be more important than in Kupffer cells in eliciting a fibrogenic response by down-regulating bone morphogenic protein (BMP) and activin membrane-bound inhibitor, a transmembrane suppressor of transforming growth factor _β_1 (TGF_β_1), which is the major fibrogenic cytokine in the liver.37 This finding has converged with evidence that specific single-nucleotide polymorphisms of TLR4 contribute to the rate of fibrosis progression in HCV infection,38 thereby linking a genetic risk marker to disease pathogenesis.
Evidence that stellate cells play a pivotal role in orchestrating hepatic immune responses extends beyond the TLR pathway and is among the most surprising findings of the past 5 years. Stellate cells produce a host of chemotactic peptides (especially chemokines) that amplify infiltration by inflammatory cells. They also interact directly with lymphocyte subsets, including natural killer cells (see Mehal and Friedman39 for detailed review). Indeed, CD8 T cells appear to be more fibrogenic towards stellate cells than CD4 cells,40 which could account for the increased rate of fibrosis in patients with HIV and HCV, in whom the CD4/CD8 ratio typically is reduced.41 In contrast, natural killer cells play an important role in clearing activated stellate cells,42,43 an activity enhanced by gamma interferon and retinoic acid44 and abrogated by ethanol.45 Intriguingly, stellate cells also are antigen-presenting cells,46 and may contribute to the liver’s immunotolerant properties through T-cell suppression.47 Finally, B lymphocytes, which comprise up to 50% of the total lymphocyte pool in liver, contribute to the fibrogenic milieu because mice lacking B cells (JH−/− animals) have attenuated fibrosis, apparently by more rapid ECM degradation after CCl4 injury, implicating an unknown interaction with pathways of matrix degradation.48
Cellular Sources of ECM in Hepatic Fibrosis: An Evolving Paradigm
The discovery of stellate cell activation—a transdifferentiation from a quiescent vitamin A–storing cell to a proliferative myofibroblast—remains among the most informative discoveries to date in unlocking the basis for hepatic fibrogenesis. However, the simple paradigm conceived 15 years ago6 that all fibrosis derives from activated stellate cells has grown far more multifaceted, both in terms of the pathways of activation and the overall contribution of stellate cells to the total fibrogenic population during liver injury. It is increasingly clear that these fibrogenic cells derive not only from resident stellate cells, but also from portal fibroblasts,49-51 circulating fibrocytes,52 bone marrow,53 and epithelial–mesenchymal cell transition.13 Although the relative contribution of each source varies, these differences are likely to reflect differing contributions with disease progression and among different etiologies (Figure 1). For example, portal fibroblasts appear to be especially important in cholestatic liver diseases and ischemia,51 where paracrine interactions between cholangiocytes and fibroblasts involve both chemokines54 and extracellular nucleotides.55 In contrast, progressive recruitment of bone-marrow– derived cells may occur over time, such that these cells can represent a substantial fraction of the total fibrogenic population in more chronic injury. Bone marrow recruitment of mesenchymal cells remains a somewhat confusing event because of contradictory findings that on the one hand, bone marrow may provide fibrogenic cells, yet on the other hand autologous bone marrow56,57 and marrow-derived endothelial progenitor cells58 can be antifibrotic. It remains unclear which cells within marrow contribute to fibrogenesis and which might be antifibrotic, and/or what mediators regulate these apparently divergent activities of bone-marrow– derived cells.
Figure 1.
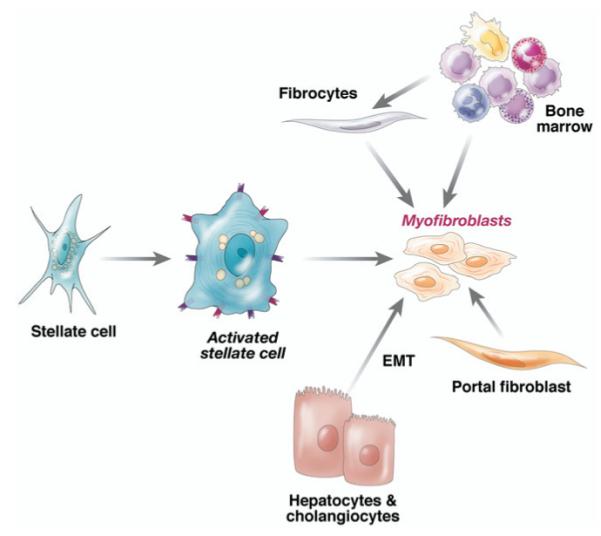
Contributions of activated stellate cells and other fibrogenic cell types to hepatic fibrosis. Quiescent stellate cell activation is initiated by a range of soluble mediators (Figure 2). The activated cell is stimulated further by key cytokines (detailed further in Figure 2) into myofibroblasts (which contain contractile filaments). Over time, however, other sources also contribute to fibrogenic populations in liver, including bone marrow (which likely gives rise to circulating fibrocytes), portal fibroblasts, and epithelial mesenchymal transition (emt) from hepatocytes and cholangiocytes. Relative contributions and the stages at which these cell types add to the myofibroblast population is likely to differ among various etiologies of liver injury (see text).
Because cellular sources of fibrogenic cells may differ among different etiologies, the relative value of particular antifibrotic therapies also may depend on the underlying disease. For example, the integrin _α_v_β_6 is up-regulated only on biliary epithelium during experimental cholestatic liver fibrosis,59 implying that antagonists to this integrin might be more rational in biliary than in parenchymal liver diseases.
Pathways of Stellate Cell Activation: A Moving Target
Initiating Pathways
Stellate cell activation unfolds progressively in sequential stages; this paradigm provides a useful framework for defining fibrogenic events after liver injury (Figure 2). In particular, the initiation phase, which refers to early events that render the quiescent stellate cell responsive to a range of growth factors, remains an important focus. Rapid induction of _β_-PDGF receptor, development of a contractile and fibrogenic phenotype, as well as modulation of growth factor signaling are the cardinal features of this early response. Initiating stimuli include paracrine signals such as reactive oxygen species from apoptotic hepatocytes and injured cholangiocytes, as detailed earlier (see Liver Injury and Inflammation section).
Figure 2.

Pathways of hepatic stellate cell activation. Features of stellate cell activation can be distinguished between those that stimulate initiation and those that contribute to perpetuation. Initiation is provoked by soluble stimuli that include oxidant stress signals (reactive oxygen intermediates), apoptotic bodies, lipopolysaccharide (lps), and paracrine stimuli from neighboring cell types including hepatic macrophages (Kupffer cells), sinusoidal endothelium, and hepatocytes. Perpetuation follows, characterized by a number of specific phenotypic changes including proliferation, contractility, fibrogenesis, altered matrix degradation, chemotaxis, and inflammatory signaling. fgf, fibroblast growth factor; et-1, endothelin-1; nk, natural killer; no, nitric oxide; mt, membrane type. Modified with permission from Friedman.7
Changes in ECM composition
Initiating events in stellate cell activation are occurring on a background of progressive changes in the surrounding ECM within the subendothelial space of Disse. Over time, the subendothelial matrix composition changes from one comprised of type IV collagen, heparan sulfate proteoglycan, and laminin (the classic constituents of a basal lamina) to one rich in fibril-forming collagens, particularly types I and III. One important yet subtle change is the deposition of a specific isoform of cellular fibronectin from sinusoidal endothelial cells, which has an activating effect on stellate cells.60 Because this response is TGF-β dependent,61 however, induction of this cytokine must occur first, either from autocrine or paracrine sources.
These progressive changes in ECM composition as fibrosis accumulates instigate several positive feedback pathways that further amplify fibrosis. First, dynamic changes in membrane receptors, in particular integrins, sense altered matrix signals that provoke stellate cell activation and migration through focal adhesion disassembly62-64 while also linking to other growth factor receptors through integrin-linked kinase.65,66 Matrix-provoked signals also engage membrane-bound guanosine triphosphate binding proteins, in particular Rho67 and Rac,68 which transduce signals to the actin cytoskeleton that promote migration and contraction. Second, activation of cellular matrix metalloproteases leads to release of growth factors from matrix-bound reservoirs in the extracellular space that may stimulate cellular growth and fibrogenesis.69,70 Third, the enhanced density of ECM leads to increasing matrix stiffness, which is a significant stimulus to stellate cell activation, at least in part through integrin signaling.71 Interestingly, however, increased stiffness caused by edema and inflammation in animal models and human beings may precede the increase in matrix content.72,73 These experimental findings nicely complement the increasing use of Fibroscan74,75 (Echosens, Paris, France) and magnetic resonance elastography,76,77 two clinical techniques that noninvasively assess hepatic stiffness as a reflection of ECM content.
Molecular mechanisms underlying stellate cell activation
Because activation occurs so rapidly, attention has focused on regulatory pathways that can respond quickly to injurious stimuli, either by activating or repressing gene transcription, by epigenetic regulation, or by posttranscriptional control. In addition, although microRNAs have emerged as important layers of regulatory control in many systems,78 their roles in liver injury and stellate cell activation have not yet been explored.
Among the many target genes of transcription factors described in stellate cells, those most comprehensively characterized include type I collagen (alpha 1 and alpha 2 chains), α_-smooth muscle actin, TGF_β_1, and TGF_β receptors, matrix metalloproteinase (MMP)-2, and tissue inhibitors of metalloproteinases (TIMPs) 1 and 2 (see Rippe and Brenner,79 Mann et al,80 and Tsukamoto et al81 for reviews).
Foxf1, JunD, and C/EBP_β_ are among the clearest examples of activating transcription factors. Deletion of one Foxf1 allele reduces stellate cell activation and fibrosis.82 Similarly, knockout of JunD, a member of the AP-1 transcription factor complex,83 protects mice from CCl4induced hepatic fibrosis, which is associated with reduced numbers of activated stellate cells and diminished expression of hepatic TIMP-1.84 Finally, phosphorylation of the transcription factor C/EBP_β_ by the RSK kinase promotes stellate cell survival.85
The LIM homeodomain protein, Lhx2, on the other hand, is a protein that preserves stellate cell quiescence, and whose loss leads to activation of stellate cells. In fact, Lhx2−/− mice develop spontaneous congenital fibrosis.86 A similar role has been uncovered for the transcription factor FoxO1; viz. Stellate cell activation and fibrogenesis are amplified in cells from mice with reduced FoxO1 activity.87
Similar to Lhx2, the peroxisome proliferator activated receptor γ nuclear receptor down-regulates stellate cell activation and reduces collagen gene expression.88 This and related observations89 have prompted the use of peroxisome proliferator activated receptor γ ligands in clinical trials not only in fibrosis associated with NASH, but also as a candidate antifibrotic in patients with HCV fibrosis. Other nuclear receptors regulating stellate cell behavior include pregnane X receptor,90,91 and retinoid receptors.92,93
Epigenetic regulation is a tightly controlled pathway that modulates stellate cell activation in part through induction of the molecules CBF1 and MeCP2.94,95 These proteins repress gene expression of the inhibitory protein inhibitor kappa beta (I_κ_B) by CpG island methylation, thereby unleashing nuclear factor κ B activity, which promotes stellate cell survival and thus increases fibrosis. Interestingly, sulfasalazine inhibits the kinase (inhibitor kappa kinase, I_κ_K) that activates I_κ_B, and thus may accelerate recovery from experimental fibrosis by clearance of activated stellate cells through apoptosis.94,96 Because these activated cells typically express high levels of TIMP-1, a metalloproteinase inhibitor, their clearance leads to increased net activity of matrix degrading proteases.
Finally, messenger RNA (mRNA) stabilization also contributes to increased gene expression during stellate cell activation. Specifically, there is a 16-fold increase in collagen alpha 1(I) mRNA stabilization during stellate cell activation as a result of interaction of a specific protein, _α_CP, to a specific sequence in the 3′ untranslated region of the mRNA,97 and also involving the interaction of a 120-kilodalton protein with the 5′ stem-loop structure.98 Similarly, there is enhanced interaction of the RNA binding protein, RBMS3, with the 3′ untranslated region of the homeobox protein Prx1, thereby increasing its mRNA stability.99
Perpetuating Pathways
The stellate cell that is activated by initiating stimuli then is primed to respond to a host of cytokines and growth factors. These signals conspire to generate scar through enhanced proliferation, contractility, fibrogenesis, matrix degradation, and proinflammatory signaling. Although earlier models suggested that the pathways of activation were identical regardless of the disease, it is now clear that there are disease-specific pathways of fibrosis (see Disease-Specific Pathways of Hepatic Fibrosis section), and, moreover, that not all cytokine pathways are necessarily activated in parallel. For example, although PDGF stimulation may drive cellular proliferation in parallel with fibrogenic stimulation in some settings, TLR9 activation blocks PDGF-mediated migration while provoking fibrogenesis,26 thereby providing a stop signal that allows activated cells to accumulate at sites of injury where they can deposit more scar.
Proliferation
Autocrine signaling by PDGF was the first cytokine loop uncovered during stellate cell activation and remains among the most potent.100 Both the PDGF ligand and the beta isoform of its receptor are rapidly induced in vivo and in culture.101,102 In addition to the well-characterized A and B chains of plateletderived growth factor (PDGF), C and D isoforms also have been discovered more recently; in fact, PDGF-D may be the most potent and physiologically relevant PDGF subunit in stellate cell activation.100 Interestingly, although both mice with transgenic expression of either PDGF-B103 or PDGF-C have hepatic fibrosis,104 the PDGF-C transgenic animals also develop hepatocellular carcinoma,104 mimicking the progression from fibrosis to cancer that occurs in human beings. Downstream consequences of PDGF signaling in stellate cells include signaling by PI3 kinase, ERK, and other pathways,105-107 as well as stimulation of Na+/H+ exchange, providing a potential site for therapeutic intervention by blocking ion transport.108 Other stellate cell mitogens include VEGF,109 thrombin and its receptor,110,111 epidermal growth factor, TGF_α_, keratinocyte growth factor,112 and basic fibroblast growth factor.113
Chemotaxis
Stellate cells can migrate towards sites of injury driven by chemoattractants that include PDGF,114,115 monocyte chemoattractant protein-1,116 and CXCR3 ligands.117 Functionally, PDGF-stimulated chemotaxis provokes cell spreading at the tip, movement of the cell body towards the stimulant, and retraction of trailing protrusions associated with transient myosin phosphorylation.118
In contrast to PDGF, adenosine blunts chemotaxis, thereby providing a counter-regulatory pathway that fixes cells at sites of injury.119 Paradoxically, enhanced adenosine signaling also may contribute to alcoholic fibrosis by stimulating stellate cell fibrogenesis,120 which not only represents a potential fibrogenic mechanism, but also may explain the protective effect of caffeine (which inhibits adenosine generation) reported in epidemiologic surveys.121
Fibrogenesis
Collagen type I is the prototype constituent of the fibril-forming matrix in fibrotic liver, and its expression is regulated both transcriptionally and posttranscriptionally as described earlier and in several reviews.122-124
TGF_β_1, derived from both paracrine and autocrine sources, remains the classic fibrogenic cytokine (see Inagaki and Okazaki124 and Breitkopf et al125 for reviews). Signals downstream of TGF_β_ converge on Smad proteins, which fine-tune and enhance the effects of TGF_β_ during stellate cell activation; Smads 2 and 3 are stimulatory whereas Smad 7 is inhibitory124,126,127 and is antagonized by Id1.128 TGF_β_1 also stimulates collagen transcription in stellate cells through a hydrogen peroxide– and C/EBP_β_-dependent mechanism.129 The response of Smads in stellate cells evolves as injury becomes chronic, further enhancing fibrogenesis.126,130
Connective tissue growth factor (CTGF/CCN2) is also a potent fibrogenic signal towards stellate cells131-133 that is up-regulated by hyperglycemia and hyperinsulinemia.134 Although stimulation of CTGF production traditionally has been considered TGF_β_-dependent,135 TGF_β_-independent regulation is increasingly likely as well.136 Moreover, TGF_β_ stimulates CTGF primarily in hepatocytes, not stellate cells,137,138 a notable exception to the general rule that cytokine signaling in stellate cell activation typically is autocrine.
Neurohumoral signaling contributes to stellate cell responses.139 In particular, cannabinoids have emerged as potent mediators of hepatic steatosis, stellate cell activation, and fibrosis (reviewed in Mallat et al140), as well as provoking the hemodynamic alterations associated with advanced liver disease.141 Two receptors, CB1 and CB2, exert opposing effects, with CB1 a fibrogenic pathway and CB2 antifibrotic. Thus, antagonism of CB1 signaling in stellate cells has emerged as a promising antifibrotic strategy, as exemplified by the clinical agent Rimonabant (Sanofi-Aventis, Paris, France).142 Conversely, agonism of CB2 receptors, which also are expressed by stellate cells, reverses fibrosis in experimental animals.143 The fundamental challenge of developing cannabinoid therapeutics for liver disease is to minimize central nervous system effects because CB1 and CB2 receptors are expressed abundantly in brain. Similarly, opioids signal in stellate cells and promote fibrogenesis,144,145 which is antagonized by naltrexone. Finally, sympathetic neurotransmitters also contribute to activation pathways.146
Contractility
Contraction of stellate cells contributes to increased portal resistance during liver fibrosis that presumably is reversible before the thickened septae, intrahepatic shunts, and lobular distortion of cirrhosis develop, leading to fixed increases in portal pressure. Even in earlier stages of fibrosis, activated stellate cells already show features of smooth muscle–like cells, characterized by expression of a number of contractile filaments including α smooth muscle actin147 and myosin,148 which generate calcium-dependent and calcium-independent contractile forces that contribute to cellular contractility.149-151 Culture models increasingly recapitulate many of these smooth muscle features, in part by restoring a more physiologic substratum, as well as by including other resident cell types in a co-culture system, especially Kupffer cells.152
As reviewed recently, stellate cells are recognized as liver-specific pericytes that contribute to angiogenesis in liver development, regeneration, and the response to injury.10 After partial hepatectomy, stellate cells migrate along with endothelial cells to establish vascular connections with hepatocytes, thereby creating new sinusoidal branches.153 Although it is unclear to what extent these responses occurring during pure regeneration are mounted during liver injury, the 2 responses—repair and regeneration— occur concurrently in chronic liver disease, and thus similar angiogenic behavior is likely to underlie repair. Moreover, progressive fibrosis with angiogenesis also contributes to tumor vascularization in which activated stellate cells play a vital role.10,154
As fibrosis advances, the collagenous bands typical of end-stage cirrhosis contain large numbers of activated stellate cells.155 These cells progressively impede portal blood flow by both constricting individual sinusoids and by contracting the cirrhotic liver, mediated by pathways that allow interaction with the ECM.156,157 At the same time, stellate cell density and coverage of the sinusoidal lumen increases.10,153 Endothelin-1 and nitric oxide are the key opposing counter-regulators that control stellate cell contractility, in addition to angiotensinogen II, eicosanoids, atrial natriuretic peptide, somatostatin, and carbon monoxide, among others (see Rockey155 and Reynaert et al158 for reviews). Progressive development of intrahepatic shunts also is likely to require angiogenic responses driven by stellate cells.159,160
The therapeutic implications of elucidating angiogenic signaling in liver fibrosis are not so clear. Although a simple paradigm would suggest that anti-angiogenic agents, for example, VEGF inhibitors, are an effective anti-inflammatory and antifibrotic strategy,161 their long-term impact on regeneration is uncertain. For example, sustained and complete inhibition of angiogenesis might compromise hepatic blood flow and oxygen delivery, especially because the liver has high metabolic demands and receives primarily venous blood. Thus, there is a delicate balance between the requirement for angiogenesis to preserve blood flow, and its negative impact on liver structure and function. Moreover, strategies to antagonize contractile proteins also may have unintended consequences. For example, mice lacking α smooth muscle actin protein in myofibroblasts have increased renal fibrosis in experimental glomerulonephritis,162 suggesting that _α_-actin induction may be a counter-regulatory response to enhanced fibrogenesis. Moreover, missense mutations of this protein have been linked to aortic aneurysms in human beings,163 indicating that the protein also may contribute to vascular integrity.
Inflammatory signaling
As reviewed earlier (see Liver Injury and Inflammation: Established Mediators and New Players section), stellate cells have emerged as central modulators of hepatic inflammation and immunity, and not just passive targets of inflammatory cytokines. In particular, a growing list of chemokines and their cognate receptors serve the dual function of provoking further fibrogenesis, as well as interacting with inflammatory cells to modify the immune response during injury.164-166
Matrix Degradation and Resolution of Fibrosis
Evidence that fibrosis and even cirrhosis are reversible has intensified interest in understanding the regulation of matrix degradation and fibrosis resolution, in hopes that therapies might exploit those endogenous pathways that reverse disease (Figure 3). Simplistically put, this response would be considered therapeutic matrix degradation, whereas early liver injury is marked by pathologic matrix degradation that disrupts hepatic homeostasis. Thus, in early liver injury matrix-degrading proteases with activity towards type IV collagen (eg, MMP-2), degrade the low-density basement membrane present in the subendothelial space. Its replacement with fibril-forming matrix has deleterious effects on differentiated cell function, in particular on hepatocytes.
Figure 3.

Pathways of matrix degradation in fibrosis progression and regression. Macrophages have assumed an important role in matrix degradation, which is profibrogenic during progression of fibrosis but antifibrotic during fibrosis resolution. Although key sources of matrix degrading activity are uncertain, it seems increasingly likely that both scar-associated macrophages and stellate cells are sources of interstitial collagenases. At the same time, decreased TIMP-1 fosters apoptosis of fibrogenic myofibroblasts. Figure adapted from studies of Duffield and Iredale177 (and accompanying editorial, pp 29–32).
Enzymes controlling matrix degradation comprise a family of matrix-metalloproteinases (also known as matrixins), which are calcium-dependent enzymes that specifically degrade collagens and noncollagenous ECM substrates.12,167
Stellate cells are a key source of the basement membrane proteases MMP-2,168 MMP-9,169 and stromelysin (MMP-3),170 as well as the interstitial collagenase MMP-13 (the rodent equivalent of MMP-1).171
A major determinant of progressive fibrosis is failure to degrade the increased fibril-forming, or interstitial, scar matrix. MMP-1 is the main protease that can degrade type I collagen, the principal collagen in fibrotic liver. However, sources of this enzyme are not as clearly established as for the type IV collagenases MMP-2 and MMP-9. Stellate cells express modest levels of MMP-1 mRNA and thus it is uncertain whether this represents the primary interstitial collagenase responsible for matrix resorption as liver fibrosis regresses. Alternative interstitial proteases might include either matrix type 1 MMP or even MMP-2, which also displays some interstitial collagenase activity.
The cross-linking of collagen by lysyl oxidase and tissue transglutaminase, and the maturation of hepatic scar through the action of a disintegrin and metalloproteinase with thrombospondin–type repeats metalloproteinase with thrombospondin type I motif (ADAMTS2) are important determinants of hepatic fibrosis reversibility. The long-standing clinical dogma that the slower the pace of injury, the less reversible the scar, is borne out by animal studies in which even advanced fibrosis of short duration is reversible, which is limited primarily by the extent of collagen cross-linking caused by tissue transglutaminase.172 Moreover, as advanced fibrosis resolves, the micronodules typical of active cirrhosis dissolve, coalescing into macronodules.172 This finding correlates remarkably well with recent clinical data showing that increased septal thickness and smaller nodule size are significant predictors of poorer clinical outcomes.173 Similar studies have been performed in knockout mice lacking ADAMTS2, which catalyzes the N-propeptide excision of procollagens I, III, and V, which allows the polymerization of collagen fibrils.174 In the ADAMTS2 knockout animals, the extent of liver injury after CCl4 was similar to that of wild-type mice, yet fibrosis reversal was slightly faster, associated with much less dense collagen fibrils, and smaller fibril diameters.175 Recently, increased proteolytic activity ascribed to ADAMS-13 also has been reported in activated stellate cells.176 However, the native substrate(s) and biological role of this protease are not known.
Hepatic macrophages are re-emerging as critical regulators of matrix remodeling. An elegant study using a genetic model that allows for the selective, timed depletion of macrophages during different stages of liver injury and resolution has shown divergent roles for these cells.177 During progression of liver fibrosis, macrophages augment fibrogenesis, whereas during resolution they hasten matrix degradation, primarily through increased production of MMP-13.178
Inactivation of proteases by binding to TIMPs also is emerging as an important locus of control12 because sustained production of these proteins during liver injury could inhibit the activity of interstitial collagenases, leading to reduced degradation of the accumulating matrix. In addition, TIMP-1 is anti-apoptotic towards stellate cells,179 and thus its sustained expression in liver injury will enlarge the population of activated stellate cells by preventing their clearance. In support of TIMP’s role in vivo, either transgenic over expression of TIMP-1 in liver, or administration of TIMP-neutralizing antibodies, delay regression of liver fibrosis in experimental animals.180 Conversely, the use of MMP-9 mutant proteins as TIMP-1 scavengers reduces fibrosis accumulation by enhancing matrix resorption.181
Stellate cells express uroplasminogen activator receptor and its inhibitor, as well as other components of the plasmin system.182-184 Collectively, these findings suggest that stellate cells contain most, if not all, of the molecules necessary to either activate or inhibit metalloproteinases.
Clearance of activated stellate cells by apoptosis remains an appealing target for antifibrotic therapy because this would use an endogenous pathway of fibrosis regression. In addition, data from cultured stellate cells185,186 and liver slices187 suggest that activated cells also can revert to a quiescent phenotype, such that this response may be an antifibrotic pathway worth exploiting.
Disease-Specific Pathways of Hepatic Fibrosis
Although key pathways of stellate cell activation are common to all forms of liver injury and fibrosis, disease-specific pathways are being unearthed as well, particularly in ASH and NASH, and in HCV disease.
NASH and ASH
The accelerating obesity epidemic is tied to an increasing prevalence of NASH and subsequent cirrhosis.188 Adipokines mediate fibrogenesis and many hepatic manifestations of obesity.189,190 Specifically, leptin, a circulating adipogenic hormone, promotes stellate cell fibrogenesis and enhances TIMP-1 expression,191-193 which is associated with increased leptin signaling.191 Concurrently, adiponectin, a counter-regulatory hormone that antagonizes the fibrogenic activity of leptin is reduced in hepatic fibrosis,194 and mice lacking adiponectin have enhanced fibrosis after toxic liver injury.195 Equally important to fibrosis is insulin resistance per se, whether associated with steatosis196 or HCV.197,198 As noted earlier, cannabinoids also mediate steatosis, and daily cannabis use is a risk factor for steatosis and fibrosis in HCV.199,200 Interestingly, a recent study directly links CB1 receptor signaling to alcoholic steatosis in rodents fed a liquid ethanol diet.201 Moreover, in these animals, activated stellate cells are a key source of the endogenous cannabinoid, 2-AG, which drives increased CB1 signaling.201 As noted earlier, oxidant stress associated with ethanol metabolism is an important stimulus to fibrogenesis. In contrast, aldehydes, although fibrogenic, are unlikely to account entirely for ethanol-induced fibrosis.15
HCV and HIV
Stellate cells express the putative HCV receptors CD80, LDL receptor, and C1q, raising the possibility of direct HCV infection in vivo, which has not yet been established. Moreover, expression of HCV nonstructural and core proteins induces stellate cell proliferation, release of inflammatory signals,202 and CTGF,203 although interaction of HCV E2 protein stimulates MMP-2 expression.204 Furthermore, hepatocytes harboring replicating HCV in culture produce fibrogenic stimuli towards stellate cells.205 In HCV-infected liver, chemokines promote lymphocyte recruitment.165 HCV proteins also may interact directly with sinusoidal endothelium.206 Remarkably, no studies have reported potential interactions between HBV and stellate cells, or HBV-specific pathways of fibrogenesis.
The increased rate of fibrosis in patients co-infected with HCV and HIV compared with those with HCV alone has been well documented.41 The reduced CD4/CD8 ratio typical of HIV infection has been invoked as a cause, because CD8 cells may be relatively fibrogenic compared with CD4 cells; however, recent preliminary data additionally suggest that hepatic stellate cells may be infected directly by HIV,207 which also might account for why fibrosis progression is slowed when HIV is suppressed by antiretroviral therapy in co-infected patients.208
Links Between Stellate and Progenitor Cells, Fibrosis, and Cancer
The remarkable phenotypic plasticity of stellate cells, combined with the recent demonstration that they express the stem cell marker CD133,209 have raised the fascinating prospect that they are true progenitor cells (Figure 4). Further studies are required to establish bona fide and robust pluripotency, but intriguing possibilities are raised. First, activated stellate cells appear to contribute to the stem cell niche based on histologic studies,210 raising the possibility that they are actually differentiating into stem cells directly. Second, could this cellular behavior provide a missing link between fibrosis and hepatocellular cancer that is derived from stem cells? Although epithelial to mesenchymal transition is well established, the possibility of mesenchymal to epithelial transition, while still quite speculative at this point, should not be ignored. Support for this suggestion includes the presence in stellate cells of both hedgehog211,212 and Wnt signaling,213 two pathways implicated in stem cell differentiation and cancer.214 Moreover, the near-absolute requirement for fibrosis to occur before hepatocellular carcinoma develops in patients with chronic HCV remains completely unexplained. Potential explanations have included the presence of secreted survival factors that prevent apoptosis of DNA-damaged hepatocytes and activated stellate cells (eg, Gas6215), reduced tumor surveillance owing to decreasing natural killer cell number and function, and/or the accelerated shortening of telomeres that accompanies progressive fibrosis. The question of how fibrosis promotes hepatocellular carcinoma is a vital one that demands clearer answers.
Figure 4.

Possible links between stellate cells, fibrosis, regeneration, and cancer. Upon liver injury, activated stellate cells release paracrine factors that may promote progenitor cell expansion, the outcome of which could be either hepatic regeneration and/or promotion of hepatocellular cancer. The possibility also exists that stellate cells may harbor the potential to transdifferentiate into progenitor cells directly, which remains speculative. Moreover, fibrosis promotes hepatocarcinogenesis through unknown mechanisms that may include release of survival signals.
Clinical and Translational Implications
The tightening links between the biology of hepatic fibrosis and clinical expression of disease attest to the importance of continued basic and translational research into mechanisms of hepatic fibrogenesis. In particular, newly uncovered correlations between matrix stiffness and fibrogenesis, ECM cross-linking and reversibility, and both cirrhotic nodule size and septal thickness with clinical outcomes, have emerged from cell culture and animal studies, yet they lead directly to new modes of diagnosis and therapy. Moreover, cirrhosis can no longer be viewed as a single, irreversible end stage of disease but rather as a much broader category subdivided by progressive stages of ECM accumulation, nodule size, portal pressure, reversibility, and clinical risk (Figure 5). More refined and rigorous characterization of cirrhosis will be essential for accurate randomization of patients in clinical trials of antifibrotic therapies and stratification of disease risk, possibly including the risk of hepatocellular carcinoma. Indeed, a key lesson from antifibrotic trials to date is that fibrosis may continue to accrue rapidly even in patients with cirrhosis when therapy is not effective.216 Based on this lesson, it is clear that trials in such cirrhotic patients will need to be lengthy, with the use of more sensitive and specific biomarkers that do not rely on biopsy alone.217 Further progress in understanding, diagnosing, and treating hepatic fibrosis will continue to rely on the exploration of fundamental mechanisms of fibrogenesis, which is certain to lead to a meaningful impact on the prognosis of patients with chronic liver disease.
Figure 5.
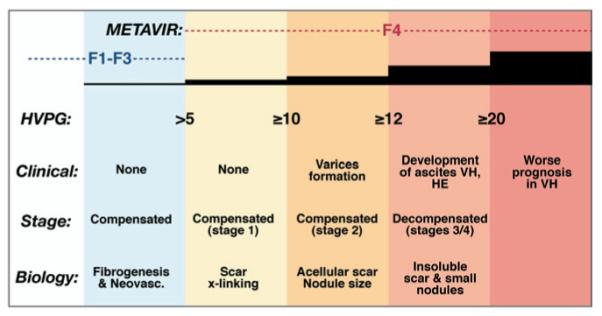
Cirrhosis is a series of progressive stages, not a single stage. Within the spectrum of cirrhosis, the disease is characterized by progressive increases in hepatic venous pressure gradient (hvpg), decompensation, and matrix cross-linking, associated with shrinking nodule size, thickening septae, and enhanced risk of decompensation. For each 1-mm increase in HVPG the risk of decompensation increases by 11%. Concepts presented here are not rigorously supported by primary data for all features, but rather are intended to convey the progressive changes that underlie deterioration in patients with chronic hepatic injury and fibrosis. Stages are based on data from D’Amico et al.218 he, hepatic encephalopathy; vh, variceal hemorrhage.
Acknowledgments
Supported by grants from the National Institutes of Health (DK37330, of DK56621), and the Feld Trust.
The author gratefully acknowledges Drs Guadalupe Garcia-Tsao and Massimo Pinzani, who contributed significantly to the concepts presented in Figure 5.
Abbreviations used in this paper
ADAMTS2
a disintegrin and metalloproteinase with thrombospondin–type repeats metalloproteinase with thrombospondin type I motif
ASH
alcoholic steatohepatitis
CTGF/CCN2
connective tissue growth factor
ECM
extracellular matrix
HIV
human immunodeficiency virus
MMP
matrix metalloproteinase
NASH
nonalcoholic steatohepatitis
PDGF
platelet-derived growth factor
TGF
transforming growth factor
TIMP
tissue inhibitors of metalloproteinase
TLR
Toll-like receptor
VEGF
vascular endothelial growth factor
Biography

References
- 1.Friedman SL, Rockey DC, Bissell DM. Hepatic fibrosis 2006: report of the Third AASLD Single Topic Conference. Hepatology. 2007;45:242–249. doi: 10.1002/hep.21459. [DOI] [PubMed] [Google Scholar]
- 2.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131:1743–1751. doi: 10.1053/j.gastro.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Bruno S, Stroffolini T, Colombo M, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: a retrospective study. Hepatology. 2007;45:579–587. doi: 10.1002/hep.21492. [DOI] [PubMed] [Google Scholar]
- 4.Veldt BJ, Heathcote EJ, Wedemeyer H, et al. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 2007;147:677–684. doi: 10.7326/0003-4819-147-10-200711200-00003. [DOI] [PubMed] [Google Scholar]
- 5.Omary MB, Lugea A, Lowe AW, et al. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 7.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 8.Schluger LK, Sheiner PA, Thung SN, et al. Severe recurrent cholestatic hepatitis C following orthotopic liver transplantation. Hepatology. 1996;23:971–976. doi: 10.1002/hep.510230505. [DOI] [PubMed] [Google Scholar]
- 9.Bonnard P, Lescure FX, Amiel C, et al. Documented rapid course of hepatic fibrosis between two biopsies in patients coinfected by HIV and HCV despite high CD4 cell count. J Viral Hepat. 2007;14:806–811. doi: 10.1111/j.1365-2893.2007.00874.x. [DOI] [PubMed] [Google Scholar]
- 10.Lee JS, Semela D, Iredale J, et al. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 11.Dev A, Patel K, Conrad A, et al. Relationship of smoking and fibrosis in patients with chronic hepatitis C. Clin Gastroenterol Hepatol. 2006;4:797–801. doi: 10.1016/j.cgh.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 12.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 14.De Minicis S, Seki E, Uchinami H, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132:1937–1946. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 15.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 16.Jaeschke H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol. 2006;290:G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 17.Castillo T, Koop DR, Kamimura S, et al. Role of cytochrome P-450 2E1 in ethanol-, carbon tetrachloride- and iron-dependent microsomal lipid peroxidation. Hepatology. 1992;16:992–996. doi: 10.1002/hep.1840160423. [DOI] [PubMed] [Google Scholar]
- 18.Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 19.De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Biophys. 2007;462:266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bataller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nussler AK, Di Silvio M, Billiar TR, et al. Stimulation of the nitric oxide synthase pathway in human hepatocytes by cytokines and endotoxin. J Exp Med. 1992;176:261–264. doi: 10.1084/jem.176.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatraman A, Shiva S, Wigley A, et al. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40:565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 23.Jaeschke H. Inflammation in response to hepatocellular apoptosis. Hepatology. 2002;35:964–966. doi: 10.1053/jhep.2002.0350964. [DOI] [PubMed] [Google Scholar]
- 24.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 25.Zhan SS, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via Toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 27.Takehara T, Tatsumi T, Suzuki T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 28.Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 29.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 30.Taimr P, Higuchi H, Kocova E, et al. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. 2003;37:87–95. doi: 10.1053/jhep.2003.50002. [DOI] [PubMed] [Google Scholar]
- 31.Wright M, Issa R, Smart D, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121:685–698. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 32.Anan A, Baskin-Bey ES, Bronk SF, et al. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–344. doi: 10.1002/hep.21036. [DOI] [PubMed] [Google Scholar]
- 33.Jaeschke H, Gujral JS, Bajt ML. Apoptosis and necrosis in liver disease. Liver Int. 2004;24:85–89. doi: 10.1111/j.1478-3231.2004.0906.x. [DOI] [PubMed] [Google Scholar]
- 34.Wagner H, Bauer S. All is not Toll: new pathways in DNA recognition. J Exp Med. 2006;203:265–268. doi: 10.1084/jem.20052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paik YH, Schwabe RF, Bataller R, et al. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 36.Hua J, Qiu de K, Li JQ, et al. Expression of Toll-like receptor 4 in rat liver during the course of carbon tetrachloride-induced liver injury. J Gastroenterol Hepatol. 2007;22:862–869. doi: 10.1111/j.1440-1746.2007.04896.x. [DOI] [PubMed] [Google Scholar]
- 37.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Shiffman ML, Friedman S, et al. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- 39.Mehal WZ, Friedman SL, Gershwin ME, Veirling JM, Manns MP. Liver immunology. Vol. 2. Humana Press; Totowa, NJ: 2007. The role of inflammation and immunity in the pathogenesis of liver fibrosis; pp. 99–109. [Google Scholar]
- 40.Safadi R, Ohta M, Alvarez CE, et al. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology. 2004;127:870–882. doi: 10.1053/j.gastro.2004.04.062. [DOI] [PubMed] [Google Scholar]
- 41.Benhamou Y, Bochet M, Di Martino V, et al. The Multivirc Group Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. Hepatology. 1999;30:1054–1058. doi: 10.1002/hep.510300409. [DOI] [PubMed] [Google Scholar]
- 42.Radaeva S, Sun R, Jaruga B, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 43.Melhem A, Muhanna N, Bishara A, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 44.Radaeva S, Wang L, Radaev S, et al. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol. 2007;293:G809–G816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 45.Jeong WI, Park O, Gao B. Abrogation of the anti-fibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–258. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liverresident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 47.Chen CH, Kuo LM, Chang Y, et al. In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology. 2006;44:1171–1181. doi: 10.1002/hep.21379. [DOI] [PubMed] [Google Scholar]
- 48.Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115:3072–3082. doi: 10.1172/JCI24798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wells RG, Kruglov E, Dranoff JA. Autocrine release of TGF-beta by portal fibroblasts regulates cell growth. FEBS Lett. 2004;559:107–110. doi: 10.1016/S0014-5793(04)00037-7. [DOI] [PubMed] [Google Scholar]
- 50.Jhandier MN, Kruglov EA, Lavoie EG, et al. Portal fibroblasts regulate the proliferation of bile duct epithelia via expression of NTPDase2. J Biol Chem. 2005;280:22986–22992. doi: 10.1074/jbc.M412371200. [DOI] [PubMed] [Google Scholar]
- 51.Beaussier M, Wendum D, Schiffer E, et al. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest. 2007;87:292–303. doi: 10.1038/labinvest.3700513. [DOI] [PubMed] [Google Scholar]
- 52.Kisseleva T, Uchinami H, Feirt N, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 53.Forbes SJ, Russo FP, Rey V, et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126:955–963. doi: 10.1053/j.gastro.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 54.Kruglov EA, Nathanson RA, Nguyen T, et al. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol. 2006;290:G765–G771. doi: 10.1152/ajpgi.00308.2005. [DOI] [PubMed] [Google Scholar]
- 55.Dranoff JA, Ogawa M, Kruglov EA, et al. Expression of P2Y nucleotide receptors and ectonucleotidases in quiescent and activated rat hepatic stellate cells. Am J Physiol. 2004;287:G417–G424. doi: 10.1152/ajpgi.00294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sakaida I, Terai S, Yamamoto N, et al. Transplantation of bone marrow cells reduces CCl(4)-induced liver fibrosis in mice. Hepatology. 2004;40:1304–1311. doi: 10.1002/hep.20452. [DOI] [PubMed] [Google Scholar]
- 57.Terai S, Ishikawa T, Omori K, et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells. 2006;24:2292–2298. doi: 10.1634/stemcells.2005-0542. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura T, Torimura T, Sakamoto M, et al. Significance and therapeutic potential of endothelial progenitor cell transplantation in a cirrhotic liver rat model. Gastroenterology. 2007;133:91–107. e1. doi: 10.1053/j.gastro.2007.03.110. [DOI] [PubMed] [Google Scholar]
- 59.Wang B, Dolinski BM, Kikuchi N, et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jarnagin WR, Rockey DC, Koteliansky VE, et al. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol. 1994;127:2037–2048. doi: 10.1083/jcb.127.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.George J, Wang SS, Sevcsik AM, et al. Transforming growth factor-beta initiates wound repair in rat liver through induction of the EIIIA-fibronectin splice isoform. Am J Pathol. 2000;156:115–124. doi: 10.1016/s0002-9440(10)64711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang C, Zeisberg M, Mosterman B, et al. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology. 2003;124:147–159. doi: 10.1053/gast.2003.50012. [DOI] [PubMed] [Google Scholar]
- 63.Zhou X, Murphy FR, Gehdu N, et al. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279:23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 64.Melton AC, Soon RK, Jr, Park JG, et al. Focal adhesion disassembly is an essential early event in hepatic stellate cell chemotaxis. Am J Physiol. 2007;293:G1272–G1280. doi: 10.1152/ajpgi.00134.2007. [DOI] [PubMed] [Google Scholar]
- 65.Shafiei MS, Rockey DC. The role of integrin-linked kinase in liver wound healing. J Biol Chem. 2006;281:24863–24872. doi: 10.1074/jbc.M513544200. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Ikegami T, Honda A, et al. Involvement of integrinlinked kinase in carbon tetrachloride-induced hepatic fibrosis in rats. Hepatology. 2006;44:612–622. doi: 10.1002/hep.21315. [DOI] [PubMed] [Google Scholar]
- 67.Yee HF., Jr. Rho directs activation-associated changes in rat hepatic stellate cell morphology via regulation of the actin cytoskeleton. Hepatology. 1998;28:843–850. doi: 10.1002/hep.510280336. [DOI] [PubMed] [Google Scholar]
- 68.Choi SS, Sicklick JK, Ma Q, et al. Sustained activation of Rac1 in hepatic stellate cells promotes liver injury and fibrosis in mice. Hepatology. 2006;44:1267–1277. doi: 10.1002/hep.21375. [DOI] [PubMed] [Google Scholar]
- 69.Schuppan D, Schmid M, Somasundaram R, et al. Collagens in the liver extracellular matrix bind hepatocyte growth factor. Gastroenterology. 1998;114:139–152. doi: 10.1016/s0016-5085(98)70642-0. [DOI] [PubMed] [Google Scholar]
- 70.Schuppan D, Ruehl M, Somasundaram R, et al. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 71.Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 72.Georges PC, Hui JJ, Gombos Z, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol. 2007;293:G1147–G1154. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 73.Vizzutti F, Arena U, Romanelli RG, et al. Liver stiffness measurement predicts severe portal hypertension in patients with HCV-related cirrhosis. Hepatology. 2007;45:1290–1297. doi: 10.1002/hep.21665. [DOI] [PubMed] [Google Scholar]
- 74.Kazemi F, Kettaneh A, N’Kontchou G, et al. Liver stiffness measurement selects patients with cirrhosis at risk of bearing large oesophageal varices. J Hepatol. 2006;45:230–235. doi: 10.1016/j.jhep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 75.Rockey DC. Noninvasive assessment of liver fibrosis and portal hypertension with transient elastography. Gastroenterology. 2008;134:8–14. doi: 10.1053/j.gastro.2007.11.053. [DOI] [PubMed] [Google Scholar]
- 76.Yin M, Talwalkar JA, Glaser KJ, et al. Assessment of hepatic fibrosis with magnetic resonance elastography. Clin Gastroenterol Hepatol. 2007;5:1207–1213. e2. doi: 10.1016/j.cgh.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Talwalkar JA, Yin M, Fidler JL, et al. Magnetic resonance imaging of hepatic fibrosis: emerging clinical applications. Hepatology. 2008;47:332–342. doi: 10.1002/hep.21972. [DOI] [PubMed] [Google Scholar]
- 78.Valencia-Sanchez MA, Liu J, Hannon GJ, et al. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 79.Rippe RA, Brenner DA. From quiescence to activation: gene regulation in hepatic stellate cells. Gastroenterology. 2004;127:1260–1262. doi: 10.1053/j.gastro.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 80.Mann J, Oakley F, Akiboye F, et al. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14:275–285. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 81.Tsukamoto H, She H, Hazra S, et al. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21(Suppl 3):S102–S105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- 82.Kalinichenko VV, Bhattacharyya D, Zhou Y, et al. Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–117. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- 83.Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 84.Smart DE, Green K, Oakley F, et al. JunD is a profibrogenic transcription factor regulated by Jun N-terminal kinase-independent phosphorylation. Hepatology. 2006;44:1432–1440. doi: 10.1002/hep.21436. [DOI] [PubMed] [Google Scholar]
- 85.Buck M, Poli V, Hunter T, et al. C/EBPbeta phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol Cell. 2001;8:807–816. doi: 10.1016/s1097-2765(01)00374-4. [DOI] [PubMed] [Google Scholar]
- 86.Wandzioch E, Kolterud A, Jacobsson M, et al. Lhx2−/− mice develop liver fibrosis. Proc Natl Acad Sci U S A. 2004;101:16549–16554. doi: 10.1073/pnas.0404678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adachi M, Osawa Y, Uchinami H, et al. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology. 2007;132:1434–1446. doi: 10.1053/j.gastro.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 88.Yavrom S, Chen L, Xiong S, et al. Peroxisome proliferator-activated receptor gamma suppresses proximal alpha1(I) collagen promoter via inhibition of p300-facilitated NF-I binding to DNA in hepatic stellate cells. J Biol Chem. 2005;280:40650–40659. doi: 10.1074/jbc.M510094200. [DOI] [PubMed] [Google Scholar]
- 89.Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 90.Haughton EL, Tucker SJ, Marek CJ, et al. Pregnane x receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology. 2006;131:194–209. doi: 10.1053/j.gastro.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 91.Marek CJ, Tucker SJ, Konstantinou DK, et al. Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem J. 2005;387:601–608. doi: 10.1042/BJ20041598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okuno M, Sato T, Kitamoto T, et al. Increased 9,13-di-cis-retinoic acid in rat hepatic fibrosis: implication for a potential link between retinoid loss and TGF-beta mediated fibrogenesis in vivo. J Hepatol. 1999;30:1073–1080. doi: 10.1016/s0168-8278(99)80262-1. [DOI] [PubMed] [Google Scholar]
- 93.Hellemans K, Grinko I, Rombouts K, et al. All-trans and 9-cis retinoic acid alter rat hepatic stellate cell phenotype differentially. Gut. 1999;45:134–142. doi: 10.1136/gut.45.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oakley F, Meso M, Iredale JP, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128:108–120. doi: 10.1053/j.gastro.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 95.Mann J, Oakley F, Akiboye F, et al. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2006;14:275–285. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 96.Habens F, Srinivasan N, Oakley F, et al. Novel sulfasalazine analogues with enhanced NF-kB inhibitory and apoptosis promoting activity. Apoptosis. 2005;10:481–491. doi: 10.1007/s10495-005-1877-0. [DOI] [PubMed] [Google Scholar]
- 97.Stefanovic B, Hellerbrand C, Holcik M, et al. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol Cell Biol. 1997;17:5201–5209. doi: 10.1128/mcb.17.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stefanovic B, Hellerbrand C, Brenner DA. Regulatory role of the conserved stem-loop structure at the 5′ end of collagen alpha1(I) mRNA. Mol Cell Biol. 1999;19:4334–4342. doi: 10.1128/mcb.19.6.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fritz D, Stefanovic B. RNA-binding protein RBMS3 is expressed in activated hepatic stellate cells and liver fibrosis and increases expression of transcription factor Prx1. J Mol Biol. 2007;371:585–595. doi: 10.1016/j.jmb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Borkham-Kamphorst E, van Roeyen CR, Ostendorf T, et al. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol. 2007;46:1064–1074. doi: 10.1016/j.jhep.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 101.Wong L, Yamasaki G, Johnson RJ, et al. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–1569. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pinzani M, Milani S, Grappone C, et al. Expression of platelet-derived growth factor in a model of acute liver injury. Hepatology. 1994;19:701–707. doi: 10.1002/hep.1840190323. [DOI] [PubMed] [Google Scholar]
- 103.Czochra P, Klopcic B, Meyer E, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 104.Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416. doi: 10.1055/s-2001-17554. [DOI] [PubMed] [Google Scholar]
- 106.Lechuga CG, Hernandez-Nazara ZH, Hernandez E, et al. PI3K is involved in PDGF-beta receptor upregulation post-PDGF-BB treatment in mouse HSC. Am J Physiol. 2006;291:G1051–G1061. doi: 10.1152/ajpgi.00058.2005. [DOI] [PubMed] [Google Scholar]
- 107.Rovida E, Navari N, Caligiuri A, et al. ERK5 differentially regulates PDGF-induced proliferation and migration of hepatic stellate cells. J Hepatol. 2008;48:107–115. doi: 10.1016/j.jhep.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 108.Di Sario A, Bendia E, Taffetani S, et al. Selective Na+/H+ exchange inhibition by cariporide reduces liver fibrosis in the rat. Hepatology. 2003;37:256–266. doi: 10.1053/jhep.2003.50028. [DOI] [PubMed] [Google Scholar]
- 109.Yoshiji H, Kuriyama S, Yoshii J, et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut. 2003;52:1347–1354. doi: 10.1136/gut.52.9.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marra F, Grandaliano G, Valente AJ, et al. Thrombin stimulates proliferation of liver fat-storing cells and expression of monocyte chemotactic protein-1: potential role in liver injury. Hepatology. 1995;22:780–787. [PubMed] [Google Scholar]
- 111.Marra F, DeFranco R, Grappone C, et al. Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury. Hepatology. 1998;27:462–471. doi: 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]
- 112.Steiling H, Muhlbauer M, Bataille F, et al. Activated hepatic stellate cells express keratinocyte growth factor in chronic liver disease. Am J Pathol. 2004;165:1233–1241. doi: 10.1016/S0002-9440(10)63383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu C, Wang F, Jin C, et al. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am J Pathol. 2003;163:1653–1662. doi: 10.1016/S0002-9440(10)63522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ikeda K, Wakahara T, Wang YQ, et al. In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology. 1999;29:1760–1767. doi: 10.1002/hep.510290640. [DOI] [PubMed] [Google Scholar]
- 115.Kinnman N, Hultcrantz R, Barbu V, et al. PDGF-mediated chemoattraction of hepatic stellate cells by bile duct segments in cholestatic liver injury. Lab Invest. 2000;80:697–707. doi: 10.1038/labinvest.3780073. [DOI] [PubMed] [Google Scholar]
- Marra F, Romanelli RG, Giannini C, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29:140–148. doi: 10.1002/hep.510290107. [DOI] [PubMed] [Google Scholar]
- 117.Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–9954. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- 118.Melton AC, Yee HF. Hepatic stellate cell protrusions couple platelet-derived growth factor-BB to chemotaxis. Hepatology. 2007;45:1446–1453. doi: 10.1002/hep.21606. [DOI] [PubMed] [Google Scholar]
- 119.Hashmi AZ, Hakim W, Kruglov EA, et al. Adenosine inhibits cytosolic calcium signals and chemotaxis in hepatic stellate cells. Am J Physiol. 2007;292:G395–G401. doi: 10.1152/ajpgi.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chan ES, Montesinos MC, Fernandez P, et al. Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol. 2006;148:1144–1155. doi: 10.1038/sj.bjp.0706812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ruhl CE, Everhart JE. Coffee and tea consumption are associated with a lower incidence of chronic liver disease in the United States. Gastroenterology. 2005;129:1928–1936. doi: 10.1053/j.gastro.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 122.Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 123.Stefanovic B, Stefanovic L, Schnabl B, et al. TRAM2 protein interacts with endoplasmic reticulum Ca2+ pump Serca2b and is necessary for collagen type I synthesis. Mol Cell Biol. 2004;24:1758–1768. doi: 10.1128/MCB.24.4.1758-1768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Inagaki Y, Okazaki I. Emerging insights into transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Breitkopf K, Godoy P, Ciuclan L, et al. TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006;44:57–66. doi: 10.1055/s-2005-858989. [DOI] [PubMed] [Google Scholar]
- 126.Liu C, Gaca MD, Swenson ES, et al. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J Biol Chem. 2003;278:11721–11728. doi: 10.1074/jbc.M207728200. [DOI] [PubMed] [Google Scholar]
- 127.Uemura M, Swenson ES, Gaca MD, et al. Smad2 and Smad3 play different roles in rat hepatic stellate cell function and alpha-smooth muscle actin organization. Mol Biol Cell. 2005;16:4214–4224. doi: 10.1091/mbc.E05-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wiercinska E, Wickert L, Denecke B, et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology. 2006;43:1032–1041. doi: 10.1002/hep.21135. [DOI] [PubMed] [Google Scholar]
- 129.Garcia-Trevijano ER, Iraburu MJ, Fontana L, et al. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 130.Dooley S, Delvoux B, Lahme B, et al. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology. 2000;31:1094–1106. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]
- 131.Paradis V, Dargere D, Bonvoust F, et al. Effects and regulation of connective tissue growth factor on hepatic stellate cells. Lab Invest. 2002;82:767–774. doi: 10.1097/01.lab.0000017365.18894.d3. [DOI] [PubMed] [Google Scholar]
- 132.Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res. 2003;26:1–9. doi: 10.1016/s1386-6346(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 133.Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem. 2004;279:8848–8855. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- 134.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 135.Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8:171–179. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 136.Brigstock DR. The CCN family: a new stimulus package. J Endocrinol. 2003;178:169–175. doi: 10.1677/joe.0.1780169. [DOI] [PubMed] [Google Scholar]
- 137.Weng HL, Ciuclan L, Liu Y, et al. Profibrogenic transforming growth factor-beta/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology. 2007;46:1257–1270. doi: 10.1002/hep.21806. [DOI] [PubMed] [Google Scholar]
- 138.Gressner OA, Lahme B, Demirci I, et al. Differential effects of TGF-beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J Hepatol. 2007;47:699–710. doi: 10.1016/j.jhep.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 139.Roskams T, Cassiman D, De Vos R, et al. Neuroregulation of the neuroendocrine compartment of the liver. Anat Rec. 2004;280:910–923. doi: 10.1002/ar.a.20096. [DOI] [PubMed] [Google Scholar]
- 140.Mallat A, Teixeira-Clerc F, Deveaux V, et al. Cannabinoid receptors as new targets of antifibrosing strategies during chronic liver diseases. Expert Opin Ther Targets. 2007;11:403–409. doi: 10.1517/14728222.11.3.403. [DOI] [PubMed] [Google Scholar]
- 141.Batkai S, Jarai Z, Wagner JA, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 142.Teixeira-Clerc F, Julien B, Grenard P, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 143.Munoz-Luque J, Ros J, Fernandez-Varo G, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324:475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ebrahimkhani MR, Kiani S, Oakley F, et al. Naltrexone, an opioid receptor antagonist, attenuates liver fibrosis in bile duct ligated rats. Gut. 2006 doi: 10.1136/gut.2005.076778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.De Minicis S, Candelaresi C, Marzioni M, et al. Role of endogenous opioids in modulating Hsc activity in vitro and liver fibrosis in vivo. Gut. 2008;57:352–364. doi: 10.1136/gut.2007.120303. [DOI] [PubMed] [Google Scholar]
- 146.Oben JA, Roskams T, Yang S, et al. Hepatic fibrogenesis requires sympathetic neurotransmitters. Gut. 2004;53:438–445. doi: 10.1136/gut.2003.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rockey DC, Boyles JK, Gabbiani G, et al. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol. 1992;24:193–203. [PubMed] [Google Scholar]
- 148.Saab S, Tam SP, Tran BN, et al. Myosin mediates contractile force generation by hepatic stellate cells in response to endothelin-1. J Biomed Sci. 2002;9:607–612. doi: 10.1159/000067289. [DOI] [PubMed] [Google Scholar]
- 149.Bataller R, Gasull X, Gines P, et al. In vitro and in vivo activation of rat hepatic stellate cells results in de novo expression of L-type voltage-operated calcium channels. Hepatology. 2001;33:956–962. doi: 10.1053/jhep.2001.23500. [DOI] [PubMed] [Google Scholar]
- 150.Yee HF., Jr. Ca2+ and rho signaling pathways: two paths to hepatic stellate cell contraction. Hepatology. 2001;33:1007–1008. doi: 10.1053/jhep.2001.23901. [DOI] [PubMed] [Google Scholar]
- 151.Laleman W, Van Landeghem L, Severi T, et al. Both Ca2+-dependent and -independent pathways are involved in rat hepatic stellate cell contraction and intrahepatic hyperresponsiveness to methoxamine. Am J Physiol Gastrointest Liver Physiol. 2007;292:G556–G564. doi: 10.1152/ajpgi.00196.2006. [DOI] [PubMed] [Google Scholar]
- 152.Fischer R, Cariers A, Reinehr R, Haussinger D. Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology. 2002;123:845–861. doi: 10.1053/gast.2002.35384. [DOI] [PubMed] [Google Scholar]
- 153.Martinez-Hernandez A, Amenta PS. The extracellular matrix in hepatic regeneration. FASEB J. 1995;9:1401–1410. doi: 10.1096/fasebj.9.14.7589981. [DOI] [PubMed] [Google Scholar]
- 154.Olaso E, Salado C, Egilegor E, et al. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003;37:674–685. doi: 10.1053/jhep.2003.50068. [DOI] [PubMed] [Google Scholar]
- 155.Rockey DC. Vascular mediators in the injured liver. Hepatology. 2003;37:4–12. doi: 10.1053/jhep.2003.50044. [DOI] [PubMed] [Google Scholar]
- 156.Pinzani M, Failli P, Ruocco C, et al. Fat-storing cells as liver-specific pericytes. Spatial dynamics of agonist-stimulated intracellular calcium transients. J Clin Invest. 1992;90:642–646. doi: 10.1172/JCI115905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Melton AC, Datta A, Yee HF., Jr. [Ca2+]i-independent contractile force generation by rat hepatic stellate cells in response to endothelin-1. Am J Physiol. 2006;290:G7–G13. doi: 10.1152/ajpgi.00337.2005. [DOI] [PubMed] [Google Scholar]
- 158.Reynaert H, Thompson MG, Thomas T, et al. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut. 2002;50:571–581. doi: 10.1136/gut.50.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sherman IA, Pappas SC, Fisher MM. Hepatic microvascular changes associated with development of liver fibrosis and cirrhosis. Am J Physiol. 1990;258:H460–H465. doi: 10.1152/ajpheart.1990.258.2.H460. [DOI] [PubMed] [Google Scholar]
- 160.Nakata M, Nakamura K, Koda Y, et al. Hemodynamics in the microvasculature of thioacetamide-induced cirrhotic rat livers. Hepatogastroenterology. 2002;49:652–656. [PubMed] [Google Scholar]
- 161.Tugues S, Fernandez-Varo G, Munoz-Luque J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46:1919–1926. doi: 10.1002/hep.21921. [DOI] [PubMed] [Google Scholar]
- 162.Takeji M, Moriyama T, Oseto S, et al. Smooth muscle alpha-actin deficiency in myofibroblasts leads to enhanced renal tissue fibrosis. J Biol Chem. 2006;281:40193–40200. doi: 10.1074/jbc.M602182200. [DOI] [PubMed] [Google Scholar]
- 163.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 164.Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci. 2002;7:d1899–d1914. doi: 10.2741/A887. [DOI] [PubMed] [Google Scholar]
- 165.Bonacchi A, Petrai I, Defranco RM, et al. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003;125:1060–1076. doi: 10.1016/s0016-5085(03)01194-6. [DOI] [PubMed] [Google Scholar]
- 166.Friedman SL. Hepatic stellate cells—protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 168.Arthur MJ, Stanley A, Iredale JP, et al. Secretion of 72 kDa type IV collagenase/gelatinase by cultured human lipocytes. Analysis of gene expression, protein synthesis and proteinase activity. Biochem J. 1992;287:701–707. doi: 10.1042/bj2870701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Han YP, Yan C, Zhou L, et al. A matrix metalloproteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J Biol Chem. 2007;282:12928–12939. doi: 10.1074/jbc.M700554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vyas SK, Leyland H, Gentry J, et al. Rat hepatic lipocytes synthesize and secrete transin (stromelysin) in early primary culture. Gastroenterology. 1995;109:889–898. doi: 10.1016/0016-5085(95)90399-2. [DOI] [PubMed] [Google Scholar]
- 171.Schaefer B, Rivas-Estilla AM, Meraz-Cruz N, et al. Reciprocal modulation of matrix metalloproteinase-13 and type I collagen genes in rat hepatic stellate cells. Am J Pathol. 2003;162:1771–1780. doi: 10.1016/S0002-9440(10)64312-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–1808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 173.Nagula S, Jain D, Groszmann RJ, et al. Histological-hemodynamic correlation in cirrhosis—a histological classification of the severity of cirrhosis. J Hepatol. 2006;44:111–117. doi: 10.1016/j.jhep.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 174.Li SW, Arita M, Fertala A, et al. Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem J. 2001;355:271–278. doi: 10.1042/0264-6021:3550271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kesteloot F, Desmouliere A, Leclercq I, et al. ADAM metallopeptidase with thrombospondin type 1 motif 2 inactivation reduces the extent and stability of carbon tetrachloride-induced hepatic fibrosis in mice. Hepatology. 2007;46:1620–1631. doi: 10.1002/hep.21868. [DOI] [PubMed] [Google Scholar]
- 176.Niiya M, Uemura M, Zheng XW, et al. Increased ADAMTS-13 proteolytic activity in rat hepatic stellate cells upon activation in vitro and in vivo. J Thromb Haemost. 2006;4:1063–1070. doi: 10.1111/j.1538-7836.2006.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 179.Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–11076. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 180.Yoshiji H, Kuriyama S, Yoshii J, et al. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology. 2002;36:850–860. doi: 10.1053/jhep.2002.35625. [DOI] [PubMed] [Google Scholar]
- 181.Roderfeld M, Weiskirchen R, Wagner S, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006;20:444–454. doi: 10.1096/fj.05-4828com. [DOI] [PubMed] [Google Scholar]
- 182.Knittel T, Fellmer P, Ramadori G. Gene expression and regulation of plasminogen activator inhibitor type I in hepatic stellate cells of rat liver. Gastroenterology. 1996;111:745–754. doi: 10.1053/gast.1996.v111.pm8780581. [DOI] [PubMed] [Google Scholar]
- 183.Zhang LP, Takahara T, Yata Y, et al. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol. 1999;31:703–711. doi: 10.1016/s0168-8278(99)80351-1. [DOI] [PubMed] [Google Scholar]
- 184.Fibbi G, Pucci M, Grappone C, et al. Functions of the fibrinolytic system in human Ito cells and its control by basic fibroblast and platelet-derived growth factor. Hepatology. 1999;29:868–878. doi: 10.1002/hep.510290343. [DOI] [PubMed] [Google Scholar]
- 185.Olaso E, Ikeda K, Eng FJ, et al. DDR2 receptor MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108:1369–1378. doi: 10.1172/JCI12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gaca MD, Zhou X, Issa R, et al. Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol. 2003;22:229–239. doi: 10.1016/s0945-053x(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 187.Guyot C, Combe C, Balabaud C, et al. Fibrogenic cell fate during fibrotic tissue remodelling observed in rat and human cultured liver slices. J Hepatol. 2007;46:142–150. doi: 10.1016/j.jhep.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 188.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 189.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74. viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 190.Ikejima K, Okumura K, Kon K, et al. Role of adipocytokines in hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S87–S92. doi: 10.1111/j.1440-1746.2007.04961.x. [DOI] [PubMed] [Google Scholar]
- 191.Ikejima K, Takei Y, Honda H, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 192.Leclercq IA, Farrell GC, Schriemer R, et al. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 193.Lin S, Saxena NK, Ding X, et al. Leptin increases tissue inhibitor of metalloproteinase I (TIMP-1) gene expression by a specificity protein 1/signal transducer and activator of transcription mechanism. Mol Endocrinol. 2006;20:3376–3388. doi: 10.1210/me.2006-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Marra F, Aleffi S, Bertolani C, et al. Adipokines and liver fibrosis. Eur Rev Med Pharmacol Sci. 2005;9:279–284. [PubMed] [Google Scholar]
- 195.Kamada Y, Tamura S, Kiso S, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 196.Lonardo A, Adinolfi LE, Loria P, et al. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586–597. doi: 10.1053/j.gastro.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 197.Fartoux L, Poujol-Robert A, Guechot J, et al. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut. 2005;54:1003–1008. doi: 10.1136/gut.2004.050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Moucari R, Asselah T, Cazals-Hatem D, et al. Insulin resistance in chronic hepatitis C: association with genotypes I and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology. 2008;134:416–423. doi: 10.1053/j.gastro.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 199.Hezode C, Roudot-Thoraval F, Nguyen S, et al. Daily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis C. Hepatology. 2005;42:63–71. doi: 10.1002/hep.20733. [DOI] [PubMed] [Google Scholar]
- 200.Hezode C, Zafrani ES, Roudot-Thoraval F, et al. Daily cannabis use: a novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134:432–439. doi: 10.1053/j.gastro.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 201.Jeong WI, Osei-Hyiaman D, Park O, et al. Paracrine activation of hepatic CB(1) receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 202.Bataller R, Paik YH, Lindquist JN, et al. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529–540. doi: 10.1053/j.gastro.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 203.Shin JY, Hur W, Wang JS, et al. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-beta1. Exp Mol Med. 2005;37:138–145. doi: 10.1038/emm.2005.19. [DOI] [PubMed] [Google Scholar]
- 204.Mazzocca A, Sciammetta SC, Carloni V, et al. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem. 2005;280:11329–11339. doi: 10.1074/jbc.M410161200. [DOI] [PubMed] [Google Scholar]
- 205.Schulze-Krebs A, Preimel D, Popov Y, et al. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology. 2005;129:246–258. doi: 10.1053/j.gastro.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 206.Pohlmann S, Zhang J, Baribaud F, et al. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tuyama A, Hong F, Mosoian A, et al. HIV entry and replication in stellate cells promotes cellular activation and fibrogenesis: implications for hepatic fibrosis in HIV/HCV co-infection. Hepatology. 2007;46:80A. [Google Scholar]
- 208.Brau N, Salvatore M, Rios-Bedoya CF, et al. for the Puerto Rico-New York Hepatitis Study G. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J Hepatol. 2006;44:47–55. doi: 10.1016/j.jhep.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 209.Kordes C, Sawitza I, Muller-Marbach A, et al. CD133+ hepatistellate in cells are progenitor cells. Biochem Biophys Res Commun. 2007;352:410–417. doi: 10.1016/j.bbrc.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 210.Roskams T. Different types of liver progenitor cells and niches. J Hepatol. 2006;45:1–4. doi: 10.1016/j.jhep.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 211.Sicklick JK, Li YX, Choi SS, et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab Invest. 2005;85:1368–1380. doi: 10.1038/labinvest.3700349. [DOI] [PubMed] [Google Scholar]
- 212.Yang L, Wang Y, Mao H, et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J Hepatol. 2008;48:98–106. doi: 10.1016/j.jhep.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Myung SJ, Yoon JH, Gwak GY, et al. Wnt signaling inhibitor activation and survival of human hepatic stellate cells. FEBS Lett. 2007;581:2954–2958. doi: 10.1016/j.febslet.2007.05.050. [DOI] [PubMed] [Google Scholar]
- 214.Taipale J, Beachy PA. The Hedgehog and Wnt signalling ways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 215.Lafdil F, Chobert MN, Couchie D, et al. Induction of Gas6 in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology. 2006;44:228–239. doi: 10.1002/hep.21237. [DOI] [PubMed] [Google Scholar]
- 216.Goodman ZD, Becker RL, Jr, Pockros PJ, et al. Progression of fibrosis in advanced chronic hepatitis C: evaluation by morphometric image analysis. Hepatology. 2007;45:886–894. doi: 10.1002/hep.21595. [DOI] [PubMed] [Google Scholar]
- 217.Pockros PJ, Jeffers L, Afdhal N, et al. Final results of a doubleblind, placebo-controlled trial of the anti-fibrotic efficacy of interisferon-gamma1b in chronic hepatitis C patients with advanced hepafibrosis or cirrhosis. Hepatology. 2007;45:569–578. doi: 10.1002/hep.21561. [DOI] [PubMed] [Google Scholar]
- 218.D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 4, 118 studies. J Hepatol. 2006;44:217–231. doi: 10.1016/j.jhep.2005.10.013. [DOI] [PubMed] [Google Scholar]