S-Nitrosylation of DRP1 Does Not Affect Enzymatic Activity and is Not Specific to Alzheimer's Disease (original) (raw)
. Author manuscript; available in PMC: 2011 Jan 1.
Published in final edited form as: J Alzheimers Dis. 2010;20(Suppl 2):S513–S526. doi: 10.3233/JAD-2010-100552
Abstract
Mitochondrial dysfunction and synaptic loss are among the earliest events linked to Alzheimer's disease (AD) and might play a causative role in disease onset and progression. The underlying mechanisms of mitochondrial and synaptic dysfunction in AD remain unclear. We previously reported that nitric oxide (NO) triggers persistent mitochondrial fission and causes neuronal cell death. A recent article claimed that S-nitrosylation of dynamin related protein 1 (DRP1) at cysteine 644 causes protein dimerization and increased GTPase activity and is the mechanism responsible for NO-induced mitochondrial fission and neuronal injury in AD, but not in Parkinson's disease (PD). However, this report remains controversial. To resolve the controversy, we investigated the effects of S-nitrosylation on DRP1 structure and function. Contrary to the previous report, S-nitrosylation of DRP1 does not increase GTPase activity or cause dimerization. In fact, DRP1 does not exist as a dimer under native conditions, but rather as a tetramer capable of self-assembly into higher order spiral- and ring-like oligomeric structures after nucleotide binding. S-nitrosylation, as confirmed by the biotin-switch assay, has no impact on DRP1 oligomerization. Importantly, we found no significant difference in S-nitrosylated DRP1 (SNO-DRP1) levels in brains of age-matched normal, AD, or PD patients. We also found that S-nitrosylation is not specific to DRP1 because S-nitrosylated optic atrophy 1 (SNO-OPA1) is present at comparable levels in all human brain samples. Finally, we show that NO triggers DRP1 phosphorylation at serine 616, which results in its activation and recruitment to mitochondria. Our data indicate the mechanism underlying nitrosative stress-induced mitochondrial fragmentation in AD is not DRP1 S-nitrosylation.
Keywords: large GTPases, mitochondrial fission and fusion, nitrosative stress, OPA1, SNO-DRP1, synapses
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder worldwide. Clinically, AD is a late-onset disease characterized by progressive loss of memory, language, and decision making skills due to loss of neurons in the hippocampus and cortex. Brain tissue of afflicted individuals exhibit two characteristic insoluble protein deposits: extracellular senile plaques composed primarily of amyloid-β (Aβ) peptide and intracellular neurofibrillary tangles consisting of hyperphosphorylated tau protein.
Oxidative stress from mitochondrial dysfunction is among the earliest changes in AD. Oxidative damage precedes the appearance of senile plaques in AD patients and AD-like transgenic models [1-3]. Oxidative stress can exacerbate intracellular Aβ accumulation by activating signal transduction pathways that induce amyloid-β protein precursor (AβPP) processing. Specifically, oxidative stress activates Jun amino-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), which increases AβPP processing [4]. Furthermore, mtDNA mutations have been found in AD brain tissue, which might contribute to reduced energy metabolism and exacerbated ROS production [5]. Axonal trafficking of mitochondria is also reduced in AD [6-8]. Finally, there is evidence that Aβ can directly injure mitochondria. For example, Aβ can block mitochondrial protein import and inhibit mitochondrial enzymes such as α-ketoglutarate dehydrogenase and cytochrome c oxidase [9-12]. Aβ also forms abnormal protein complexes with mitochondrial proteins such as Aβ-binding alcohol dehydrogenase (ABAD) or the serine protease OMI [13, 14], both of which are thought to contribute to neuronal injury and cell death in AD.
Nitric oxide (NO) is an important neurotransmitter and neuromodulator and is produced in glia and neurons by enzymes called nitric oxide synthases (NOS). When generated in excess and under pathophysiological conditions, NO becomes neurotoxic by creating reactive nitrogen species (RNS) that can react with cysteine residues in proteins forming covalent thiol derivates (R-SNO). Such protein modification, known as S-nitrosylation, alters protein structure and function and can result in protein aggregation, a hallmark of many chronic neurodegenerative disorders. Nitrosative stress caused by RNS is not specific to AD but is thought to play an important role in many neurodegenerative disorders including Parkinson's disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and stroke [15]. Nitrosative stress in AD can originate from inflammation, glial cell activation, and cytokine release that activate inducible nitric oxide synthase (iNOS) or from accumulation of excitatory amino acids that over-stimulate glutamate receptors leading to neuronal nitric oxide synthase (nNOS) activation [15].
Neurons are highly dependent on mitochondrial function for energy supply. Mitochondria in neurons are dynamic and migrate, divide, and fuse. These processes are thought to facilitate energy distribution throughout neuronal projections and to sites of high energy demand such as synapses, to compensate for mtDNA mutations, to mix metabolites, and to maintain bioenergetic functionality. Mitochondrial fission and fusion is regulated by a family of large GTPases, with dynamin-related protein 1 (DRP1) directing fission and the mitofusins (MFN1, MFN2) and optic atrophy 1 (OPA1) directing fusion [16-23]. Excessive mitochondrial fission or insufficient mitochondrial fusion causes mitochondrial fragmentation, which decreases respiration and energy production, results in loss of mitochondrial membrane potential and mtDNA, and increases vulnerability to neuronal injury and cell death [24-26].
We previously showed that Aβ peptide induces mitochondrial fragmentation in cortical neurons [27], which was later confirmed by others [28]. Furthermore, using an animal model of acute ischemic brain injury we detected mitochondrial fragmentation prior to neuronal cell death in vivo. In addition, nitrosative stress is known to play an important role in ischemic stroke triggered mitochondrial fragmentation in isolated neurons [27, 29]. Using time-lapse microscopy, we observed that the NO-induced mitochondrial fragmentation can be reversible and is insufficient to cause cell death. Thus, short-term NO-induced fragmentation and mitophagy might reflect a stress response to repair or eliminate damage and thereby increase cell survival [27, 29, 30]. However, long-term, persistent mitochondrial fragmentation results in bio-energetic failure, additional oxidative stress, and secondary activation of signaling pathways, all of which cause neuronal cell death [27, 29, 30].
The precise mechanism underlying NO-induced mitochondrial fragmentation remains unclear. Expanding on our original line of investigation, a recent report proposed that S-nitrosylation of DRP1 increased DRP1 GTPase activity and was the mechanism of mitochondrial fragmentation in AD [31]. While this finding was of potentially great interest, the report raises several concerns in relation to previously published work in the field including the relative enzymatic activity of dominant negative mutant DRP1K38A and the oligomeric state of active DRP1. In addition, it is puzzling that all AD cases in this study exhibited elevated SNO-DRP1 levels while none of the PD or control cases did. Such a distribution is unusual for a sporadic disease like AD and would imply that SNO-DRP1 is the initiating event in all AD cases [31]. Finally, nitrosative stress is not specific to AD; therefore, it is a mystery why no SNO-DRP1 was observed in sporadic PD patient samples.
Because of these concerns and to reconcile the conflicting reports, we set out to investigate the effects of NO in AD. We found that, contrary to the previous report, S-nitrosylation of DRP1 does not alter GTPase activity. Furthermore, we found that DRP1 is mostly a tetramer both in vitro and in vivo that assembles into ring- and spiral-like structures under experimental conditions, independently of S-nitrosylation. In addition, we observed that S-nitrosylation of DRP1 was not specific to AD as human postmortem brain samples from PD and even normal patients exhibited comparable SNO-DRP1 levels. Furthermore, we also observed S-nitrosylation of the mitochondrial fusion protein OPA1. In sum, our data indicate that S-nitrosylation of DRP1 is not the underlying mechanism of mitochondrial fragmentation in AD and we speculate that alternative pathways likely mediate the process.
Materials and Methods
Materials, chemicals, and antibodies
Neocuproine, S-methyl methanethiosulfonate (MMTS), TBS/0.05%Tween20, and sodium ascorbate were obtained from Sigma. NeutrAvidin agarose resin, N-[6-(biotinamide)hexyl]-3′-(2′-pyridyldithio)propionamide (EZ-Link Biotin-HPDP), SuperSignal West-Pico, -Dura, and -Femto chemiluminescent substrates were purchased from Thermo Scientific. NuPAGE LDS sample buffer, reducing agent, NuPAGE 4%-12% Bis-Tris gel, 3-12% acrylamide NativePAGE™ Novex Bis-Tris Gel System, Coomassie R-250, NativePAGE™ G-250 sample additive, and 1× NativePAGE sample buffer were purchased from Invitrogen. Hybond-ECL nitrocellulose membranes and ECL anti-mouse and anti-rabbit horseradish peroxidase conjugated IgG antibodies were purchased from GE Healthcare. Complete protease inhibitor cocktail was purchased from Roche. S-Nitrosylated Protein Detection Assay kit was purchased from Cayman Chemical Company. Mouse monoclonal anti-DRP1 (clone 8/DLP1) and mouse monoclonal anti-OPA1 (clone 18/OPA1) antibodies were purchased from BD Bioscience. Mouse anti-actin (clone JLA20) antibodies were purchased from Calbiochem. Rabbit anti-phospho-DRP1 Ser616 and anti-COX IV antibodies were purchased from Cell Signaling.
Plasmids and recombinant proteins
Human DRP1 cDNAs encoding the wild-type (699 amino acid muscle-expressed isoform, NCBI accession NM_005690.3, in pcDNA3 [23]) or mutant K38A were subcloned into the bacterial expression vectors pD13(pET30a) and pw12(pET17b), to create 80 kDa recombinant proteins with a MGSSHHHHHHSSGLVPRGSH N-terminus tag. The recombinant proteins were purified with Ni-beads and a Superdex S200 column and stored in 50 mM Hepes pH 7.5, 250 mM NaCl, 10% glycerol at -70°C. The human DRP1 cDNA (736 amino acid brain-specific isoform, NCBI accession NM_012062.3) was purchased from GeneCopoeia and subcloned into the baculovirus expression vector pFastBac (Invitrogen) to create an 84 kDa recombinant protein with a KGENLYFQGHHHHHH C-terminus tag. This recombinant protein was purified with Ni-beads, a Superdex S200 column, dialyzed and stored in 25 mM Hepes, 25 mM Pipes, 200 mM NaCl, pH 7.0 at -70°C.
Postmortem brain tissue
Human brain tissue from the frontal cortex of normal, AD, and PD (diffuse Lewy body disease) patients were from the Brain Bank of the Alzheimer's Disease Research Center, UCSD.
GTPase assay
Recombinant proteins were incubated for 10 min at room temperature in the dark with either 200 μM fresh SNOC, aged SNOC, or left untreated. Fresh SNOC was prepared as described [32]. GTPase activity of 0.1 mg/ml DRP1 (699 aa isoform) was measured with the continuous, regenerative coupled GTPase assay [33] and increasing GTP concentrations (0, 10, 20, 30, 60, 100, 150, 250, 500, and 1000 μM). Absorbance (340 nm) was measured at 30°C, pH 7.0 for 100 cycles of approximately 60 seconds per cycle with a FLUOstar Galaxy plate reader (BMG Labtechnologies). After plotting absorbance versus time, the steady state GTPase velocities and activities were calculated as described [33]. For analysis of the lag phase, the GTPase continuous assays were performed with 800 μM NADH, 500 μM GTP, 20 U/ml pyruvate kinase/lactate dehydrogenase, 1 mM PEP, 5 mM MgCl2, 187.5 mM NaCl, 7.5 mM KCl in 25 mM Hepes, 25 mM Pipes pH 7.0, 30°C.
Biotin-switch assay of recombinant DRP1 protein
After the GTPase assay, the DRP1 protein was collected from three wells of the 96-well plate and subjected to biotin-switch assay using a commercial kit. First, recombinant proteins were acetone precipitated and air-dried. The blocking step was performed at 50°C for 20 min. At the end only a small fraction (1/200) was separated on NuPAGE Novex 4-12% Bis-Tris minigels without prior heating or DTT addition [34]. After immunoblotting, biotinylated proteins were detected with avidin-HRP and SuperSignal West-Pico chemiluminescent substrate.
Biotin-switch assay of human brain tissue
To detect SNO-DRP1 in human brain, we used the biotin-switch assay as described [34, 35]. Approximately 1 g of tissue was lysed in 13 ml of HEN buffer plus Triton X-100 (250 mM Hepes pH 7.7, 1 mM EDTA, 0.1 mM Neocuproine plus 1% Triton X-100) and homogenized with a 15 ml Teflon-glass dounce and 10 strokes at 4°C. Lysates were pre-cleared overnight with NeutrAvidin-agarose beads at 4°C. Free thiols were blocked with 2 volumes of blocking buffer (40 mM MMTS: methyl methanethiosulfonate, 2.5% SDS in HEN buffer) and incubated for 20 min at 50°C. MMTS was removed by acetone precipitation and pellets were resuspended in HENS buffer (25 mM Hepes pH 7.7, 0.1 mM EDTA, 10 μM neocuproine, 1% SDS). Nitrosothiols were reduced to thiols with 1 mM ascorbate and biotinylated with 4 mM N-[6-(biotinamide)hexyl]-3′-(2′-pyridyldithio)propionamide (Biotin-HPDP) labeling solution in dimethylformamide. Biotin-HPDP was removed by acetone precipitation and pellets were resuspended in HENS buffer. Biotinylated proteins were collected with NeutrAvidin agarose resin for 1 h at 4°C under gentle agitation and washed five times with buffer (20 mM Hepes, 600 mM NaCl, 1 mM EDTA, 0.5% TritonX-100). Biotinylated proteins were separated on NuPAGE Novex 4-12% Bis-Tris minigels and SNO-DRP1 or SNO-OPA1 was detected by immunoblotting.
Subcellular fractionation
HEK293 cells were treated with 300 μM SNOC for different time periods, scraped gently, and centrifuged at 200 g for 5 min at 4°C. The cell pellets were washed twice with cold PBS (pH 7.4) and resuspended with 500 μl Buffer A (250 mM sucrose, 20 mM Hepes, pH 7.4, 10 mM KCl, 1.5 mM EGTA, 1.5 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM NaF, complete protease inhibitors). Cells were homogenized with a glass dounce and a B-type pestle for 50 strokes after 30 min incubation on ice. Nuclei and unbroken cells were removed as pellet after centrifugation at 800 g for 10 min at 4°C. Supernatants were then centrifuged at 22,000 g for 15 min at 4°C, and the resulting supernatants were collected as cytosolic fractions. The mitochondrial pellets were resuspended with 200 μl Buffer B [50 mM Hepes, pH 7.4, 1 % (v/v) NP-40, 10 % (v/v) glycerol, 1 mM EDTA, 2 mM DTT, 1 mM NaF, complete protease inhibitors] and incubated on ice for 20 min. After centrifugation for 15 min at 22,000 g at 4°C, the supernatants containing mitochondria were stored as mitochondrial fractions.
Immunoblotting
Samples were separated with NuPAGE Novex 4-12% Bis-Tris minigels with or without reducing agent and transferred to a Hybond-ECL membrane (0.45 μm pores) for 2 h at 30 volts. Membranes were blocked with 5% nonfat milk in PBS or TBS with 0.05%Tween20 for 3 h at room temperature and incubated with mouse anti-DRP1 (clone 8/DLP1) antibodies (1:1000) overnight at 4°C. After three washes, membranes were incubated for 1 h at room temperature with anti-mouse horseradish peroxidase-conjugated secondary antibodies (1:2000) in blocking solution. Immunocomplexes were detected using the SuperSignal West-Pico,-Dura, or-Femto chemiluminescent substrates. To detect SNO-OPA1, membranes from the biotin-switch assay were incubated with mouse monoclonal anti-OPA1 (1:1000) antibodies. To detect phospho-DRP1 Ser616 membranes were incubated with rabbit polyclonal anti-DRP Ser616 (1:1000) antibodies.
Native gel electrophoresis
Recombinant DRP1 was separated on 3-12% Acrylamide NativePAGE™ Novex Bis-Tris Gel System and stained with either Coomassie R-250 or transferred to a PVDF membrane (Invitrolon™) following manufacturer's instructions (Invitrogen). Human brain extracts (15 μg) were mixed with 0.34% NativePAGE™ G-250 sample additive and 1× NativePAGE sample buffer at room temperature and processed as described above.
Transmission electron microscopy (TEM)
Recombinant bacterial DRP1 protein (699 aa isoform) was diluted to 0.5 mg/ml in buffer (20 mM Hepes pH 7.4, 100 mM NaCl, 1 mM MgCl2) and treated with or without fresh SNOC (200 μM) for 10 min at room temperature and protected from light. To obtain oligomerization, protein samples were incubated with 1 mM non-hydrolyzable GTP analog guanosine 5′-(β-γ-imido) triphosphate (GMP-PNP) for about 3 h at room temperature. Samples were adsorbed to Formvar coated copper grids and negatively stained with 1% uranyl acetate. Electron micrographs were obtained using a LEO 912 transmission electron microscope (Zeiss) with an in-column spectrophotometer. The TEM was operated at 120 kV and micrographs were acquired at zero energy loss filtering.
Results
S-nitrosylation has no effect on DRP1 GTPase activity
A recent report claimed that S-nitrosylation increases DRP1 GTPase activity and is the mechanism responsible for mitochondrial fragmentation, bioenergetic compromise, and synaptic damage in a cellular model of AD [31]. However, the article conflicts with previous work. First, the authors investigated whether S-nitrosylation of DRP1 affects its GTPase activity. However, they mistakenly equated absorbance at a single time point and one GTP concentration with enzyme activity instead of determining a steady state activity with multiple time points and substrate concentrations. Second, the GTPase-defective dominant negative DRP1K38A mutant, which was previously shown as having very low if any enzymatic activity, is a critical negative control for DRP1 activity determination [36]. However, Cho et al. reported an unexpected DRP1K38A absorbance (used as an indicator of activity) that was more than half as great as wild-type DRP1, which casts doubts on the validity of their GTPase assay [31]. Based on this assay, the authors proposed a two-fold increase in DRP1 GTPase activity upon S-nitrosylation at cysteine 644, a cornerstone of their report.
To get to the bottom of these contradictory findings, we investigated the effect of S-nitrosylation on recombinant bacterial DRP1 using nine GTP concentrations and more than eighty time points to determine the steady state accurately. Wild-type DRP1 protein or the dominant negative mutant DRP1K38A were left untreated or treated with fresh NO donor S-nitrocysteine (SNOC) or aged SNOC control, in which all NO was released due to its instability. As shown in Figure 1A and summarized in Table 1, neither fresh nor aged SNOC increased DRP1 GTPase activity. In our experiments, DRP1K38A exhibited the expected 5 to 15-fold reduction in GTPase activity compared to wild-type DRP1 protein indicating that our GTPase assay does not harbor any artifacts. To test whether DRP1 S-nitrosylation reduces the lag phase of enzyme activity before steady state, we measured the initial rate of GTP hydrolysis after DRP1 was exposed to either aged or fresh SNOC. As shown in Figure 1C, we did not observe a change in the lag phase of DRP1 GTP hydrolysis. It could be argued that our recombinant DRP1 protein was already maximally oxidized. In such a scenario, the protein cysteines might already be modified, preventing their S-nitrosylation by NO donors, and thereby providing an explanation for the lack of detectable increased DRP1 activity. To rule out this possibility, we tested whether the same DRP1 protein we used in our GTPase assay was indeed S-nitrosylated using the biotin-switch assay, an assay which detects S-nitrosylated proteins. In the biotin-switch assay, nitrosylated cysteines are converted into biotinylated cysteines. Using this assay, we readily detected SNO-DRP1, which would not have been possible if DRP1 was oxidized during the protein isolation (Figure 1B). Similar results were obtained with several independent bacterial as well as baculovirus protein preparations. In sum, our data provides strong evidence that S-nitrosylation of DRP1 does not increase its GTPase activity.
Figure 1.
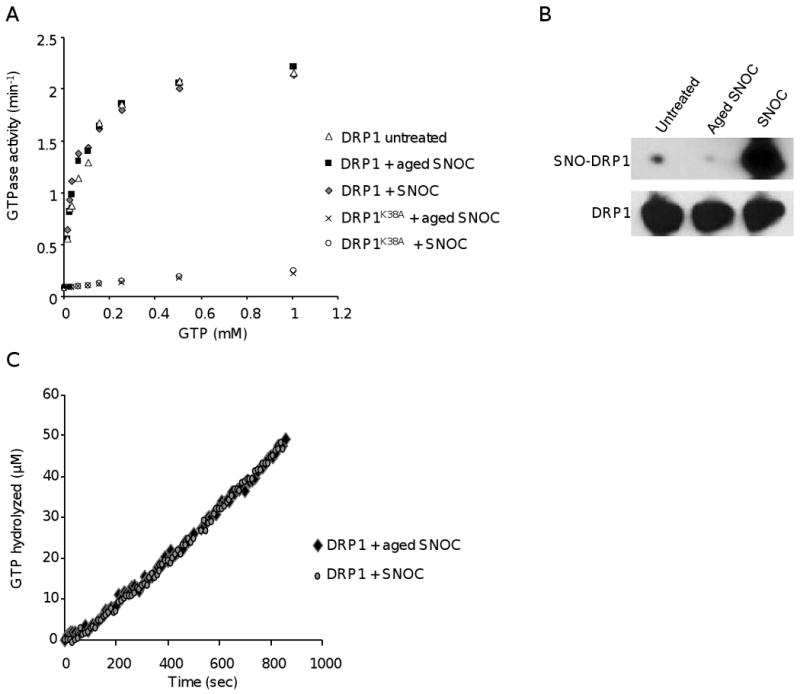
S-Nitrosylation of DRP1 does not alter GTPase activity. A) Kinetics of DRP1 GTPase activity. Recombinant wild-type DRP1 or mutant DRP1K38A were pre-treated with either 200 μM of fresh SNOC, aged (decayed) SNOC, or left untreated and then subjected to the GTPase assay. Data indicate mean steady state values (n=3). B) Detection of SNO-DRP1. Biotin-switch assay of the DRP1 protein after the GTPase assay of the experiment shown in (A). Proteins were separated by SDS-PAGE and immunoblots were probed with either avidin-horseradish peroxidase for detection of SNO-DRP1 (top) or with anti-DRP1 antibodies (bottom). C) Initial rate of GTP hydrolysis at 500 μM GTP in the presence of DRP1 pretreated with either 200 μM fresh or aged SNOC.
Table 1.
DRP1 GTPase activity per minute at three different GTP concentrations of the experiment shown in Figure 1A. Values are ± SEM (n=3).
| GTP(mM) | DRP1 Untreated(min-1) | DRP1 + aged SNOC(min-1) | DRP1 + SNOC(min-1) | DRP1K38A + aged SNOC(min-1) | DRP1K38A + SNOC(min-1) |
|---|---|---|---|---|---|
| 0.01 | 0.57 ± 0.075 | 0.561 ± 0.085 | 0.656 ± 0.059 | 0.0986 ± 0.019 | 0.104 ± 0.011 |
| 0.15 | 1.69 ± 0.118 | 1.65 ± 0.108 | 1.62 ± 0.148 | 0.128 ± 0.0072 | 0.140 ± 0.0148 |
| 1 | 2.17 ± 0.325 | 2.22 ± 0.118 | 2.15 ± 0.014 | 0.229 ± 0.0194 | 0.262 ± 0.0825 |
DRP1 is mostly a tetramer under native conditions
The recent article by Cho et al. proposed that S-nitrosylation of cysteine 644 enhances GTP hydrolysis via DRP1 dimerization [31]. However, this is in stark disagreement with the current mammalian DRP1 literature [37, 38]. There are no reports that support a dimer-stimulated DRP1 activation. Instead, cross linking and gel filtration studies have shown that DRP1 is a tetramer and that self-assembly into higher order oligomers, rings, and spirals stimulates GTP hydrolysis [37, 38]. Therefore, DRP1 dimerization would reflect the opposite, disassembly and decreased GTP hydrolysis.
To resolve this apparent contradiction, we performed native gel electrophoresis of two bacterial DRP1 protein isoforms before treatment or after treatment with either aged or fresh SNOC. The first isoform is 699 amino acids, expressed predominantly in muscle, and used by Cho et al. [31], and the second isoform is 736 amino acids and brain-specific. Both DRP1 isoforms were fused with a polyhistidine tag used for purification. In all cases, DRP1 migrated mostly as a tetramer or a higher molecular weight unit thereof (Figure 2A). Importantly, SNO-DRP1 (Figure 2A, lane 3 and 6) did not exhibit greater assembly into higher molecular weight tetrameric units when compared to DRP1 either untreated or treated with aged SNOC (Figure 2A, lane 1, 2 and 4, 5 respectively). Furthermore, addition of reducing agent dithiothreitol (DTT) at room temperature did not alter the tetramers (Figure 2B). Thus, DRP1 is predominantly a tetramer or higher order oligomer in its native condition and S-nitrosylation of DRP1 has no effect on its oligomerization state. Finally, we found that DRP1 is present mostly as a tetramer in three groups of three human postmortem brain samples of normal, AD, and PD patients (Figure 2C), confirming our in vitro data with recombinant protein and previous cell line studies [37, 38]. Cho et al. reported that S-nitrosylation of cysteine 644 increases DRP1 disulfide-linked dimerization using SDS-PAGE in the absence of reducing agent DTT [31]. It is puzzling to us how SNO-DRP1 could undergo the alleged disulfide-linked dimerization considering that the critical cysteine (Cys644) is reportedly blocked by S-nitrosylation. To explore this further, we performed SDS-PAGE of DRP1 and SNO-DRP1 in the presence or absence of DTT. Using SDS-PAGE, we show that under these denaturing conditions both DRP1 and SNO-DRP1 exhibit monomers and dimers in the absence of DTT, which are released only as monomers in the presence of DTT (Figure 2D). Thus, dimerization under denaturing conditions does not depend on S-nitrosylation. In summary, our data do not support a dimer-mediated DRP1 activation model as proposed by Cho et al. and are in strong agreement with the current literature indicating that DRP1 is mostly a tetramer and higher order oligomerization promotes GTP hydrolysis.
Figure 2.
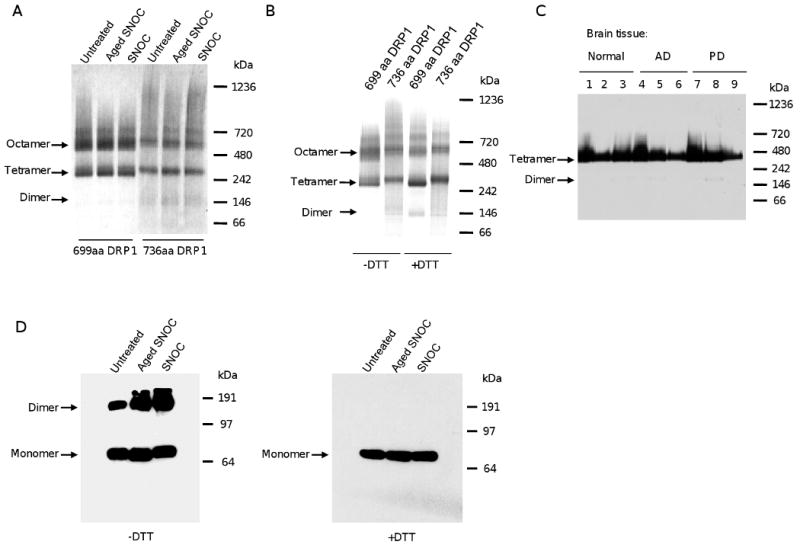
Recombinant and brain DRP1 form tetramers under native conditions irrespective of S-nitrosylation. A) DRP1 runs as tetramer or higher order oligomer in native gels. Recombinant DRP1 isoforms were left untreated or pre-treated with aged or fresh SNOC (200 μM), separated on native gels and stained with Coomassie blue. B) Reducing conditions do not break the tetramers in native gels. Recombinant DRP1 isoforms were separated under non-reducing (-DTT) or reducing conditions (+DTT) on native gels and Coomassie blue stained. C) Human brain DRP1 is mostly a tetramer. Brain extracts from normal controls (lanes 1, 2, 3), AD (lanes 4, 5, 6) or PD patients (lanes 7, 8, 9) were separated on native gels and immunoblots were probed with anti-DRP1 antibodies. D) S-nitrosylation of DRP1 does not trigger DRP1 dimerization. Recombinant DRP1 (699 amino acid isoform) was left untreated or treated with aged or fresh SNOC (200 μM), heat denatured under non-reducing (-DTT) or reducing conditions (+DTT) and separated by SDS-PAGE, immunoblotted, and probed with anti-DRP1 antibodies.
S-nitrosylation does not stimulate DRP1 self-assembly
Similar to yeast DNM1, human DRP1 assembles into higher order oligomers, forming curved filaments and occasional rings in the presence of low salt conditions or rings and spiral-like structures in the presence of non-hydrolyzable nucleotides [23, 39]. To test whether S-nitrosylation of DRP1 increases its ability to self-assemble, which would be consistent with increased activation, we used negative stain and transmission electron microscopy to visualize oligomerization. We found that DRP1 and SNO-DRP1 assemble into similar large spiral-like structures in the presence of the non-hydrolyzable GTP analogue guanosine GMP-PNP (Figure 3A and 3B). Similarly, in low salt conditions and in the absence of nucleotides, curved filaments and rings were observed both with DRP1 and SNO-DRP1 (Figure 3C and 3D). Thus, S-nitrosylation did not enhance DRP1 oligomerization. Of note is that the diameter of the rings was about 35 nm, similar to previously described human DRP1 rings [23], which is smaller than the 110 nm diameter of DNM1 rings [40].
Figure 3.
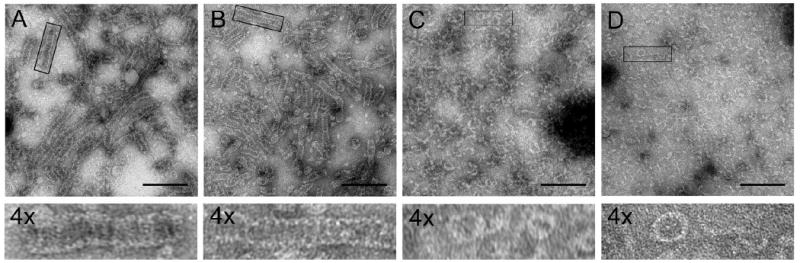
S-Nitrosylation has no effect on DRP1 assembly. Electron micrographs of negatively stained recombinant DRP1 (699aa isoform) in the presence of non-hydrolyzable GTP analog GMP-PNP and corresponding 4× zoom of regions of interest (A) − SNOC and (B) + SNOC. C) Control at 100 mM NaCl with neither nucleotide nor SNOC. D) Control at 100 mM NaCl with 200 μM SNOC without nucleotide. Scale bar, 200 nm.
SNO-DRP1 is not increased in the AD brain
Using the biotin-switch assay to detect S-nitrosylated proteins, SNO-DRP1 was reported to be increased in brain tissue of Tg2576 AD mice and AD patients [31]. However, important controls were missing in the original report. To test whether SNO-DRP1 levels are indeed increased in AD, brain tissue lysates of normal age-matched individuals and AD and PD patients were prepared and subjected to the biotin-switch assay. Our samples were obtained from the same brain bank as some, if not all, of the samples described by Cho et al. To remove endogenously biotinylated proteins that interfere with the biotin-switch assay, we pre-cleared the brain extracts with NeutrAvidin agarose beads. Unexpectedly, we found biotinylated DRP1 protein in all brain samples (Figure 4A). Thus, a small fraction of DRP1 is modified by biotinylation in aged individuals. When running the pre-cleared brain lysates we found reduced DRP1 protein in the AD patient relative to the normal sample (Figure 4B), which is consistent with previous reports [41].
Figure 4.
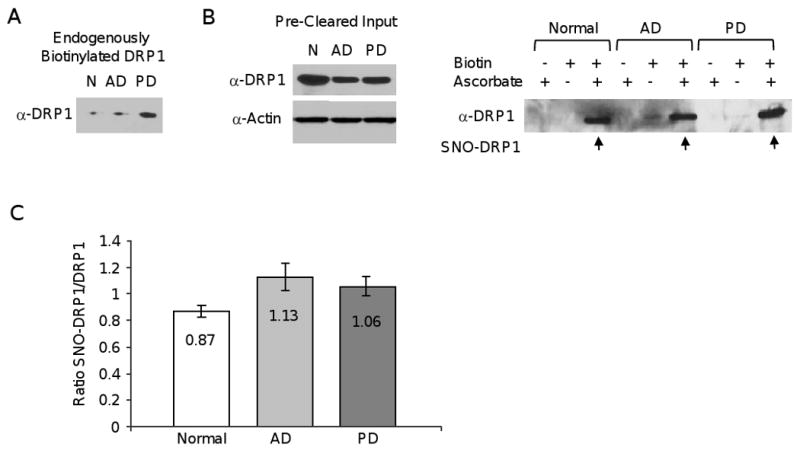
SNO-DRP1 is not specific to AD. A) DRP1 is endogenously biotinylated. Immunoblot of NeutrAvidin agarose from normal (N), AD or PD human brain lysates probed with anti-DRP1 antibodies. B) Immunoblots of pre-cleared input brain DRP1 protein used in the biotin-switch assay, with actin serving as loading control. C) SNO-DRP1 (arrows) in human normal, AD, or PD brain including the biotin and ascorbate controls using the biotin-switch assay and DRP1 immunoblotting. ID number of postmortem samples is indicated above each blot. D) Ratios of SNO-DRP1 to DRP1 in normal (n=4), AD (n=4), and PD (n=4) brain samples. Ratios were calculated from densitometric values using the ImageJ Gel Analysis software. The SNO-DRP1 signals were corrected for the signals obtained in ascorbate controls. Data are ± S.E.M..
We next used the NeutrAvidin agarose pre-cleared extracts in the three step biotin-switch assay. In the first step, the free R–SH groups are blocked by S-methylation. In the second step, ascorbate is used to reduce the R-SNO groups to R-SH. In the third step, the newly formed R-SH groups are biotinylated, the R-S-S-biotin proteins are collected by incubation with NeutrAvidin agarose resin, and bound proteins are separated by SDS-PAGE. As shown in Figure 4C, omission of the biotinylating agent in the presence of ascorbate revealed little or no endogenously biotinylated DRP1 protein left after pre-clearing. Furthermore, omission of ascorbate and prevention of R-SNO reduction indicated that most free R-SH groups were effectively blocked by S-methylation. Finally, performing all steps of the biotin-switch assay revealed SNO-DRP1 (Figure 4C). However, contrary to the previous report we found no consistent increase in SNO-DRP1 levels in the AD patient samples. The PD and even normal patient samples exhibited comparable SNO-DRP1 levels. We also found no evidence for a significant increase in the ratio of SNO-DRP1 to DRP1 in AD patients (Figure 4D). In summary, our data suggest that DRP1 S-nitrosylation is not specific to AD, but occurs in all aged individuals.
SNO-OPA1 is found in the human brain
To determine whether other proteins associated with mitochondrial fission and fusion might be targets of S-nitrosylation, we re-examined the human postmortem brain samples for signs of S-nitrosylation of OPA1, a mitochondrial fusion factor localized to the mitochondrial inner membrane. The biotin-switch assay of normal, AD, and PD patient samples all indicated the presence of SNO-OPA1 (Figure 5). Thus, it appears that DRP1 is not the only mitochondrial fission/fusion GTPase that is S-nitrosylated in the human brain. However, we also found, similar to a previous report, that OPA1 protein levels were reduced in the AD sample [41]. Whether S-nitrosylation of OPA1 modifies its function remains to be determined.
Figure 5.
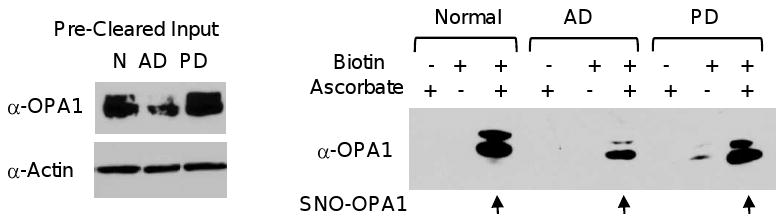
Presence of SNO-OPA1 in normal, AD, and PD brains. Immunoblot of input proteins used in the biotin-switch assay probed with anti-OPA1 or anti-Actin controls (left panels). SNO-OPA1 (arrows) in human normal (N), AD, or PD brain, including the biotin and ascorbate controls, was detected by the biotin-switch assay and OPA1 immunoblotting (right panel).
NO triggers DRP1 phosphorylation and recruitment to mitochondria
Because S-nitrosylation does not increase DRP1 activity and is therefore not the mechanism underlying NO-mediated mitochondrial fission, we speculated that NO stress might activate kinases, which in turn phosphorylate and activate DRP1. There is evidence that NO stress activates several kinases that participate in NO-mediated neuronal injury and cell death [42]. Furthermore, cdk1 phosphorylates DRP1 at serine 616, which allows translocation of DRP1 to mitochondria and stimulates mitochondrial fission [43, 44]. Interestingly, several cell cycle kinases are abnormally elevated in AD, a phenomenon that is thought to cause an abortive cell cycle entry and neuronal cell death [42, 45]. To test whether NO induces cdk1-mediated DRP1 phosphorylation, we exposed HEK293 cells to either aged or fresh SNOC and isolated the cytosolic and mitochondrial fraction at various time points. Immunoblotting using a phospho-DRP1 Ser616 specific antibody revealed a rapid increase of cytosolic phospho-DRP1 Ser616 after one hour of SNOC exposure, which was followed by an accumulation of phospho-DRP1 Ser616 at mitochondria after three hours of SNOC exposure (Figure 6A). To test whether this observation might be of relevance to human AD, we determined the ratio of phospho-DRP1 Ser616 to DRP1 of five groups of normal, AD, and PD brain samples (summarized in Table 2). Figure 6C shows that there is a trend of increased phospho-DRP1 Ser616 in AD samples, although it was not a statistically significant difference relative to normal samples. These results are consistent with a previous report indicating an increase in phospho-DRP1 Ser616 in the AD brain [41]. Thus, our data suggest that NO stress induces rapid DRP1 phosphorylation at serine 616 and activates mitochondrial fission, offering an alternative mechanism of NO-mediated mitochondrial fragmentation in AD.
Figure 6.
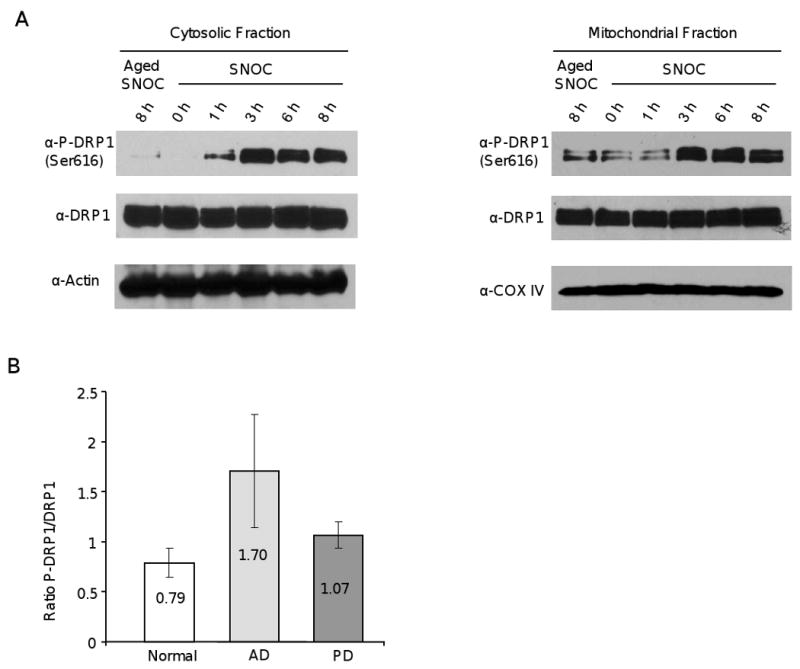
NO triggers DRP1 Ser616 phosphorylation and recruitment to mitochondria. A) Immunoblots of Ser616 phosphorylated DRP1 and total DRP1 in the cytosolic or mitochondrial fractions. Actin and COX IV were used as loading controls and markers for the cytosolic and mitochondrial fractions, respectively. HEK293 cells were exposed either to aged or fresh SNOC (300 μM) and the cytosolic and mitochondrial fractions were isolated after various time points. B) Immunoblots of phospho-DRP1 (upper panel), total DRP1 (middle panel) and actin (lower panel) in human normal, AD, or PD brain samples. C) Ratios of p-DRP1 Ser616 to DRP1 of normal (n=5), AD (n=5), or PD (n=5) brains. Ratios were obtained by densitometric measurements of immunoblots using the ImageJ Gel Analysis software. Samples were normalized with the actin loading control.
Table 2.
Human brain samples analyzed in this study. AD: Alzheimer's disease, PD: Parkinson's disease, M: male, F: female, PMH: postmortem hours, nd: not done.
| Patient | Fig 2C lane # | Fig 6B lane # | Fig 5 samples | Diagnosis | Age (years) | PMH | Gender | SNO-DRP1 | Relative ratio SNO-DRP1/DRP1 for Fig 4C | Relative ratio phospho-DRP1/DRP1 for Fig 6B |
|---|---|---|---|---|---|---|---|---|---|---|
| 5130 | Normal | 71 | 2 | M | + | nd | nd | |||
| 5347 | 1 | Normal | Normal | 86 | M | + | 0.818 | 0.576 | ||
| 5248 | 4 | Normal | 93 | 18 | F | + | 0.866 | 1.018 | ||
| 5341 | 3 | 2 | Normal | 77 | 12 | F | + | 0.81 | 0.609 | |
| 5167 | 1 | 5 | Normal | 83 | M | + | 0.987 | 1.257 | ||
| 5404 | AD | 82 | 10 | M | + | nd | nd | |||
| 5391 | AD | 91 | 6 | M | + | nd | nd | |||
| 5276 | AD | 93 | 5 | M | + | nd | nd | |||
| 5457 | 8 | AD | AD | 77 | 8 | M | + | 0.934 | 1.282 | |
| 5446 | 7 | AD | 90 | 12 | F | + | 0.998 | 3.789 | ||
| 5464 | 6 | 6 | AD | 73 | 9 | M | + | 1.372 | 1.534 | |
| 5454 | 4 | 9 | AD | 71 | 12 | M | + | 1.216 | 0.634 | |
| 5445 | 12 | PD | PD | 86 | 12 | M | + | 1.272 | 0.586 | |
| 5357 | 14 | PD | 94 | 12 | M | + | 1.05 | 1.349 | ||
| 5386 | 9 | 13 | PD | 74 | 27 | M | + | 1.005 | 1.102 | |
| 5353 | 7 | 15 | PD | 78 | 12 | M | + | 0.917 | 1.033 | |
| 5302 | 2 | 3 | Normal | 83 | 72 | M | nd | nd | 0.47 | |
| 5451 | 5 | 10 | AD | 71 | 8 | M | nd | nd | 1.19 | |
| 5216 | 8 | 11 | PD | 74 | M | nd | nd | 1.255 |
Discussion
A recent study proposed that S-nitrosylation of DRP1 in AD induces mitochondrial fission by a dimer-mediated increase in GTPase activity and results in bioenergetic failure, synaptic loss, and neuronal injury characteristic of the disease [31]. Here we provide strong evidence contesting this claim. First, we show that S-nitrosylation does not increase the GTPase activity of DRP1. We arrived at this conclusion using two independent GTPase assays, the continuous assay (Figure 1A) and malachite green-based assay (unpublished results), and several independent bacterial and baculovirus DRP1 protein preparations. Second, we find that S-nitrosylation of DRP1 does not cause dimerization or activation. In fact, most recombinant DRP1 and human brain DRP1 do not exist as monomers and dimers under native conditions, but rather as tetramers or higher order oligomers thereof. This finding is in agreement with the mammalian DRP1 literature [37, 38]. Third, we show that S-nitrosylation of DRP1 does not enhance assembly into oligomeric spiral-like structures. Fourth, SNO-DRP1 is not increased in the postmortem human AD brain and is also readily detected in PD brain and even normal brain. Finally, we demonstrate that S-nitrosylation is not specific to DRP1 as the mitochondrial fusion GTPase OPA1 also undergoes S-nitrosylation.
In evaluating our data, it is critical to point out that the recombinant DRP1 protein we used was not over-oxidized. The following observations argue against this claim. First, we easily detected SNO-DRP1 with the biotin-switch assay. Thus, over-oxidation and consequent failure to S-nitrosylate DRP1 did not occur. Second, mass spectrometry did not reveal any hints of oxidation (unpublished results). Third, our protein is fully functional and able to form spiral-like structures. Collectively, our data strongly indicate that the mechanism underlying NO-mediated mitochondrial fragmentation in AD and likely other neurodegenerative disorders is not DRP1 S-nitrosylation. Additional investigations are needed to unravel the mechanism of NO-mediated mitochondrial fragmentation.
Excess NO mediates cell death by inhibiting multiple metabolic and cellular pathways, all of which could cause mitochondrial fragmentation and morphology changes. For example, NO and peroxynitrite are potent inhibitors of the mitochondrial respiratory chain [46-48]. Thus, NO-mediated respiratory inhibition, similar to inhibition caused by rotenone or 3-NP, might elicit mitochondrial fragmentation [27, 30]. Alternatively, NO might break down the cytoskeleton and microtubule organization, which could detach mitochondria from motor proteins and microtubule fibers, resulting in conversion of mitochondrial morphology to the more rounded phenotype, arrest in trafficking of mitochondria along axons and dendrites, and ultimately a decline in mitochondrial fusion. Defects in cytoskeletal-mediated axonal organelle trafficking have been observed in AD [6].
Another possible mechanism of NO-mediated mitochondrial fragmentation in AD is that peroxynitrite nitrates tyrosines in the peroxisome proliferator-activated receptor gamma (PPARγ), preventing its translocation from the cytoplasm to the nucleus and thus preventing mitochondrial biogenesis [49]. PPARγ is a nuclear hormone receptor which regulates mitochondrial biogenesis by increasing expression of nuclear respiratory factor (NRF) and mitochondrial transcription factor A (mtTFA). Both factors increase the expression of nuclear and mitochondrial genes that encode components of the respiratory chain, antioxidant defense mechanism, and mitochondrial DNA replication system. PPARγ expression has been shown to protect neurons from Aβ-mediated toxicity [50]. Interestingly, MFN2 is regulated by PPARγ. Hence, peroxynitrite-mediated inhibition of PPARγ and mitochondrial biogenesis might contribute to bio-energetic defects and an imbalance in expression of mitochondrial fission and fusion GTPases.
Nitrosative stress activates kinases that in turn might phosphorylate DRP1 resulting in its activation [44]. For example, p38MAP kinase is activated by peroxynitrite [51] and DRP1 phosphorylation by Cdk1/cyclinB at serine 616 promotes mitochondrial fission in mitotic cells [44]. We show here that DRP1 is rapidly phosphorylated at serine 616 and recruited to mitochondria upon exposure to the NO donor SNOC. Whether Cdk1/cyclinB is the serine kinase that mediates the NO effect remains unclear; however, activation of cell cycle kinases including Cdk1/cyclinB is thought to participate in AD pathogenesis [42]. In addition, a recent study reported DRP1 Ser616 phosphorylation in the cytosolic and mitochondrial fraction of the AD patient brain [41]. Another kinase that might get activated by nitrosative stress is Ca2+/calmodulin-dependent kinase Iα (CaMKIα). NO blocks mitochondrial respiration, which could lead to a decline in energy metabolism, inhibition of the Na+/K+ ATPase pump, plasma membrane depolarization, and activation of voltage-dependent Ca2+ channels (VDCC). An impaired Ca2+ homeostasis has been reported in AD [52]. Activation of VDCC by elevated extracellular K+ was found to increase intracellular Ca2+ levels and to activate CaMKIα kinase, which in turn phosphorylates DRP1 at serine 600 causing increased binding to Fis1 and mitochondrial fragmentation in neurons and other cell types [53]. Yet another scenario is that altered Ca2+ homeostasis might cause activation of the Ca2+-dependent phosphatase calcineurin, which dephosphorylates DRP1 at specific sites and causes its activation [54, 55].
Last, nitrosative and oxidative stress may cause processing, misfolding, oligomerization, and accumulation of Aβ peptide in mitochondria. Whether toxic Aβ oligomers can abnormally interact with mitochondrial fission and fusion factors like DRP1, OPA1, or MFN2 has not been tested; however, abnormal Aβ interaction with mitochondrial proteins has been documented and is thought to contribute to AD pathogenesis [56, 57].
The pathways here are just a few potential mechanisms that might account for NO-mediated mitochondrial fragmentation in AD, which underscores the pleiotropic nature of the cellular responses to nitrosative stress. Thus, it is very unlikely that NO mediates mitochondrial fragmentation by directly modulating a single protein target such as DRP1. Accordingly, while Cho et al. showed that the DRP1 C644A mutant, in which the critical cysteine has been mutated preventing S-nitrosylation, still induces mitochondrial fragmentation as strongly as wild-type DRP1, it appeared to prevent NO-mediated cell death by a mechanism distinct from dominant negative interference. It is difficult to envision how this mutant could mediate neuronal survival while still inducing mitochondrial fragmentation.
Furthermore, whether mitochondrial fragmentation is the cause or consequence of the bioenergetic defects and the synaptic loss in AD remains to be shown. Presently, not a single in vivo study has conclusively demonstrated that mitochondrial fragmentation occurs early in AD and is the primary event causing mitochondrial defects and synaptic changes. In fact, all data are based on overexpression systems or treatment of cultured cells with Aβ. In addition, existing studies have produced conflicting results. For example, human fibroblasts of AD patients show the opposite phenotype, more elongated and fewer mitochondria, of that observed in human brain tissue, cultured neurons, and animal models [58].
It is plausible that in AD the bioenergetic defects are primarily the result of other mechanisms including Ca2+ overload, mitochondrial permeability transition and loss of mitochondrial membrane potential, nitrosative or oxidative stress and inhibition of key mitochondrial enzymes, and direct interference of toxic oligomeric Aβ with mitochondrial function and transport, with impaired mitochondrial fission and fusion being a secondary event. For example, only mitochondria that have a high mitochondrial membrane potential can fuse and mitochondria with low potential are selectively degraded by mitophagy. Thus, the hypothesis that the mitochondrial defects in a sporadic neurodegenerative conditions like AD are the result of a dysfunction in the fission/fusion machinery is premature and likely too simplistic [59] and would require more conclusive experimental support using appropriate in vivo models.
Acknowledgments
We thank Dr. A.M. van der Bliek (UCLA) for the human WT-DRP1 and DRP1K38A cDNAs that encode the 699 amino acid DRP1 isoform. This work is supported by NIH grants R01EY016164 and R01NS055193 to EBW.
Footnotes
References
- 1.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 2.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 3.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 5.Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan Z, Sun X, Hou FS, Oh HW, Hilgenberg LG, Hol EM, van Leeuwen FW, Smith MA, O'Dowd DK, Schreiber SS. Mutant ubiquitin found in Alzheimer's disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 2007;14:1721–1732. doi: 10.1038/sj.cdd.4402180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109 1:153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 11.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 12.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 13.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 14.Park HJ, Seong YM, Choi JY, Kang S, Rhim H. Alzheimer's disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi. Neurosci Lett. 2004;357:63–67. doi: 10.1016/j.neulet.2003.11.068. [DOI] [PubMed] [Google Scholar]
- 15.Knott AB, Bossy-Wetzel E. Nitric oxide in health and disease of the nervous system. Antioxid Redox Signal. 2009;11:541–554. doi: 10.1089/ars.2008.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 19.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 20.Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 21.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 22.Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, Guillou E, Delettre C, Valette A, Hamel CP, Ducommun B, Lenaers G, Belenguer P. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–176. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- 23.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 30.Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 33.Ingerman E, Nunnari J. A continuous, regenerative coupled GTPase assay for dynamin-related proteins. Methods Enzymol. 2005;404:611–619. doi: 10.1016/S0076-6879(05)04053-X. [DOI] [PubMed] [Google Scholar]
- 34.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 35.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 36.Yoon Y, Pitts KR, McNiven MA. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell. 2001;12:2894–2905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin HW, Takatsu H, Mukai H, Munekata E, Murakami K, Nakayama K. Intermolecular and interdomain interactions of a dynamin-related GTP-binding protein, Dnm1p/Vps1p-like protein. J Biol Chem. 1999;274:2780–2785. doi: 10.1074/jbc.274.5.2780. [DOI] [PubMed] [Google Scholar]
- 38.Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–35974. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
- 39.Mears JA, Hinshaw JE. Visualization of dynamins. Methods Cell Biol. 2008;88:237–256. doi: 10.1016/S0091-679X(08)00413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naylor K, Ingerman E, Okreglak V, Marino M, Hinshaw JE, Nunnari J. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J Biol Chem. 2006;281:2177–2183. doi: 10.1074/jbc.M507943200. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, Petersen RB, Bajic V, Smith MA. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem Int. 2009;54:84–88. doi: 10.1016/j.neuint.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang CR, Blackstone C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. 2007;8:1088–1089. doi: 10.1038/sj.embor.7401118. author reply 1089-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 45.Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolanos JP, Peuchen S, Heales SJ, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- 47.Brown GC, Bolanos JP, Heales SJ, Clark JB. Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett. 1995;193:201–204. doi: 10.1016/0304-3940(95)11703-y. [DOI] [PubMed] [Google Scholar]
- 48.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 49.Shibuya A, Wada K, Nakajima A, Saeki M, Katayama K, Mayumi T, Kadowaki T, Niwa H, Kamisaki Y. Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line, RAW 264. FEBS Lett. 2002;525:43–47. doi: 10.1016/s0014-5793(02)03059-4. [DOI] [PubMed] [Google Scholar]
- 50.Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, Inestrosa NC, Bronfman M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- 51.Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- 52.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 57.Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258–267. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 58.Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer's and Parkinson's diseases. Apoptosis. 2010 doi: 10.1007/s10495-010-0476-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]