An Accurate, Clinically Feasible Multi-Gene Expression Assay for Predicting Metastasis in Uveal Melanoma (original) (raw)
Abstract
Uveal (ocular) melanoma is an aggressive cancer that often forms undetectable micrometastases before diagnosis of the primary tumor. These micrometastases later multiply to generate metastatic tumors that are resistant to therapy and are uniformly fatal. We have previously identified a gene expression profile derived from the primary tumor that is extremely accurate for identifying patients at high risk of metastatic disease. Development of a practical clinically feasible platform for analyzing this expression profile would benefit high-risk patients through intensified metastatic surveillance, earlier intervention for metastasis, and stratification for entry into clinical trials of adjuvant therapy. Here, we migrate the expression profile from a hybridization-based microarray platform to a robust, clinically practical, PCR-based 15-gene assay comprising 12 discriminating genes and three endogenous control genes. We analyze the technical performance of the assay in a prospective study of 609 tumor samples, including 421 samples sent from distant locations. We show that the assay can be performed accurately on fine needle aspirate biopsy samples, even when the quantity of RNA is below detectable limits. Preliminary outcome data from the prospective study affirm the prognostic accuracy of the assay. This prognostic assay provides an important addition to the armamentarium for managing patients with uveal melanoma, and it provides a proof of principle for the development of similar assays for other cancers.
Most cancer deaths are the result of metastatic spread from the primary tumor to distant organs. Once metastatic disease has advanced to the point that it can be identified by clinical imaging techniques, the tumor burden is so great, and the cancer cells so genetically deranged, that therapy is often ineffective. Uveal (ocular) melanoma is an aggressive cancer that epitomizes this problem. Uveal melanoma spreads preferentially to the liver in up to half of patients, and the metastatic disease is almost always fatal.1 Improvements in survival are unlikely to arise from new therapies for advanced metastatic disease but, rather, from early detection of metastatic disease at a less advanced stage that is more amenable to therapy or from adjuvant therapy before detection of metastatic disease. An accurate test for identifying high-risk patients would allow for such personalized management, as well as for stratification of high-risk patients into clinical trials for adjuvant therapy. Further, most patients desire to have accurate prognostic information to allow them to make personal decisions regarding their future.2
We previously identified a gene expression profile derived from primary uveal melanomas that distinguished between low metastatic risk (class 1 signature) and high metastatic risk (class 2 signature).3 Further, we showed that this gene expression profile could be performed using a small number of discriminating genes on archival specimens and fine needle biopsy samples, and that it was more accurate than the clinical, pathological, and chromosomal prognostic factors that we evaluated.3,4,5,6 Two independent groups have confirmed the superior prognostic accuracy of gene expression profiling compared with the other prognostic biomarkers currently available.7,8
Here, we migrated the gene expression profile from a high-density hybridization-based microarray platform to a 15-gene PCR-based assay that can be used on a routine clinical basis on very small samples obtained by fine needle aspiration and archival formalin fixed specimens. We provide results from a multicenter prospective study showing that the assay can be performed on samples sent from distant locations with a very low failure rate, and we show preliminary outcome data affirming the prognostic accuracy of the 15-gene assay.
Materials and Methods
Microarray-Based Gene Expression Profiling
Gene expression profile data generated from high-density commercial microarray platforms were published and described elsewhere.3,5,6,9 The data were collected on one or more of the following platforms: Affymetrix U133A GeneChip (28 cases), U133Av2 GeneChip (11 cases), and Illumina Ref8 Beadchip array (26 cases).
Preparation of RNA Samples
Tumor RNA samples were obtained by fine needle aspirate biopsy (FNAB; 553 samples) or immediately after enucleation (56 samples). Samples were obtained from Washington University (188 samples) and from 11 collaborating sites (421 samples). For all patient samples collected at Washington University and the other sites, Institutional Review Board approval and patient informed consent were obtained. Fine needle biopsies were performed using a 25-gauge needle on untreated uveal melanomas, as previously described.5,10 For each FNAB, the first sample was used for cytologic examination and the second for RNA analysis. Each needle pass was directed into the same location, toward the center of the tumor. Cytology reports and patient outcomes were available only on samples collected at Washington University.
FNAB samples for RNA analysis were expelled into an empty RNase-free tube in the operating room. The empty syringe was filled with 200 μl of extraction buffer (XB) from the PicoPure RNA isolation kit (Molecular Devices, Sunnyvale, CA), which was flushed through the syringe to collect any additional tumor cells lodged in the needle hub. The tube was then snap-frozen in liquid nitrogen in the operating room before transportation. On arrival in the laboratory, the samples were logged and stored in a freezer at −80°C until they could be processed. RNA was isolated using the PicoPure kit (including the optional DNase step), which yielded about 100 ng to 1.5 μg total RNA per aspirate using the NanoDrop 1000 system (NanoDrop Products, Wilmington, DE). For samples obtained at enucleation, the eye was taken away from the operative field and sectioned along the pupil-optic nerve axis using a razor blade. A thin section of tumor was then sliced off and placed on Whatman paper, on which the tumor orientation was marked (A, apex; B, base; C and D, the anterior and posterior edges, respectively). The tumor was then scored using the razor blade to facilitate later removal of small fragments from different parts of the tumor to assess for tumor heterogeneity without having to thaw the entire tumor specimen. The specimen was then wrapped in foil and immediately snap-frozen in liquid nitrogen in the operating room. Total RNA was obtained using TRIzol (Invitrogen, Carlsbad, CA), including the optional isolation step, which is performed to rid the sample of any insoluble material and purified using RNeasy kits (QIAGEN, Valencia, CA) according to manufacturers' instructions. RNA quality was assessed on the NanoDrop 1000 system. For formalin-fixed paraffin-embedded (FFPE) samples, five 10-μm sections were obtained from tissue blocks, and tumor tissue was dissected away from surrounding normal material. Total RNA was isolated using the RecoverAll Total Nucleic Acid Isolation kit (Ambion, Austin, TX) following the manufacturer's protocol. RNA samples were stored at −80°C. For specimens sent to St. Louis from other centers, tubes were placed on dry ice, mailed by overnight courier, and handled as described above. No RNA degradation was observed for samples handled according to protocol.
Real-Time PCR Analysis
All RNA samples were converted to cDNA using the High Capacity cDNA Reverse Transcription kit from Applied Biosystems (Applied Biosystems Inc., Foster City, CA) and following the manufacturer's protocol. For samples of sufficient quantity, 1.0 μg of RNA was converted to cDNA. For less concentrated samples, for instance RNA collected from FNAB or FFPE samples, the entire 10 μl of RNA was used. Complete conversion of RNA to cDNA was assumed. For samples of low RNA quantity (FNAB) or quality (FFPE), cDNA was amplified for 14 cycles with pooled TaqMan Gene Expression Assays and TaqMan Pre-Amp Master Mix following manufacturer's protocol. Preamplified samples were diluted 20-fold into sterile TE buffer and stored at −20°C until needed. Expression of mRNA for individual genes was quantified using the 7900HT Real-Time PCR System with Applied Biosystems TaqMan Gene Expression Assays and Gene Expression Master Mix following manufacturer's protocol. TaqMan Microfluidics Expression Arrays were custom ordered to include our 12 class discriminating genes, three endogenous control genes, and 18S rRNA as a manufacturer's control, so that each assay would be run in triplicate for each sample loaded. Ct values were calculated using the manufacturer's software, and mean Ct values were calculated for all triplicate sets. ΔCt values were calculated by subtracting the mean Ct of each discriminating gene from the geometric mean of the mean Ct values of the three endogenous control genes.11
Biostatistical Analyses
Selection of endogenous control genes was performed using geNorm software (http://medgen.ugent.be/genorm), which identifies stable combinations of genes from a pool of potential controls. Rank order of discriminating probesets to be entered stepwise into the predictive test for cross validation were determined with minimum redundancy/maximum relevance (mRMR) software, using mutual information difference as the feature selection scheme and ± 0.5 standard deviations as a threshold for discretizing expression values. Significance of discriminating probeset overlap was determined using hypergeometric probability using the PROBHYBR function of SAS 9.0 statistical software as previously described.12 Molecular class assignments were made by entering the 12 ΔCt values of each sample into the machine learning algorithm GIST 2.3 Support Vector Machine (SVM) (http://bioinformatics.ubc.ca/svm). SVM was trained using a set of 28 well-characterized uveal melanomas of known molecular class and clinical outcome. SVM creates a hyperplane between the training sample groups (here, class 1 and class 2), then places unknown samples on one or the other side of the hyperplane based on their gene expression profiles. Confidence is measured by discriminant score, which is inversely proportional to the proximity of the sample to the hyperplane. Kaplan–Meier analysis was used to assess metastasis-free survival. Statistical analyses were performed using Partek Genomics Suite software, version 6.4 and MedCalc software, version 9.3.6.0.
Results
Identification of Candidate Discriminating and Control Genes
In the current study, we sought to identify a small number of the most highly discriminating genes and invariant control genes to migrate to a PCR-based clinically practical platform. The flow chart describing this process is summarized in Figure 1. First, we identified a set of genes that were highly robust discriminators between class 1 and class 2 across multiple microarray platforms. For this analysis, we used our previously published datasets obtained from the Affymetrix U133A and U133Av2 GeneChips and the Illumina Ref8 Beadchip.3,5,9 Genes were filtered according to signal-to-noise (S2N) ratio and then by fold change. We chose the top 38 genes, including 23 genes that were up-regulated in Class 1 and 15 genes that were up-regulated in Class 2. Based on our earlier estimates that a small number of discriminating genes would be sufficient to maintain full prognostic accuracy,5 we expected that these 38 candidate genes would be an ample pool of candidates from which to select the final set of genes based on their qPCR performance.
Figure 1.
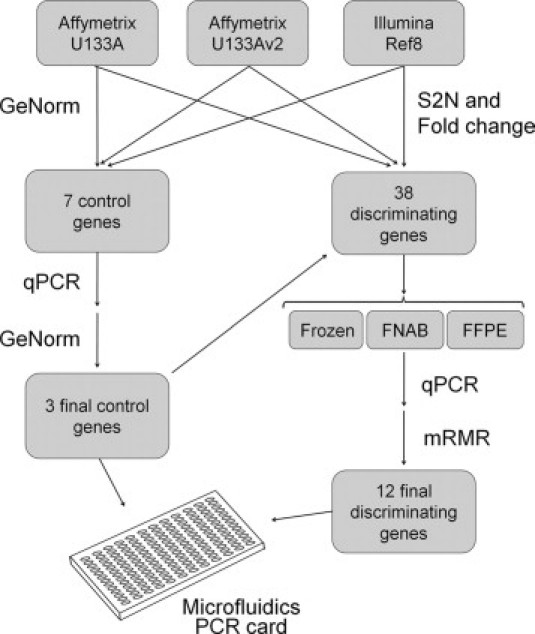
Flow diagram summarizing the process for migrating the gene expression profile from a hybridization-based microarray platform to a 15-gene qPCR-based assay performed on a microfluidics card.
In parallel, we used geNorm software to analyze the microarray expression data to identify genes to be used as controls whose expression varied minimally across all of the class 1 and class 2 tumor samples. This analysis ranked the top candidate genes and estimated that three control genes would be optimal for the internal structure of the dataset. We selected the seven top candidate genes to validate by qPCR, with the ultimate goal of identifying the three best control genes.
All candidate genes were evaluated further by qPCR using predesigned TaqMan Gene Expression Assays that were run in triplicate. The genes were tested on our well-characterized training set of 28 uveal melanomas (15 class 1 and 13 class 2) with long clinical follow-up of at least five years. Snap-frozen tissue was available on all samples, and FFPE tissue was available on 19 of these specimens (10 class 1 and nine class 2).
Selection of Control Genes
The candidate control genes were initially analyzed on unfixed, snap-frozen samples. Among the candidate control genes, MRPS21, RBM23, and SAP130 exhibited the least variable expression by geNorm analysis. These three genes were thus selected as the control genes. The geometric mean of the average Ct values of these genes was calculated for each sample, and this value was used as an internal control to normalize the ΔCt values obtained for the 38 candidate discriminating genes.11
Selection of Discriminating Genes
The candidate discriminating genes were initially analyzed on unfixed snap-frozen samples from the training set. Sixteen genes were eliminated because they were not validated by qPCR-based expression, which did not correlate with their microarray-based expression or with tumor class assignments. The remaining 22 genes were evaluated further to identify an optimum set of genes that would yield accurate class assignments in FNAB and FFPE samples.
To approximate the amount of cDNA obtained from FNAB specimens, cDNA from the snap-frozen training samples were diluted to ≤100 ng per sample, pre-amplified for 14 cycles using a pool of all 22 candidate discriminating genes and the three control genes, then analyzed by qPCR, using the geometric mean of the three control genes for normalization.
To test the candidate genes on archival tissue, FFPE tissue samples from the training set tumors were deparaffinized, and RNA was extracted. The RNA samples were converted to cDNA and then preamplified and analyzed as described above. In two of the 19 FFPE cDNA samples, multiple genes were undetectable by qPCR, and these samples were not used for subsequent studies.
Using the expression data from the three sample training sets, the 22 candidate discriminating genes were ranked by mRMR based on their weighted contribution, or relevance, to the overall gene expression signature and their lack of redundancy with other discriminating genes. The rank lists for each of the three training sets (snap-frozen, FNAB, and FFPE) were merged to generate a final list of 12 discriminating genes.
Development of the 15-Gene Assay
Using SVM as the machine learning algorithm, we analyzed 609 samples with the 15-gene assay. Preliminary outcome data were available on FNAB samples collected from 172 patients from the Washington University center. In this group of patients with a median follow up of 16 months, the 15-gene assay accurately identified which patients would develop metastatic disease (log rank test, P value = 1.9 × 10−6; Figure 2). We compared SVM to the Weighted Voting, Regularized Discriminant Analysis and the Predictive Analysis of Microarrays machine learning algorithms and none performed better than SVM in survival analyses (data not shown).
Figure 2.
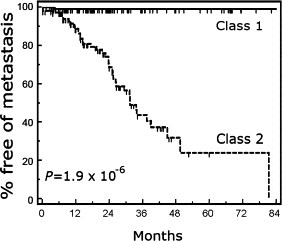
Kaplan-Meier analysis showing metastasis-free survival as a function of molecular class in 172 uveal melanoma patients. Statistical significance determined by the log rank test is shown.
Performance of Smaller Gene Sets
We next wished to determine the smallest number of genes necessary to maintain the accuracy of the assay. First, we used mRMR to rank the 12 discriminating genes in order of greatest contribution to the SVM classification with the least redundancy (Figure 3A). Next, we reclassified the samples using only the top 11 genes, the top 10 genes, etc. (Figure 3B). Compared with the class assignments using all 12 genes, the 11-gene set made no errors, the 10-gene set made one error (0.3%), and further reductions in the number of genes led to a sharp rise in the number of errors. When only the top gene (HTR2B) was used, 45 errors (13%) were made. As a result of these findings, coupled with the fact that there was no logistical advantage in reducing the number of genes below the 12 discriminating genes and three control genes because of the design of the TaqMan® microfluidics card, we retained all 12 discriminating genes in the final prognostic assay.
Figure 3.
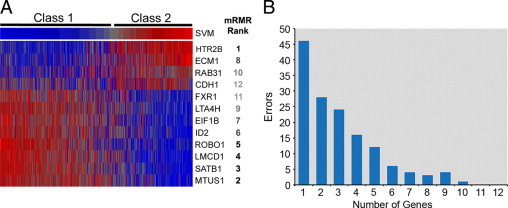
Analysis of top genes. A: Heatmap showing normalized expression of the 12 discriminating genes compared with SVM discriminant score in a set of prospectively collected samples (blue = low relative expression, red = high relative expression, gray = intermediate expression). B: Classification errors as a function of the number of discriminating genes in the assay. Genes were removed stepwise according to mRMR rank. See text for details.
Effect of RNA Quality and Quantity on Assay Performance
To evaluate the technical performance of the 15-gene assay in FNAB and fresh frozen samples, we analyzed 609 prospectively collected samples by FNAB (553 samples) or immediately after enucleation (56 samples) as part of a multicenter prospective study. Because the multicenter study focused on FNAB and fresh frozen samples, the technical performance for FFPE samples will be reported in a separate article.
For this analysis, a gene was said to be undetectable if its transcript was undetectable (i.e., no Ct value) after 40 qPCR cycles. A sample was said to have failed if one or more endogenous controls was undetectable. By this definition, sample failure occurred in 32 samples (5.2%). To determine the cause of these failures, we analyzed RNA quantity and quality. With regard to quantity, there was no relationship between sample failure and the concentration of RNA in the original sample as measured by NanoDrop (data not shown), indicating that the assay could detect RNA transcripts below the limits of the NanoDrop instrument.
Because the amount of RNA in these samples was too low to measure RNA quality using conventional electrophoretic methods, we used the geometric mean of the Ct values of the three endogenous controls as a measure of intact RNA template available for amplification in each sample. This was based on: (1) the Ct values were inversely proportional to the log of the number of available RNA templates in the sample (i.e., the more intact RNA present, the lower the Ct value), and (2) the endogenous controls should be expressed at constant levels across all uveal melanomas, a high Ct value should be a technical rather than biological aberration.
Most samples exhibited the expected linear relationship between RNA concentration and the geometric mean of the control Ct values (Figure 4A). Samples that were shifted below this expected linear range contained very low RNA concentrations, whereas those shifted to the right of the linear range were interpreted to contain poor quality RNA (i.e., detectable RNA concentration but diminished intact transcript available for amplification). This accounted for 29 (91%) of the 32 failures (Figure 4B). The only factor that we identified that was statistically associated with sample failure was incorrect shipping and/or handling of the sample in a manner that deviated from our study protocol (Figure 4B). Sample failure occurred in nine (36%) of the 25 samples that were handled incorrectly, compared with 25 (4.4%) of samples that were handled correctly, and this was statistically significant (Fisher exact test, P < 0.0001). There was no correlation between sample failure and molecular class assignment or cytologic diagnosis (Figure 4, C and D). Remarkably, the 15-gene assay failed on only one (2.0%) of 51 samples with a cytologic diagnosis of QNS (“quantity not sufficient,” indicating that there were too few cells on the cytology specimen for diagnosis), demonstrating that the assay is sufficiently sensitive to generate a class assignment in the vast majority of cases that are handled properly, even when few cells are seen on the cytology specimen.
Figure 4.
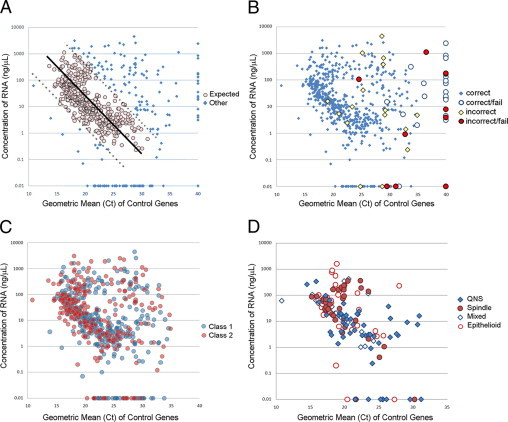
Relationship between RNA concentration and RNA quality. A: Scatter plot of RNA concentration versus the geometric mean of the Ct values for the three endogenous control genes. Black line indicates linear regression of values within the expected range; dashed lines indicate 90% prediction interval for regression. B: The same plot as in A, showing samples that were handled/shipped incorrectly and samples that failed the assay (defined as one or more endogenous controls undetectable after 40 qPCR cycles). C: The same plot as in A, showing samples according to their molecular class assignment. D: A subset of 148 cases from the Washington University center in which cytologic diagnosis was available. QNS indicates quantity not sufficient (too few cells to make a diagnosis).
Effect of Tumor Heterogeneity on Gene Expression Profile
Intratumoral heterogeneity for monosomy 3 has been estimated to occur in at least 14 to 18% of uveal melanomas,13,14,15 and this heterogeneity may be an important cause of reduced prognostic accuracy for chromosomal analysis. Indeed, it has been postulated that monosomy 3 tumor cells in heterogeneous tumors can be masked from detection by the more numerous disomy 3 cells.16 We investigated the effect intratumoral genetic heterogeneity on the 15-gene assay in two ways. First, we compared the expression profile of the 12 discriminating genes in eight fresh frozen tumor samples and matched postenucleation FNAB samples from the same tumor. The former were cross-sectional slices encompassing the base, center, apex, and edges of the tumor. The FNAB samples were obtained according to the FNAB protocol described in the Materials and Methods, with the needle directed toward the center of the tumor. The SVM class assignment was the same for all of the matched samples (Table 1). To investigate further the issue of heterogeneity, we analyzed the gene expression profile from 33 sections of seven different tumors obtained from enucleated eyes (Table 2). For all of these tumors, we obtained samples from the apex, base, and both tumor edges of cross-sectional tumor slices, as described in the Materials and Methods. For one of these tumors (MM110) we analyzed an additional four sections through different regions of the center of the tumor. Only one of the 33 regions that we analyzed showed discordance with the other regions within the same tumor. In MM116, all samples exhibited the class 2 signature except the posterior edge sample, which exhibited the class 1 signature. Importantly, however, this one discordant sample had a low discriminant score, which reduced the confidence in this result. We concluded from these experiments that intratumoral heterogeneity for the gene expression profile is uncommon.
Table 1.
Analysis of Heterogeneity of Gene Expression Signature in Fresh Frozen Tumors and Matching FNAB Samples
| Tumor number | SVM class | Discriminant score |
|---|---|---|
| 78_MB | Class 1 | −0.032 |
| 78_MM | Class 1 | −0.054 |
| 80_MB | Class 2 | 0.238 |
| 80_MM | Class 2 | 0.296 |
| 82_MB | Class 1 | −0.109 |
| 82_MM | Class 1 | −0.164 |
| 85_MB_Center | Class 1 | −0.069 |
| 85_MB_Edge | Class 1 | −0.041 |
| 85_MM | Class 1 | −0.086 |
| 86_MB | Class 1 | −0.193 |
| 86_MM | Class 1 | −0.195 |
| 87_MB | Class 2 | 0.290 |
| 87_MM | Class 2 | 0.294 |
| 89_MB | Class 1 | −0.137 |
| 89_MM | Class 1 | −0.153 |
| 91_MB | Class 2 | 0.284 |
| 91_MM | Class 2 | 0.271 |
Table 2.
Analysis of Regional Heterogeneity for Gene Expression Signature
| Tumor number | Location | SVM class | Discriminant score |
|---|---|---|---|
| MM 109 | Apex | Class 1 | −0.507 |
| MM 109 | Base | Class 1 | −0.770 |
| MM 109 | Anterior | Class 1 | −0.521 |
| MM 109 | Posterior | Class 1 | −0.674 |
| MM 110 | Edge/apex | Class 2 | 1.812 |
| MM 110 | Edge/base | Class 2 | 1.799 |
| MM 110 | Edge/anterior | Class 2 | 1.728 |
| MM 110 | Edge/posterior | Class 2 | 1.738 |
| MM 110 | Center/apex | Class 2 | 1.818 |
| MM 110 | Center/base | Class 2 | 1.717 |
| MM 110 | Center/anterior | Class 2 | 1.723 |
| MM 110 | Center/posterior | Class 2 | 1.824 |
| MM 112 | Apex | Class 2 | 0.257 |
| MM 112 | Base | Class 2 | 0.018 |
| MM 112 | Anterior | Class 2 | 0.172 |
| MM 112 | Posterior | Class 2 | 0.694 |
| MM 116 | Apex | Class 2 | 0.991 |
| MM 116 | Base | Class 2 | 0.665 |
| MM 116 | Anterior | Class 2 | 1.084 |
| MM 116 | Posterior | Class 1 | −0.102 |
| MM 120 | Apex | Class 2 | 0.899 |
| MM 120 | Base | Class 2 | 1.134 |
| MM 120 | Anterior | Class 2 | 0.961 |
| MM 120 | Posterior | Class 2 | 1.054 |
| MM 121 | Apex | Class 2 | 0.687 |
| MM 121 | Base | Class 2 | 0.765 |
| MM 121 | Anterior | Class 2 | 0.407 |
| MM 121 | Posterior | Class 2 | 0.454 |
| MM 122 | Apex | Class 1 | −0.639 |
| MM 122 | Base | Class 1 | −0.194 |
| MM 122 | Anterior | Class 1 | −0.567 |
| MM 122 | Posterior | Class 1 | −0.606 |
Because the work from our lab and others has shown that gene expression profiling appears to be a more accurate prognostic marker than chromosomal analysis6,7,8 and because the gene expression profile showed less intratumoral heterogeneity than monosomy 3, we speculated that the 15-gene assay may be more sensitive at detecting class 2 tumor cells in heterogeneous tumors where the majority of cells are class 1. To test this hypothesis, we mixed various ratios of RNA from three pairs of class 1 and class 2 tumors and analyzed the mixtures with the 15-gene assay (Figure 5). The assay correctly classified all pure class 1 and class 2 samples. Interestingly, samples containing as little as 25% contribution from class 2 tumor cells were identified as class 2 by the assay, indicating that the assay is very sensitive for detecting the class 2 signature in heterogeneous tumors.
Figure 5.
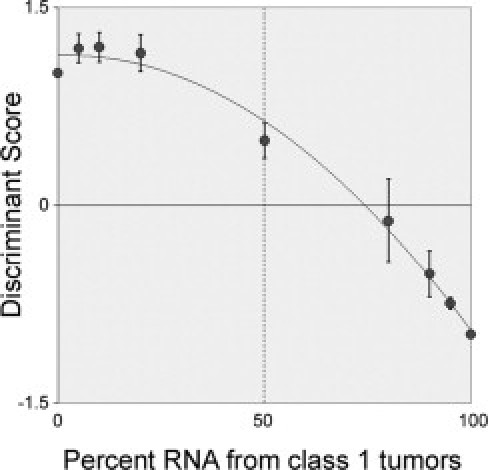
Dilution series of RNA from class 1 and class 2 tumors analyzed by the 15-gene assay. Discriminant scores for SVM (y axis) are plotted against contribution of class 1 RNA (x axis). The 15-gene assay classified mixtures as class 2 even when class 2 RNA was in the minority. Negative scores indicate class 1; positives scores, class 2. Closest fit trendline included for clarity.
Discussion
In this study, we migrated our gene expression profile for predicting metastasis in uveal melanoma from a high-density hybridization-based microarray platform to a PCR-based platform TaqMan microfluidics card, which was chosen because of its acceptance as a clinical testing standard around the world. The assay was optimized for use as a routine clinical assay on FNAB and FFPE samples. In this report, we focused on the results using the assay to analyze FNAB samples. Preliminary results of a prospective multicenter study of FNAB samples showed that samples shipped from long distances can easily be analyzed without loss of accuracy, and that following proper protocol is important for minimizing sample failures. In a subset of patients enrolled from our center, the 15-gene assay exhibited excellent prognostic accuracy. It will be important to reassess the prognostic accuracy from all collaborating centers after longer follow-up has been obtained.
We selected SVM as the machine learning algorithm for this assay based on our previous favorable experience with the algorithm,17 because it is widely accepted as among the most highly robust machine learning algorithms, and because it outperformed other similar algorithms in our dataset. SVM requires a training set of samples with known molecular class. We have generated such a training set of samples from patients with very long follow-up. SVM inputs the gene expression data of the training set as two sets of vectors (class 1 and class 2) in _n_-dimensional space, then constructs a separating hyperplane in that space to maximize the margin between the two data sets.18 SVM then classifies test samples by placing them on one or the other side of this hyperplane. The proximity of the sample to the hyperplane is inversely proportional to the discriminant score, a measure of confidence. To date, we have not identified a discriminant score below which the predictive accuracy decreases (data not shown), but future studies will be performed to address this question as greater numbers of patients and longer follow-up are obtained.
Although the biological functions of the discriminating genes are not important to their value as biomarkers, it is interesting that many of these genes have been shown previously to be involved in cancer. HTR2B is a serotonin receptor that signals through the heterotrimeric small GTPase, GNAQ,19 which is mutated in half of uveal melanomas.20 The bHLH inhibitor ID2 functions as a tumor suppressor in uveal melanoma and some other cancers.9,21,22 MTUS1 is a tumor suppressor protein.23 ECM1 is an extracellular matrix protein that plays an important role in angiogenesis and is overexpressed in some tumors.24 ROBO1 is an axonal guidance cue receptor that has been implicated in angiogenesis, as well as migration of neural crest and tumor cells.25,26 SATB1 regulates higher order chromatin organization, and its disruption could lead to broad epigenetic changes.27 LTA4H is involved in leukotriene synthesis, which influences immune modulation and leukocytic tumor infiltration.28,29 EIF1B is a translation initiation factor that is involved in protein synthesis and is down-regulated in some cancers such as hepatocellular carcinomas.30 RAB31 is a member of the RAS family of small GTPases and is thought to play a role in mitogenic signaling through the vesicular transport machinery.31 FXR1 is a nucleolar targeting factor that has been linked to fragile-X mental retardation and has been identified as a possible tumor suppressor in colon carcinoma.32 LMCD1 is a GATA transcription factor inhibitor that regulates migration and differentiation of mesenchymal cells.33 Various individual biomarkers have been suggested for prognostication in uveal melanoma, including nibrin,17 autotaxin,34 osteopontin,35 and TIMP3.36 However, our findings here indicate that the simultaneous measurement of expression of several highly discriminating genes provides superior prognostic accuracy compared with individual biomarkers.
Our findings suggest that intratumoral heterogeneity for gene expression profile is uncommon and may explain why the prognostic accuracy of gene expression profiling appears to be superior to chromosomal analysis. The 15-gene assay identifies the class 2 signature even when class 2 tumor cells represent a minority of the specimen, implying that only a small proportion of class 2 cells need to be present in the tumor to convey a high risk for metastasis. This finding could have far-reaching implications for understanding the biology of metastasis in uveal melanoma. However, a relatively small number of tumors were analyzed for heterogeneity, and a much larger study is underway to provide a better understanding of heterogeneity and its impact on clinical testing.
Acknowledgements
We thank the Collaborative Ocular Oncology Group (comprising 15 cooperating ocular oncology centers) for their contributions to this study. Group members include Thomas M. Aaberg, Jr., Michael M. Altaweel, James J. Augsburger, David S. Bardenstein, Devron H. Char, Zelia M. Correa, Paul T. Finger, Brenda L. Gallie, Hans E. Grossniklaus, Peter G. Hovland, Robert N. Johnson, Peter E. Liggett, Prithvi Mruthyunjaya, Rand Simpson, David J. Wilson, and William J. Wirostko.
Footnotes
Supported by grants (to J.W.H.) from the National Cancer Institute (R01 CA125970), Barnes-Jewish Hospital Foundation, Kling Family Foundation, Tumori Foundation, Horncrest Foundation, and a Research to Prevent Blindness David F. Weeks Professorship. This work was also supported by awards to the Department of Ophthalmology and Visual Sciences at Washington University from a Research to Prevent Blindness, Inc. Unrestricted grant, and the National Institutes of Health Vision Core grant P30 EY02687c.
CME Disclosure: J. William Harbour and Washington University may receive income based on a license of related technology by the University to Castle Biosciences, Inc. This work was not supported by Castle Biosciences, Inc. The other authors are employees of Washington University; they did not disclose any other relevant financial relationships.
References
- 1.Harbour JW. In: Clinical overview of uveal melanoma: introduction to tumors of the eye. Albert DM, Polans A, editors. Marcel Dekker; New York: 2003. pp. 1–18. [Google Scholar]
- 2.Cook SA, Damato B, Marshall E, Salmon P. Psychological aspects of cytogenetic testing of uveal melanoma: preliminary findings and directions for future research. Eye. 2008;23:581–585. doi: 10.1038/eye.2008.54. [DOI] [PubMed] [Google Scholar]
- 3.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang SH, Worley LA, Onken MD, Harbour JW. Prognostic biomarkers in uveal melanoma: evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008;18:191–200. doi: 10.1097/CMR.0b013e3283005270. [DOI] [PubMed] [Google Scholar]
- 5.Onken MD, Worley LA, Davila RM, Char DH, Harbour JW. Prognostic testing in uveal melanoma by transcriptomic profiling of fine needle biopsy specimens. J Mol Diagn. 2006;8:567–573. doi: 10.2353/jmoldx.2006.060077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Worley LA, Onken MD, Person E, Robirds D, Branson J, Char DH, Perry A, Harbour JW. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007;13:1466–1471. doi: 10.1158/1078-0432.CCR-06-2401. [DOI] [PubMed] [Google Scholar]
- 7.Petrausch U, Martus P, Tonnies H, Bechrakis NE, Lenze D, Wansel S, Hummel M, Bornfeld N, Thiel E, Foerster MH, Keilholz U. Significance of gene expression analysis in uveal melanoma in comparison to standard risk factors for risk assessment of subsequent metastases. Eye. 2008;22:997–1007. doi: 10.1038/sj.eye.6702779. [DOI] [PubMed] [Google Scholar]
- 8.van Gils W, Lodder EM, Mensink HW, Kilic E, Naus NC, Bruggenwirth HT, van Ijcken W, Paridaens D, Luyten GP, de Klein A. Gene expression profiling in uveal melanoma: two regions on 3p related to prognosis. Invest Ophthalmol Vis Sci. 2008;49:4254–4262. doi: 10.1167/iovs.08-2033. [DOI] [PubMed] [Google Scholar]
- 9.Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006;66:4602–4609. doi: 10.1158/0008-5472.CAN-05-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faulkner-Jones BE, Foster WJ, Harbour JW, Smith ME, Daivila RM. Fine needle aspiration biopsy with adjunct immunohistochemistry in intraocular tumor management. Acta Cytol. 2005;49:297–308. doi: 10.1159/000326153. [DOI] [PubMed] [Google Scholar]
- 11.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. research0034.0031-research0034.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wackerly DD, Mendenhall W, Scheaffer RL. Mathematical statistics with applications. Belmont Press; Duxbury: 1996. [Google Scholar]
- 13.Schoenfield L, Pettay J, Tubbs RR, Singh AD. Variation of monosomy 3 status within uveal melanoma. Arch Pathol Lab Med. 2009;133:1219–1222. doi: 10.5858/133.8.1219. [DOI] [PubMed] [Google Scholar]
- 14.Maat W, Jordanova ES, van Zelderen-Bhola SL, Barthen ER, Wessels HW, Schalij-Delfos NE, Jager MJ. The heterogeneous distribution of monosomy 3 in uveal melanomas: implications for prognostication based on fine-needle aspiration biopsies. Arch Pathol Lab Med. 2007;131:91–96. doi: 10.5858/2007-131-91-THDOMI. [DOI] [PubMed] [Google Scholar]
- 15.Mensink HW, Vaarwater J, Kilic E, Naus NC, Mooy N, Luyten G, Bruggenwirth HT, Paridaens D, de Klein A. Chromosome 3 intratumor heterogeneity in uveal melanoma. Invest Ophthalmol Vis Sci. 2009;50:500–504. doi: 10.1167/iovs.08-2279. [DOI] [PubMed] [Google Scholar]
- 16.Damato B, Coupland SE. Translating uveal melanoma cytogenetics into clinical care. Arch Ophthalmol. 2009;127:423–429. doi: 10.1001/archophthalmol.2009.40. [DOI] [PubMed] [Google Scholar]
- 17.Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res. 2005;11:1849–1853. doi: 10.1158/1078-0432.CCR-04-2054. [DOI] [PubMed] [Google Scholar]
- 18.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Jr, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cussac D, Boutet-Robinet E, Ailhaud M-C, Newman-Tancredi A, Martel J-C, Danty N, Rauly-Lestienne I. Agonist-directed trafficking of signalling at serotonin 5-HT2A, 5-HT2B and 5-HT2C-VSV receptors mediated Gq/11 activation and calcium mobilisation in CHO cells. Eur J Pharmacol. 2008;594:32–38. doi: 10.1016/j.ejphar.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 20.Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, Harbour JW. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell RG, Lasorella A, Dettin LE, Iavarone A. Id2 drives differentiation and suppresses tumor formation in the intestinal epithelium. Cancer Res. 2004;64:7220–7225. doi: 10.1158/0008-5472.CAN-04-2095. [DOI] [PubMed] [Google Scholar]
- 22.Tsunedomi R, Iizuka N, Tamesa T, Sakamoto K, Hamaguchi T, Somura H, Yamada M, Oka M. Decreased ID2 promotes metastatic potentials of hepatocellular carcinoma by altering secretion of vascular endothelial growth factor. Clin Cancer Res. 2008;14:1025–1031. doi: 10.1158/1078-0432.CCR-07-1116. [DOI] [PubMed] [Google Scholar]
- 23.Di Benedetto M, Bièche I, Deshayes F, Vacher S, Nouet S, Collura V, Seitz I, Louis S, Pineau P, Amsellem-Ouazana D, Couraud PO, Strosberg AD, Stoppa-Lyonnet D, Lidereau R, Nahmias C. Structural organization and expression of human MTUS1, a candidate 8p22 tumor suppressor gene encoding a family of angiotensin II AT2 receptor-interacting proteins. ATIP. Gene. 2006;380:127–136. doi: 10.1016/j.gene.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Yu J, Ni J, Xu X-M, Wang J, Ning H, Pei X-F, Chen J, Yang S, Underhill CB, Liu L, Liekens J, Merregaert J, Zhang L. Extracellular matrix protein 1 (ECM1) is over-expressed in malignant epithelial tumors. Cancer Letters. 2003;200:57–67. doi: 10.1016/s0304-3835(03)00350-1. [DOI] [PubMed] [Google Scholar]
- 25.Jia L, Cheng L, Raper J. Slit/Robo signaling is necessary to confine early neural crest cells to the ventral migratory pathway in the trunk. Dev Biol. 2005;282:411–421. doi: 10.1016/j.ydbio.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Legg J, Herbert J, Clissold P, Bicknell R. Slits and Roundabouts in cancer, tumour angiogenesis and endothelial cell migration. Angiogenesis. 2008;11:13–21. doi: 10.1007/s10456-008-9100-x. [DOI] [PubMed] [Google Scholar]
- 27.Kumar PP, Bischof O, Purbey PK, Notani D, Urlaub H, Dejean A, Galande S. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol. 2007;9:45–56. doi: 10.1038/ncb1516. [DOI] [PubMed] [Google Scholar]
- 28.Brock TG, Maydanski E, McNish RW, Peters-Golden M. Co-localization of leukotriene A4 hydrolase with 5-lipoxygenase in nuclei of alveolar macrophages and rat basophilic leukemia cells but not neutrophils. J Biol Chem. 2001;276:35071–35077. doi: 10.1074/jbc.M105676200. [DOI] [PubMed] [Google Scholar]
- 29.Okano-Mitani H, Ikai K, Imamura S. Human melanoma cells generate leukotrienes B4 and C4 from leukotriene A4. Arch Dermatol Res. 1997;289:347–351. doi: 10.1007/s004030050203. [DOI] [PubMed] [Google Scholar]
- 30.Lian Z, Pan J, Liu J, Zhang S, Zhu M, Arbuthnot P, Kew M, Feitelson MA. The translation initiation factor, hu-Sui1 may be a target of hepatitis B X antigen in hepatocarcinogenesis. Oncogene. 1999;18:1677–1687. doi: 10.1038/sj.onc.1202470. [DOI] [PubMed] [Google Scholar]
- 31.Ng EL, Wang Y, Tang BL. Rab22B's role in trans-Golgi network membrane dynamics. Biochem Biophys Res Comm. 2007;361:751–757. doi: 10.1016/j.bbrc.2007.07.076. [DOI] [PubMed] [Google Scholar]
- 32.Ivanov I, Lo KC, Hawthorn L, Cowell JK, Ionov Y. Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene. 2006;26:2873–2884. doi: 10.1038/sj.onc.1210098. [DOI] [PubMed] [Google Scholar]
- 33.Rath N, Wang Z, Lu MM, Morrisey EE. LMCD1/Dyxin is a novel transcriptional cofactor that restricts gata6 function by inhibiting DNA binding. Mol Cell Biol. 2005;25:8864–8873. doi: 10.1128/MCB.25.20.8864-8873.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh AD, Sisley K, Xu Y, Li J, Faber P, Plummer SJ, Mudhar HS, Rennie IG, Kessler PM, Casey G, Williams BG. Reduced expression of autotaxin predicts survival in uveal melanoma. Br J Ophthalmol. 2007;91:1385–1392. doi: 10.1136/bjo.2007.116947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadkol SS, Lin AY, Barak V, Kalickman I, Leach L, Valyi-Nagy K, Majumdar D, Setty S, Maniotis AJ, Folberg R, Pe'er J. Osteopontin expression and serum levels in metastatic uveal melanoma: a pilot study. Invest Ophthalmol Vis Sci. 2006;47:802–806. doi: 10.1167/iovs.05-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Velden PA, Zuidervaart W, Hurks MH, Pavey S, Ksander BR, Krijgsman E, Frants RR, Tensen CP, Willemze R, Jager MJ, Gruis NA. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer. 2003;106:472–479. doi: 10.1002/ijc.11262. [DOI] [PubMed] [Google Scholar]