Distribution of Genes for Virulence and Ecological Fitness among Diverse Vibrio cholerae Population in a Cholera Endemic Area: Tracking the Evolution of Pathogenic Strains (original) (raw)
Abstract
The pathogenic strains of Vibrio cholerae that cause acute enteric infections in humans are derived from environmental nonpathogenic strains. To track the evolution of pathogenic V. cholerae and identify potential precursors of new pathogenic strains, we analyzed 324 environmental or clinical V. cholerae isolates for the presence of diverse genes involved in virulence or ecological fitness. Of 251 environmental non-O1, non-O139 strains tested, 10 (3.9%) carried the toxin coregulated pilus (TCP) pathogenicity island encoding TCPs, and the CTX prophage encoding cholera toxin, whereas another 10 isolates carried the TCP island alone, and were susceptible to transduction with CTX phage. Most V. cholerae O1 and O139 strains carried these two major virulence determinants, as well as the Vibrio seventh pandemic islands (VSP-1 and VSP-2), whereas 23 (9.1%) non-O1, non-O139 strains carried several VSP island genes, but none carried a complete VSP island. Conversely, 30 (11.9%) non-O1, non-O139 strains carried type III secretion system (TTSS) genes, but none of 63 V. cholerae O1 or O139 strains tested were positive for TTSS. Thus, the distribution of major virulence genes in the non-O1, non-O139 serogroups of V. cholerae is largely different from that of the O1 or O139 serogroups. However, the prevalence of putative accessory virulence genes (mshA, hlyA, and RTX) was similar in all strains, with the mshA being most prevalent (98.8%) followed by RTX genes (96.2%) and hlyA (94.6%), supporting more recent assumptions that these genes imparts increased environmental fitness. Since all pathogenic strains retain these genes, the epidemiological success of the strains presumably depends on their environmental persistence in addition to the ability to produce major virulence factors. Potential precursors of new pathogenic strains would thus require to assemble a combination of genes for both ecological fitness and virulence to attain epidemiological predominance.
Introduction
The natural habitat of the gram-negative bacterium Vibrio cholerae is the aquatic ecosystems, although some strains of this species are associated with severe enteric infections in humans (Colwell and Spira, 1992; Faruque et al., 1998a, 2004a). Toxigenic strains of V. cholerae belonging to the O1 and O139 serogroups cause cholera, a severe diarrheal disease that occurs frequently as epidemics in many developing countries. Strains belonging to other serogroups, collectively referred to as non-O1, non-139, have also been implicated as etiologic agents of moderate to severe human gastroenteritis (Morris and Black, 1985; Janda et al., 1988), although the vast majority of the non-O1, non-O139 strains are presumed to be nonpathogenic bacteria constituting part of the normal aquatic flora.
V. cholerae O1 and O139 strains are commonly known to carry a set of virulence genes necessary for pathogenesis in humans (Kaper et al., 1995; Faruque et al., 1998a, 2004a). The major virulence factors of V. cholerae include cholera toxin (CT), which is responsible for the profuse watery diarrhea, and a pilus colonization factor known as toxin coregulated pilus (TCP). In addition to CT and TCP, cholera pathogenesis is presumed to depend on the synergistic effect of a number of putative accessory virulence-associated factors. These factors include the mannose-sensitive hemagglutinin (MSHA) pilus, the RTX toxin, hemolysins, as well as a few other accessory toxins (Kaper et al., 1995; Faruque et al., 1998a, 2004a; Faruque and Mekalanos, 2003a). However, the roles of the accessory virulence factors in cholera pathogenesis are not well established, and recent studies are beginning to reveal that at least some of these factors also play a role in environmental fitness of the pathogen (Watnick et al., 1999; Chiavelli et al., 2001).
The pathogenic strains of V. cholerae have evolved from nonpathogenic environmental strains, and horizontal transfer of virulence-related gene clusters play a major role in the process (Faruque et al., 1998b; Faruque and Mekalanos, 2003a). The ctxAB genes encoding CT reside in the genome of a lysogenic filamentous phage, CTXΦ (Waldor and Mekalanos, 1996), whereas genes encoding the major colonization factor, TCP, are part of a large cluster of genes also referred to as the TCP pathogenicity island (Kovach et al., 1996; Faruque and Mekalanos, 2003a). To track the evolutionary events in the origination of pathogenic V. cholerae from their non-pathogenic progenitors, it is important to identify intermediate strains that are likely to carry some of the virulence-related genes, but fall short of the complete set of genes required for pathogenesis and epidemic spread. For example, occasionally, environmental non-O1, non-O139 vibrios have been found to carry one or a few of the virulence-associated genes or their homologs (Mukhopadhyay et al., 2001; Faruque et al., 2004a). However, the general prevalence of virulence-associated genes among non-O1, non-O139 serogroups of V. cholerae, and the selection pressures for environmental V. cholerae carrying putative virulence genes are not clear.
Recent studies have also identified new putative virulence-related gene clusters in V. cholerae including genes for a type III secretion system (TTSS) (Dziejman et al., 2005), and the Vibrio seventh pandemic islands (VSP-1 and VSP-2) (Dziejman et al., 2002), the distribution of which in environmental V. cholerae strains, is largely unknown. In the present study, we conducted extensive analyses of V. cholerae O1 and non-O1 isolated from the aquatic environment in Bangladesh, and compared with strains from diarrhea patients for the presence of diverse genes associated with virulence or possible environmental fitness. Here we show that although the prevalence of major virulence genes is quite asymmetric among V. cholerae non-O1, non-O139 and O1 or O139 serogroup strains, the accessory virulence genes that now appear to have functions in the environment are uniformly distributed among both pathogenic and nonpathogenic strains.
Materials and Methods
Environmental sampling and bacterial strains
A total of 370 water samples collected between July 2003 and August 2005 from 11 different previously described sampling sites in Dhaka (Faruque et al., 2005a) were analyzed to obtain the environmental strains included in this study. Clinical strains used as controls or for comparison with the environmental strains were either from patients who reported to the Dhaka Hospital of the International Centre for Diarrhoeal Disease Research, Bangladesh (ICDDR,B), or from our culture collection. Of 324 V. cholerae strains analyzed in this study, 261 were non-O1, non-O139 strains (251 environmental and 10 from clinical sources), 46 were O1 strains (17 clinical and 29 environmental), and the remaining 17 strains (8 clinical and 9 environmental) belonged to the O139 serogroup.
Culture of environmental samples
Water samples were cultured to isolate V. cholerae by previously described methods (Faruque et al., 2004b). Briefly, 25 mL aliquots of water were added to 225 mL of alkaline peptone water (peptone 1% [w/v] NaCl 1% [w/v] pH8.5) and incubated at 37°C for 6 h. Dilutions of this enriched culture were plated on TTGA plates ( Monsur, 1961), and grown overnight at 37°C. Suspected Vibrio colonies were picked and subjected to biochemical and serological tests to identify V. cholerae belonging to the O1, O139, and non-O1, non-O139 serogroups (World Health Organization, 1974). All V. cholerae O1 or O139 isolates, as well as one non-O1, non-O139 isolate from each of 251 V. cholerae O1 or non-O1-positive water samples, were saved for further analyses.
Probes and PCR assays
Presence of virulence genes was determined by using specific DNA probes or PCR assays (Keasler and Hall, 1993; Faruque et al., 2003b, 2004b). Probes for the Vibrio seventh pandemic island (VSP-1 and VSP-2) genes (Dziejman et al., 2002) were generated by PCR from V. cholerae strain N16961 using primers based on published sequence of the respective genes (Heidelberg et al., 2000). The gene probe for the TTSS was a mixture of PCR products derived from four TTSS-related genes, including vcsV2, vspD, vcsN2, and vcsC2 of strain AM 19226 (Dziejman et al., 2005). Probes were labeled using a random primers DNA labeling kit (Invitrogen, Carlsbad, CA) and [α-32P]-deoxycytidine triphosphate (3000 Ci/mmol; Amersham Biosciences, Uppsala, Sweden). Colony blots or Southern blots were prepared using nylon filters (Hybond; Amersham Biosciences) and hybridized with the labeled probes following standard methods (Maniatis et al., 1982).
PCR assays used in this study for different virulence-associated gene clusters have been described previously. This included PCR assays specific for the tcpA, tcpI, and acfB genes of the TCP pathogenicity island (Keasler and Hall, 1993; Faruque et al., 1999), environmental tcpA variant (Mukhopadhyay et al., 2001), different variants of the repressor gene rstR of CTX phage, hemolysin gene (hlyA), RTX toxin genes (rtxA and rtxC), and mshA for mannose-sensitive hemagglutinin (Chow et al., 2001; Mukhopadhyay et al., 2001; Rivera et al., 2001; Faruque et al., 2003b). PCR reagents and kits were obtained from either Perkin-Elmer (Norwalk, CT) or Invitrogen, and PCR was done essentially as described previously.
Infection of V. cholerae with CTX-KmΦ
The susceptibility of V. cholerae strains to CTXΦ infection was assayed using a genetically marked phage, CTX-KmΦ (Waldor and Mekalanos, 1996; Faruque et al., 1998b). The marked phage was initially derived from strain SM44, in which a kanamycin resistance (KmR) determinant was introduced by marker exchange disrupting the ctxAB operon (Waldor and Mekalanos, 1996). CTX-KmΦ isolated from a mitomycin C–induced culture of strain SM44 was used to infect the classical V. cholerae O1 strain O395. CTX-KmΦ was prepared for the present study from a culture of strain O395 carrying RF of the phage O395 (pCTX-Km), using previously described methods (Waldor and Mekalanos, 1996; Faruque et al, 1998b). Susceptibility of V. cholerae strains to CTX-KmΦ was assayed under laboratory conditions and inside the intestines of infant mice as described previously (Faruque et al., 1998b). In brief, recipient strains were grown in LB at 37°C; the cells were precipitated by centrifugation and washed in fresh LB. The recipient cells and phage particles were mixed in LB to make an approximate final concentration of 106 bacterial cells and 106 phage particles per mL. For the in vitro assay the mixture was incubated for 16 h at 30°C, and aliquots of the culture were diluted and plated on LA plates containing kanamycin (50 μg/mL) to select for kanamycin-resistant colonies and on plates devoid of kanamycin to determine the total number of colonies.
For the in vivo assay, the same mixtures of phage and recipient cells were used to gastrointestinally inoculate groups of 5-day-old Swiss Albino mice obtained from the breeding facilities of the Animal Resources Branch of ICDDR,B. For each strain and phage combination, at least five mice were inoculated. Animals were sacrificed after 16 h, and their intestines were removed and homogenized in 10 mM phosphate-buffered saline (PBS) pH 7.2. The homogenate was centrifuged briefly to precipitate debris, the supernatant was then centrifuged at 4000 g for 10 min to precipitate bacterial cells, and the pellet was resuspended in PBS. The suspension was then screened for the presence of KmR V. cholerae colonies. The ratio of KmR colonies to total colonies was calculated and expressed as the percentage of recipient cells carrying the phage genome. Representative infected colonies were grown overnight in LB containing kanamycin (50 μg/mL), and cells were precipitated by centrifugation. Total DNA or plasmids were extracted from bacterial pellets by standard methods (Maniatis et al., 1982), and purified using microcentrifuge filter units (Sigma Chemical, St. Louis, MO). Presence of the phage genome was verified by comparative Southern blot analysis of total DNA and plasmid preparations from the phage-infected and the corresponding native strains.
Results
Diversity of CTX prophages and TCP genes
The distribution of CTX prophage and TCP pathogenicity island genes among diverse V. cholerae strains analyzed in this study is presented in Table 1. Of 251 non-O1, non-O139 strains isolated from surface water, 10 (3.9%) were positive for the CTX prophage and the TCP island-specific genes, including tcpA, tcpI, and acfB. This study also identified another 10 non-O1, non-O139 strains that were positive for the TCP island but did not carry any CTX prophage. All 9 environmental O139 strains and 26 of 29 environmental O1 strains studied were positive for both CTX prophage and the TCP island, whereas the remaining 3 O1 strains were positive for TCP but negative for CTX. All clinical isolates of V. cholerae O1 or O139 carried both the CTX prophage and TCP island. Interestingly, none of the 10 clinical isolates of V. cholerae non-O1, non-O139 analyzed in this study were positive for either CTX or TCP. However, as described later, some of these strains carried other virulence-associated genes, including those for a TTSS, RTX, and hemolysin.
Table 1.
Diversity of rstR Gene of the CTX Prophage and tcpA Gene of the TCP Pathogenicity Island Carried by Environmental and Clinical V. cholerae Strains in Bangladesh
| | | | | Type of rstR geneb | Type of tcpA geneb | | | | | | | | | -------------------- | ---------------------------- | -------- | ---------------------------- | ---------------------------- | - | ----------- | -------- | --------- | --------- | ----------- | -------- | | V. cholerae strain | Number of strainsa | Source | Presence of CTX | | | rstRClass | rstRET | rstRCal | rstREnv | tcpAClass | tcpAET | | Non-O1, non O139 | 3 | Water | + | − | − | − | + | + | − | | | | | 4 | Water | + | + | − | − | + | + | − | | | | | | 3 | Water | + | − | − | + | + | + | − | | | | | | 10 | Water | − | − | − | − | − | + | − | | | | | O139 | 9 | Water | + | − | + | + | − | − | + | | | | O139 | 8 | Patient | + | − | + | + | − | − | + | | | | O1 El Tor | 26 | Water | + | − | + | − | − | − | + | | | | O1 El Tor | 3 | Water | − | − | − | − | − | − | + | | | | O1 El Tor | 17 | Patient | + | − | + | − | − | − | + | | |
PCR or DNA probe assays were conducted to identify the repressor genes (rstR) carried by the CTX prophages resident in various strains. All toxigenic O1 strains were found to carry CTX prophages with the El Tor-type rstRET gene, whereas the non-O1, non-O139 strains carried prophages with diverse rstR genes. One or more rstR genes, including rstRClass, rstREnv, and rstRCalc, in different combinations were found among these strains (Table 1).All O139 strains analyzed carried both rstRCal and rstRET categories of the CTX phage repressor gene.
The tcpA genes carried by the TCP-positive strains were further differentiated into the El Tor and classical alleles using specific PCR assays. Whereas the O1 El Tor and O139 strains carried the El Tor allele, the non-O1, non-O139 strains were found to carry the classical allele of the tcpA gene (Keasler and Hall, 1993).
Distribution of VSP island genes among non-O1, non-O139 strains
The Vibrio seventh pandemic islands (VSP-1 and VSP-2) are two clusters of genes found initially in the seventh pandemic El Tor strains and were absent from the classical strains, as well as pre-seventh pandemic El Tor strains (Dziejman et al., 2002). To determine the general distribution of these genes, mixed probes consisting of PCR amplicons derived from VSP-1-specific genes VC175, VC178, VC181, and VC184, and VSP-2-specific genes VC490, VC493, and VC497 (Heidelberg et al., 2000; Dziejman et al., 2002) were used for initial screening of all V. cholerae strains. V. cholerae O1 and O139 strains, regardless of their source of isolation, were positive for VSP-1 and VSP-2 in the probe assays. However, out of 251 non-O1, non-O139 environmental strains tested, 22 (8.8%) reacted with the mixed probes, and of 10 clinical non-O1, non-O139 strains; 1 strain (10%) was positive with the probes (Tables 2 and 3). These 23 VSP-positive non-O1, non-O139 strains and 15 toxigenic O1 or O139 strains were extensively analyzed for the presence of 18 different ORF of VSP-1 and VSP-2 gene cluster using specific PCR assays for each ORF. The O1 and O139 strains were positive for all ORFs analyzed. However, the non-O1, non-O139 strains carried several of the ORFs of either VSP-1 or VSP-2, or both, but none of the strains carried all ORFs of both the VSP islands (Table 2).
Table 2.
Diverse Combination of Vibrio Seventh Pandemic Island ORFs Identified in V. cholerae Non-O1, Non-O139 Strains in Bangladesh
| | Presence of VSP-1a–specific ORFs | Presence of VSP-2–specific ORFs | | | | | | | | | | | | | | | | | ----------------------------------------------- | --------------------------------- | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ------ | ----------------- | | Strain | VC 176 | VC 179 | VC 180 | VC 181 | VC 182 | VC 183 | VC 184 | VC 185 | VC 490 | VC 491 | VC 492 | VC 493 | VC 494 | VC 496 | VC 497 | No. of isolates | | V. cholerae non-O1, non-O139b | + | + | + | + | + | + | + | + | − | − | − | − | − | − | − | 3 | | | − | + | + | + | + | + | + | + | − | − | − | − | − | − | − | 1 | | | | + | + | + | + | + | + | − | + | − | − | − | − | − | − | − | 1 | | | | − | − | − | − | − | − | − | − | + | + | + | + | + | + | + | 1 | | | | − | − | − | − | − | − | − | − | + | + | + | + | + | + | + | 2 | | | | − | − | − | − | − | − | − | − | − | − | − | − | + | + | + | 5 | | | | − | − | − | − | − | − | − | − | − | − | − | − | + | − | + | 6 | | | | − | − | − | − | − | − | − | − | − | − | − | − | − | − | + | 1 | | | | + | + | + | + | + | + | − | − | − | − | − | + | + | + | + | 1 | | | | + | + | + | + | + | + | + | + | − | − | − | − | + | + | + | 1 | | | | + | + | + | + | + | + | − | + | − | − | − | − | − | − | + | 1 | | | V. cholerae O1c | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 10 | | V. cholerae O139c | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 5 |
Table 3.
Distribution of Virulence-Associated Genes among 289 V. cholerae Strains Isolated from Surface Water and 35 Strains Isolated from Cholera Patients in Bangladesh
| | | Presence of virulence-associated genes and gene clustersa | | | | | | | | | | | | -------------------------- | ------------------------------------------------------------------- | ----- | ---- | ----- | ------ | ------ | ------ | ------ | -------- | -------- | ------ | | Serogroup | No. of isolates | CTX | rstC | TCP | rtx_A_ | rtx_C_ | hly_A_ | msh_A_ | _VSP_-1 | _VSP_-2 | TTSS | | (A) Environmental isolates | | | | | | | | | | | | | O1 | 24 | + | + | + | + | + | + | + | + | + | | | | 2 | + | + | + | + | + | − | + | + | + | − | | | O139 | 3 | − | − | + | + | − | − | + | + | + | − | | | 7 | + | + | + | + | + | + | + | + | + | − | | | | 2 | + | + | + | + | + | − | + | + | + | − | | | Non-O1, non-O139 | 2 | + | − | + | + | + | − | + | − | + | − | | | 5 | + | − | + | + | + | + | + | − | + | − | | | | 3 | + | − | + | + | + | + | + | − | − | − | | | | 10 | − | − | + | + | + | + | + | − | − | | | | | 3 | − | − | − | + | + | + | + | + | + | − | | | | 5 | − | − | − | + | + | + | + | + | − | − | | | | 5 | − | − | − | + | + | + | + | − | + | − | | | | 19 | − | − | − | + | + | + | + | − | − | | | | | 1 | − | − | − | − | − | + | + | − | − | + | | | | 152 | − | − | − | + | + | + | + | − | − | − | | | | 8 | − | − | − | − | + | + | + | − | − | − | | | | 14 | − | − | − | + | − | + | + | − | − | − | | | | 9 | − | − | − | − | − | + | + | − | − | − | | | | 1 | − | − | − | + | + | + | − | − | − | − | | | | 7 | − | − | − | + | + | − | + | − | − | − | | | | 2 | − | − | − | + | − | − | + | − | − | − | | | | 1 | − | − | − | + | + | − | − | − | − | − | | | | 1 | − | − | − | + | + | + | − | − | − | − | | | | 7 | − | − | − | + | + | − | + | − | − | − | | | | 2 | − | − | − | + | − | − | + | − | − | − | | | | 1 | − | − | − | + | + | − | − | − | − | − | | | | 1 | − | − | − | − | + | − | + | − | − | − | | | (B) Clinical isolates | | | | | | | | | | | | | O1 | 17 | + | + | + | + | + | + | + | + | + | − | | O139 | 8 | + | + | + | + | + | + | + | + | + | | | Non-O1, non-O139 | 3 | − | − | − | + | + | + | + | − | − | + | | | 3 | − | − | − | + | + | + | + | − | − | − | | | | 1 | − | − | − | − | + | + | + | − | − | + | | | | 1 | − | − | − | + | − | + | + | − | + | − | | | | 1 | − | − | − | + | − | + | + | − | − | − | | | | 1 | − | − | − | − | − | + | + | − | − | − | |
Prevalence of TTSS genes
Of 251 environmental V. cholerae non-O1, non-O139 strains analyzed in the present study, 30 (11.9%) were positive with the TTSS gene probes (Table 3).None of the 46 V. cholerae O1 and 17 V. cholerae O139 strains tested were positive for TTSS genes. Ten strains of V. cholerae non-O1, non-O139 that were positive for TTSS also carried the TCP island genes. All these 10 strains carried the classical-type tcpA allele, similar to other environmental TCP-positive non-O1, non-O139 strains.
Susceptibility of TCP-positive environmental strains to CTX phage
The pilus colonization factor TCP is also used by CTXΦ as its receptor for infecting V. cholerae cells. We, therefore, tested the susceptibility of all TCP-positive but CTX-negative environmental V. cholerae strains to CTXΦ. This included 3 TCP-positive CTX-negative O1 strains, and the 10 CTX-negative non-O1, non-O139 strains that were positive for both TTSS and TCP genes. The susceptibility was determined by using a KmR-labeled phage CTX-KmΦ, so that the infected strains showed a KmR phenotype (Waldor and Mekalanos, 1996; Faruque et al., 1998b). The results showed that all 3 O1 strains, and 8 of the 10 non-O1, non-O139 strains that carried both TTSS and TCP were infected by CTXΦ in the intestines of infant mice (mean frequency of infection between 5.9×10−5 and 3.7×10−4). However, none of the strains were infected in the in vitro assays. All KmR-infected strains were found to carry the CTX phage genome in subsequent assays (data not shown).
Prevalence of putative accessory virulence genes
In addition to the CTX prophage, and TCP pathogenicity island-specific genes, all V. cholerae strains were screened for the presence of putative accessory virulence genes. These included hlyA encoding hemolysin, r_txA_, and rtxC of RTX (repeat in toxin) gene cluster, and the mshA gene for mannose-sensitive hemagglutinin (MSHA). The distribution of these genes among V. cholerae strains is summarized in Table 3. All environmental or clinical O1 and O139 strains, and 98.8% of the environmental non-O1, non-O139 strains were positive for mshA. The RTX genes were also widely prevalent, and overall 96.2% of V. cholerae strains tested carried rtxA or rtxC or both these genes (Table 3).The hlyA gene was also common, and 247 (94.6%) of 261 non-O1, non-O139 strains tested were found to carry the hlyA gene. Among the O1 and O139 strains all clinical strains, and 79% of environmental strains were positive for the hlyA gene. Thus, overall mshA gene was most prevalent (98.8%) followed by RTX genes (96.2%) and hlyA (94.6%) among all V. cholerae strains tested. On the other hand, although CTX and TCP island genes are mostly confined to clinical strains and lesser number of environmental strains, the TCP- and CTX-positive strains invariably carry the mshA, rtx, and hlyA genes. This study identified at least two strains in which mshA is the only putative virulence-associated gene, and another 10 strains that carry mshA and RTX genes (Table 3).
Discussion
Understanding the evolution of bacterial pathogens from their nonpathogenic progenitors is challenging. Toxigenic V. cholerae, the etiologic agent of cholera, represents a paradigm for this process in that this organism evolved from environmental nonpathogenic V. cholerae by acquisition of virulence genes. The aquatic environment of a cholera endemic area harbors strains with various virulence gene profile, and thus constitute a reservoir of diverse virulence genes (Chakraborty et al., 2000). The ecological settings presumably favors extensive genetic interactions among V. cholerae mediated by phages and mobile genetic elements (Faruque and Mekalanos, 2003a) as well as selection of pathogenic clones leading to clustering of a critical combination of genes required for the emergence of an epidemiologically thriving pathogenic strain. We predict that the results described herein may help to better understand the significance of the distribution of major and accessory virulence genes, and genes for ecological fitness among diverse environmental and clinical strains of V. cholerae, and the steps toward the emergence of new pathogenic strains from possible evolutionary intermediates.
Environmental gene pool and hybrid strains
It is not clear what determines the emergence of pathogenic strains with particular CTX phage or tcpA types, but recent studies are beginning to reveal the presence of unconventional combination of virulence gene alleles in clinical V. cholerae strains (Nair et al., 2002; Ansaruzzaman et al, 2004). We presume that the diversity of virulence genes, for example, different tcpA alleles, or different CTX phages dispersed among V. cholerae strains in the environment contribute to this process. In a typical CTXΦ genome, the 4.5 kb core region comprises several ORFs, including ctxAB, zot, ace, and orfU, and encodes CT as well as the functions that are required for virion morphogenesis, whereas the 2.5 kb RS2 region encodes the regulation, replication, and integration functions of the CTXΦ genome (Waldor et al., 1997). The existence of at least four diverse repressor genes _(rstR_class, _rstR_ET, _rstR_Calc, and _rstR_Env) carried by different CTX phages, that is, CTXΦClass, CTXΦET, CTXΦCalc, and CTXΦEnv, respectively, have been recognized (Davis et al., 1999; Mukhopadhyay et al., 2001; Faruque et al., 2003b). This diversity of rstR genes constitutes the molecular basis for heteroimmunity among CTX phages (Kimsey and Waldor, 1998; Kimsey et al., 1998).
It has been established that due to heteroimmunity, toxigenic classical strains of V. cholerae O1 can be superinfected by CTXΦET, whereas toxigenic El Tor strains are resistant to further infection by the same phage (Kimsey and Waldor, 1998; Kimsey et al., 1998). The emergence of strains with multiple CTX phage types as shown in the present study thus might have resulted from superinfection of a toxigenic strain with a CTXΦ different from the resident CTX prophage in the strain.
Besides the rstR genes of CTX phages, different homologs of TCP island genes carried by environmental V. cholerae strains have been described previously (Mukhopadhyay et al., 2001). At least two tcpA alleles, including El Tor–type and classical-type tcpA genes, normally carried by El Tor and classical biotype strains, respectively, have been recognized. Interestingly, in the present study all toxigenic non-O1, non-O139 strains from the environment carried tcpA gene of the classical biotype (Table 1).The classical biotype strain of V. cholerae O1 that caused the sixth pandemic of cholera is now extinct. However, our results suggest that virulence-associated genetic material associated with the classical biotype of V. cholerae O1 including the classical type tcpA and the CTXClass prophage continues to exist as part of the environmental gene pool, and this may contribute to the genetic reassortment that produce new pathogenic V. cholerae strains. It is interesting to note that the isolation of unusual El Tor biotype strains possessing the classical-type tcpA gene from cholera patients in Bangladesh has been reported (Nair et al., 2002). Further, an El Tor biotype strain that unusually carries the CTXClass prophage has recently been associated with clinical cases of cholera in Mozambique (Ansaruzzaman et al., 2004).
Potential for the emergence of new pathogenic strains
The distribution of major virulence genes in the non-O1, non-O139 serogroups of V. cholerae was found to be largely different from that of the O1 or O139 serogroups (Tables 2 and 3). While genes for CT and TCP were present in most O1 or O139 strains, none of these strains carried the TTSS genes. On the other hand, TTSS genes were present in 11% of non-O1, non-O139 strains. Dziejman et al. (2005) recently showed the presence of TTSS similar to TTSS2 of V. parahaemolyticus in a clinical strain of V. cholerae non-O1, non-O139. The TTSS constitute a well-known category of virulence apparatus in diverse genera of gram-negative enteric pathogen, including Salmonella, Shigella, Escherichia, Yersinia, Pseudomonas, and Aeromonas species (Park et al., 2004). These bacteria encode specialized TTSS that deliver bacterial proteins (effector molecules) into the plasma membrane and cytoplasm of eukaryotic cells (Makino et al., 2003, Marlovits et al., 2004). The TTSS2 genes of V. parahaemolyticus have been shown to contribute to enterotoxicity of the pathogen.
Interestingly, in the present study we identified 10 environmental strains of V. cholerae non-O1, non-O139 that were positive for both TTSS and the TCP island genes, and 8 of these strains were infected by CTXΦ in the intestine of infant mice. Previous studies showed that the efficiency of CTXΦ infection was considerably higher in vivo, and this was attributed to more adequate expression of the phage receptor TCP in vivo than under laboratory conditions (Waldor and Mekalanos, 1996; Faruque et al., 1998b). In the present study, we did not detect any KmR transductants of the environmental strains in the in vitro assays, and infection with CTXΦ was detectable only in the infant mouse assay. This suggested that infection of these V. cholerae strains by CTXΦ was possibly TCP dependent. Hence, these TTSS-positive environmental strains of V. cholerae carry TCP island genes, and these genes are functional and capable of producing TCP pili; thus, these strains are potentially susceptible to CTXΦ infection under natural conditions, that is, in the human host or other environments where TCP is functionally expressed. We propose that these strains are intermediates in the evolution of a group of pathogenic V. cholerae strains that carry TTSS in addition to producing TCP and CT. This assumption is further supported by a previous study in which clinical TCP+ CT+ strains belonging to O141 serogroup have been found positive for the TTSS (Dziejman et al., 2005).
The Vibrio seventh pandemic islands (VSP-1 and VSP-2) are two clusters of genes found initially in the seventh pandemic El Tor strains and were absent from the classical strains, as well as pre-seventh pandemic El Tor strains (Dziejman et al., 2002). It has been suggested that these genes might have been involved in the epidemiological success of the seventh pandemic clone. The average GC content of VSP-1 and VSP-2 genes is lower (41%) than the average GC content (47%) of the rest of the V. cholerae chromosome, suggesting that like CTX and TCP island genes, these regions in the seventh pandemic strain were also acquired by horizontal transfer. The presence of incomplete VSP islands in environmental strains (Table 2)may represent VSP island precursors that are intermediates in the process of clustering of VSP ORFs into functional VSP islands. Alternatively, the presence of incomplete VSP islands in these strains might have resulted from multiple gene deletion events, which remain to be confirmed by further analysis.
Genes for ecological fitness and pathogenesis may be evolutionarily linked
While V. cholerae is autochthonous to the aquatic environment, it has been suggested previously that acquisition of virulence genes including the TCP pathogenicity island and the CTX prophage allowed specific strains of V. cholerae to become adapted to the human intestinal environment (Faruque et al., 1998a). In view of a recent report that TCP pili can promote biofilm formation on chitin surfaces (Reguera and Kolter, 2005), the function of TCP could also be to increase environmental persistence of the strains by using TCP to adhere to aquatic flora.
The finding that the CTXΦ that encodes CT uses TCP as its receptor for infecting new strains, and thus these two horizontally moving elements are linked evolutionarily, provides impetus for examining the evolutionary linkages among possible other genes supposedly related to virulence or environmental fitness. Possible evolutionary lineages for the emergence of pathogenic V. cholerae strains are shown in Figure 1. Although the role of MSHA pilus in V. cholerae virulence is controversial (Thelin and Taylor, 1996), recent studies have indicated that the MSHA pilus is used by environmental V. cholerae to adhere to plankton or to form biofilms on abiotic surfaces (Watnick et al., 1999; Chiavelli et al., 2001). Given the role of MSHA in environmental persistence, possession of mshA gene would provide a strong selective advantage to any V. cholerae recipient in the aquatic environment. This assumption is reflected in the widespread prevalence of mshA in V. cholerae (Table 3).This finding together with recent studies showing that MSHA can also act as a receptor for diverse filamentous phages (Jouravleva et al., 1998; Faruque et al, 2005b) further suggests that possession of mshA gene might have been a significant event in early V. cholerae evolution, since filamentous phages can facilitate horizontal gene transfer among V. cholerae (Faruque and Mekalanos, 2003).
FIG. 1.
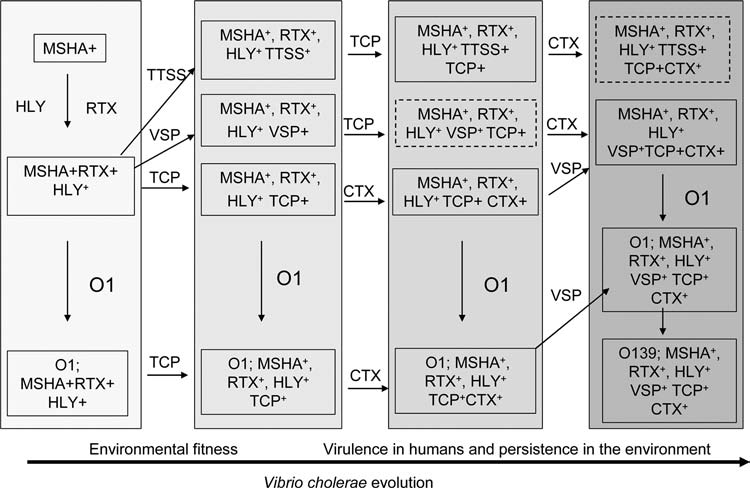
Schematic diagram of possible evolutionary lineages for the emergence of pathogenic V. cholerae strains. Lineages are based on the identification of intermediate strains carrying combination of genes for virulence or environmental fitness and other considerations (see text). Solid boxes describe strains that have been identified in this or other studies, whereas boxes with broken lines designate strains that could hypothetically exist but have not been isolated. Abbreviations: CTX, cholera toxin phage; TCP, toxin coregulated pilus gene cluster; MSHA, mannose-sensitive hemagglutinin; HLY, hemolysin; O1 and O139, various O antigen gene clusters; RTX, repeat in toxin gene cluster; VSP, Vibrio seventh pandemic island; TTSS, type III secretion system genes.
It has previously been suggested that the VSP islands could have been responsible for the success of the seventh pandemic strain, possibly by providing increased environmental fitness to strains carrying these genes (Dziejman et al., 2002). Similarly, the role in virulence of the newly discovered TTSS in non-O1 V. cholerae is not clearly known, but it has been suggested that the TTSS might also have functions in the environment (Dziejman et al, 2005), since V. cholerae are found in contact with eukaryotic cells not only within the human host but also in the marine environment where the bacteria can associate with insects, plankton, copepods, and shrimp. V. cholerae that reside in environmental reservoirs also survive predation by unicellular eukaryotes, and a recently described type VI secretion system (T6SS) in V. cholerae appears to be involved in the process (Pukatzki et al, 2006). The genes of the T6SS referred to as virulence-associated secretion (VAS) genes are required for cytotoxicity of V. cholerae cells toward an amoebae Dictyostelium discoideum, by a contact-dependent mechanism (Pukatzki et al., 2006).
Since cholera is a waterborne disease and the species is part of the normal aquatic flora, increased survival in the environment may also favor epidemic or pandemic spread of the pathogen. As shown, many of the accessory virulence genes are also present in environmental strains, which do not carry the major virulence genes, that is, CTX and TCP island genes. Thus the accessory genes, which have been described as putative virulence genes in the context of human infection, may also have a role in surviving unfavorable environmental conditions. Therefore, possession of these genes may provide a selective advantage to the bacteria in the aquatic environment. Interestingly, all pathogenic strains that carry CTX and TCP island also carry most of these genes. Thus, acquisition of genes for both virulence and environmental fitness may be required for the successful emergence of a pathogenic strain of V. cholerae from its nonpathogenic progenitor.
Acknowledgments
This research was funded in part by the National Institutes of Health grants 2RO1-GM068851-5 and AI070963-01A1 and AI 39129 (08) under different sub-agreements between the Harvard Medical School and ICDDR,B, and the Johns Hopkins University. The ICDDR,B is supported by countries and agencies that share its concern for the health problems of developing countries. M. Hasibur Rahman was a recipient of a doctoral fellowship from the University Grants Commission, Bangladesh.
References
- Ansaruzzaman M. Bhuiyan N.A. Nair G.B. Sack D.A. Lucas M. Deen J.L. Ampuero J. Chaignat C.L. Cholera in Mozambique, variant of Vibrio cholerae. Emerg Infect Dis. 2004;10:2057–2059. doi: 10.3201/eid1011.040682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S. Mukhopadhyay A.K. Bhadra R.K. Ghosh A.N. Mitra R. Shimada T. Yamasaki S. Faruque S.M. Takeda Y. Colwell R.R. Nair G.B. Virulence genes in environmental strains of Vibrio cholerae. Appl Environ Microbiol. 2000;66:4022–4028. doi: 10.1128/aem.66.9.4022-4028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavelli D.A. Marsh J.W. Taylor R.K. The mannose-sensitive hemagglutinin of Vibrio cholerae promotes adherence to zooplankton. Appl Environ Microbiol. 2001;67:3220–3225. doi: 10.1128/AEM.67.7.3220-3225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow K.H. Ng T.K. Yuen K.Y. Yam W.C. Detection of RTX toxin gene in Vibrio cholerae by PCR. J Clin Microbiol. 2001;39:2594–2597. doi: 10.1128/JCM.39.7.2594-2597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell R.R. Spira W.M. The ecology of Vibrio cholerae. In: Barua D., editor; Greenough W.B. III, editor. Cholera. Plenum Medical Book; New York, NY: 1992. pp. 107–127. [Google Scholar]
- Davis B.M. Kimsey H.H. Chang W. Waldor M.K. The Vibrio cholerae O139 Calcutta bacteriophage CTXΦ is infectious and encodes a novel repressor. J Bacteriol. 1999;181:6779–6787. doi: 10.1128/jb.181.21.6779-6787.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziejman M. Balon E. Boyd D. Fraser C.M. Heidelberg J.F. Mekalanos J.J. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci USA. 2002;99:1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziejman M. Serruto D. Tam V.C. Sturtevant D. Diraphat P. Faruque S.M. Rahman M.H. Heidelberg J.F. Decker J. Li L. Montgomery K. Grills G. Kucherlapati R. Mekalanos J.J. Genomic characterization of non-O1 non-O139 Vibrio cholerae reveals genes for a novel type III secretion system. Proc Natl Acad Sci USA. 2005;102:3465–3470. doi: 10.1073/pnas.0409918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Mekalanos J.J. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. 2003a;11:505–510. doi: 10.1016/j.tim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Faruque S.M. Albert M.J. Mekalanos J.J. Epidemiology, genetics, and ecoology of toxigenic Vibrio cholerae. Microbiol Mol Biol Rev. 1998a;62:1301–1314. doi: 10.1128/mmbr.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Asadulghani Saha M.N. Alim A.R.M.A. Albert M.J. Islam K.M.N. Mekalanos J.J. Analysis of clinical and environmental strains of nontoxigenic Vibrio cholerae for susceptibility to CTXΦ; molecular basis for the origination of new strains with epidemic potential. Infect Immun. 1998b;66:5819–5825. doi: 10.1128/iai.66.12.5819-5825.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Chowdhury N. Kamruzzaman M. Ahmad Q.S. Faruque A.S.G. Salam M.A. Ramamurthy T. Nair G.B. Weintraub A. Sack D.A. Reemergence of epidemic Vibrio cholerae O139, Bangladesh. Emerg Infect Dis. 2003b;9:1116–1121. doi: 10.3201/eid0909.020443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Nair G.B. Mekalanos J.J. Genetics of stress adaptation and virulence in toxigenic Vibrio cholerae. DNA Cell Biol. 2004a;11:723–741. doi: 10.1089/dna.2004.23.723. [DOI] [PubMed] [Google Scholar]
- Faruque S.M. Chowdhury N. Kamruzzaman M. Dziejman M. Rahman M.H. Sack D.A. Nair G.B. Mekalanos J.J. Genetic diversity and virulence potential of environmental Vibrio cholerae population in a cholera epidemic area. Proc Natl Acad Sci USA. 2004b;101:2123–2128. doi: 10.1073/pnas.0308485100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Islam M.J. Ahmad Q.S. Faruque A.S.G. Sack D.A. Nair G.B. Mekalonas J.J. Self-limiting nature of seasonal cholera epidemics: role of host-mediated amplification of phage. Proc Natl Acad Sci USA. 2005a;102:6119–6124. doi: 10.1073/pnas.0502069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Nasser I.B. Fujihara K. Diraphat P. Chowdhury N. Kamruzzaman M. Qadri F. Yamasaki S. Mekalanos J.J. Genomic sequence and receptor for the Vibrio cholerae phage KSF-1Φ: evolutionary divergence among filamentous vibriophages mediating lateral gene transfer. J Bacteriol. 2005b;187:4095–4103. doi: 10.1128/JB.187.12.4095-4103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque S.M. Siddique A.K. Saha M.N. Asadulghani Rahman M. Zaman K. Albert M.J. Sack D.A. Sack R.B. Molecular characterization of a new ribotype of Vibrio cholerae O139 Bengal associated with an outbreak of cholera in Bangladesh. J Clin Microbiol. 1999;37:1313–1318. doi: 10.1128/jcm.37.5.1313-1318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg J.F. Elsen J.A. Nelson W.C. Clayton R.J. Gwinn M.L. Dodson R.J. Haft D.H. Hickey E.K. Peterson J.D. Umayam L. Gill S.R. Nelson K. Read T.D. Tettelin H. Richardson D. Ermolaeva M.D. Vamathevan J. Bass S. Qin H. Dragoi I. Sellers P. McDonald L. Utterback T. Fleishmann R.D. Nierman W.C. White O. Salzberg S.H.O. Colwell R.R. Mekalanos J.J. Venter J.C. Fraser C. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda J.M. Powers C. Bryant R.G. Abbott S.L. Current perspective on the epidemiology and pathogenesis of clinically significant Vibrio spp. Clin Microbiol Rev. 1988;1:245–267. doi: 10.1128/cmr.1.3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouravleva E.A. McDonald G.A. Marsh J.W. Taylor R.K. Finkelstein M.B. Finkelstein R.A. The Vibrio cholerae mannose-sensitive hemagglutinin is the receptor for a filamentous bacteriophage from V. cholerae O139. Infect Immun. 1998;66:2535–2539. doi: 10.1128/iai.66.6.2535-2539.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper J.B. Morris J.G. Levine M.M. Cholera. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasler S.P. Hall R.H. Detection and biotyping Vibrio cholerae O1 with multiplex polymerase chain reaction. Lancet. 1993;341:1661. doi: 10.1016/0140-6736(93)90792-f. [DOI] [PubMed] [Google Scholar]
- Kimsey H.H. Waldor M.K. CTXΦ immunity: application in the development of cholera vaccines. Proc Natl Acad Sci USA. 1998;95:7035–7039. doi: 10.1073/pnas.95.12.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimsey H.H. Nair G.B. Ghosh A. Waldor M.K. Diverse CTXΦ and evolution of new pathogenic Vibrio cholerae. Lancet. 1998;352:457–458. doi: 10.1016/S0140-6736(05)79193-5. [DOI] [PubMed] [Google Scholar]
- Kovach M.E. Shaffer M.D. Peterson K.M. A putative integrase gene defines the distal end of large cluster of ToxR-regulated colonization genes in Vibrio cholerae. Microbiology. 1996;142:2165–2174. doi: 10.1099/13500872-142-8-2165. [DOI] [PubMed] [Google Scholar]
- Makino K. Oshima K. Kurokawa K. Yokoyama K. Uda T. Tagomori K. Iijima Y. Najima M. Nakano M. Yamashita A. Kubota Y. Kimura S. Yasunaga T. Honda T. Shinagawa H. Hattori M. Iida T. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- Maniatis T. Fritch E.F. Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Plainview, NY: 1982. [Google Scholar]
- Marlovits T.C. Kubori T. Sukhan A. Thomas D.R. Galan J.E. Unger V.M. Structural insights into the assembly of the type III secretion needle complex. Science. 2004;306:1040–1042. doi: 10.1126/science.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsur K.A. A highly selective gelatine-taurocholoatetellurite medium for the isolation of Vibrio cholerae. Trans R Soc Trop Med Hyg. 1961;55:440–442. doi: 10.1016/0035-9203(61)90090-6. [DOI] [PubMed] [Google Scholar]
- Morris J.G., Jr. Black R.E. Cholera and other vibrios in the United States. N Eng J Med. 1985;312:343–350. doi: 10.1056/NEJM198502073120604. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A.K. Chakraborty S. Takeda Y. Nair G.B. Berg D.E. Characterization of VPI pathogenicity island and CTXΦ prophage in environmental strains of Vibrio cholerae. J Bacteriol. 2001;183:4737–4746. doi: 10.1128/JB.183.16.4737-4746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair G.B. Faruque S.M. Bhuiyan N.A. Kamruzzaman M. Siddique A.K. Sack D.A. New variants of Vibrio cholerae O1 biotype El Tor with attributes of the classical bio-type from hospitalized patients with acute diarrhoea in Bangladesh. J Clin Microbiol. 2002;40:3296–3299. doi: 10.1128/JCM.40.9.3296-3299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.S. Ono T. Rokuda M. Jang M.H. Okada K. Iida T. Honda T. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun. 2004;72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S. Ma A.T. Sturtevant D. Krastins B. Sarracino D. Nelson W.C. Heidelberg J.F. Mekalanos J.J. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera G. Kolter R. Virulence and the environment: a novel role for Vibrio cholerae toxin-coregulated pili in biofilm formation on chitin. J Bacteriol. 2005;187:3551–3555. doi: 10.1128/JB.187.10.3551-3555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera I.N.G. Chun J. Huq A. Sack R.B. Colwell R.R. Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl Environ Microbiol. 2001;67:2421–2429. doi: 10.1128/AEM.67.6.2421-2429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelin K.H. Taylor R.K. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun. 1996;64:2853–2856. doi: 10.1128/iai.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor M.K. Mekalanos J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Waldor M.K. Rubin E.J. Pearson G.D. Kimsey H. Mekalanos J.J. Regulation, replication, and integration functions of the Vibrio cholerae CTXΦ are encoded by region RS2. Mol Microbiol. 1997;24:917–926. doi: 10.1046/j.1365-2958.1997.3911758.x. [DOI] [PubMed] [Google Scholar]
- Watnick P.I. Fullner K.J. Kolter R. A role for the mannose sensitive hemagglutinin in biofilm formation by Vibrio cholerae El Tor. J Bacteriol. 1999;181:3606–3609. doi: 10.1128/jb.181.11.3606-3609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. World Health Organization guideline for the laboratory diagnosis of cholera. Bacterial Disease Unit, World Health Organization; Geneva: 1974. [Google Scholar]