The sigma-1 receptor chaperone as an inter-organelle signaling modulator (original) (raw)
. Author manuscript; available in PMC: 2011 Dec 1.
Published in final edited form as: Trends Pharmacol Sci. 2010 Oct 1;31(12):557–566. doi: 10.1016/j.tips.2010.08.007
Abstract
Inter-organelle signaling plays important roles in many physiological functions. Endoplasmic reticulum (ER)-mitochondrion signaling affects intra-mitochondrial calcium (Ca2+) homeostasis and cellular bioenergetics. ER-nucleus signaling attenuates ER stress. ER-plasma membrane signaling regulates cytosolic Ca2+ homeostasis, and ER-mitochondrion-plasma membrane signaling regulates hippocampal dendritic spine formation. Here we propose that the sigma-1 receptor (Sig-1R), an ER chaperone protein, acts as an inter-organelle signaling modulator. Sig-1Rs normally reside at the ER-mitochondrion contact called the MAM (mitochondrion-associated ER membrane), where Sig-1Rs regulate ER-mitochondrion signaling and the ER-nucleus cross-talk. When cells are stimulated by ligands or undergo prolonged stress, Sig-1Rs translocate from the MAM to the ER reticular network and plasmalemma/plasma membrane to regulate a variety of functional proteins, including ion channels, receptors, and kinases. Thus, the Sig-1R serves as an inter-organelle signaling modulator locally at the MAM and remotely at the plasmalemma/plasma membrane. Many pharmacological/physiological effects of Sig-1Rs may relate to this unique action of Sig-1Rs.
Introduction
Originally mistaken for a subtype of opioid receptors, the sigma-1 receptor (Sig-1R) [1–3] is now recognized as a non-opioid receptor residing specifically at the endoplasmic reticulum (ER)-mitochondrion interface called the MAM [4]. At the MAM, the Sig-1R not only regulates the stability of inositol 1,4,5-trisphosphate (IP3) receptors to ensure proper Ca2+ signaling between the ER and mitochondrion [4] but also controls the dendritic spine arborization in neurons by increasing Rac-GTP on the plasma membrane (PM) through regulation of the level of reactive oxygen species (ROS) at the ER [5]. In addition, through the ROS, Sig-1Rs at the MAM control gene expression in the nucleus of an anti-apoptotic protein Bcl-2 by activating Nuclear Factor-KappaB (NFkB) [6]. Sig-1Rs reside specifically on ceremide- and cholesterol-rich lipid microdomains at the MAM [7], where, either upon stimulation by ligands such as cocaine and (+)pentazocine [8,9] or when under prolonged cellular stress [4], Sig-1Rs translocate to other areas of the cell. Those areas include the extended ER reticular network, including proximities right under the PM (i.e., the plasmalemmal area) or the PM, where Sig-1Rs interact and regulate the function of a variety of ion channels, receptors, or kinases [10–12]. We propose that the Sig-1R acts as an inter-organelle signaling modulator, not only locally at the MAM, where the receptor affects ER-mitochondrion and ER-nucleus signaling, but also remotely at the ER-PM interface, where it regulates functional proteins at the PM. Evidence to support this notion is reviewed and presented in this article.
A brief overview of the pharmacology of Sig-1Rs
Since the inception of the concept of the sigma-1 receptor, confusion over its identity and even existence lasted over a period of quite a few years until the receptor was cloned. The 223 amino acid Sig-1R that has been cloned from several mammalian species [13–17] contains 90% identical and 95% similar amino acid sequences across species. This receptor shares 30% identity and 67% similarity with a yeast sterol C8-C7 isomerase (ERG2), which is involved in postsqualene sterol synthesis [18]. Unlike the yeast sterol isomerase, however, the Sig-1R does not contain sterol isomerase activity [13] and shares no sequence homology with any known mammalian proteins, including the mammalian C8-C7 sterol isomerase, the emapomil binding protein (EBP). Though EBP was able to recover the ability to convert Δ8-sterol into Δ7-sterol in ERG2-deficient yeast Saccharomyces cerevisiae [19], the Sig-1R was unable to rescue C8-C7 isomerization [13]. Hydropathy analyses have indicated that the Sig-1R contains three hydrophobic domains (amino acids 11-29, 91-109 and 176-194), and is topologically similar to the yeast sterol isomerase. TMBase analysis (http://www.ch.embnet.org/software/tmbase/TMBASE_doc.html) predicts the first two hydrophobic domains (11-29 and 91-109) to be transmembrane-spanning helices with a 50 amino acid loop between, and a 125 amino acid C terminus [20]. In the ER of Chinese Hamster Ovary (CHO) cells, the topological model of Sig-1R [4] generally corroborates the two-transmembrane model initially proposed by Aydar et al. [20] (Figure 1).
Fig. 1. Model of the Sig-1R binding site.

The Sig-1R ligand binding region, highlighted by the circular area, includes the three predicted hydrophobic domains. The SBDLI (steroid binding domain-like 1) and SBDLII (steroid binding domain like II) regions are so named because of sequence homology to yeast sterol isomerase. Aspartate 188 (D188) in SBDLII has been identified as part of cocaine binding site [22]. Though the exact structure is unknown at this time, the three hydrophobic regions are depicted as alpha helical cylinders.
In addition to the amino acid sequence, the ligand binding domains of Sig-1Rs have been identified. Identification of the Sig-1R binding site has been substantially aided by Sig-1R protein purification [21] and the use of photoaffinity labels [22–25]. The SBDLI, SBDLII and the N-terminal TM1 domains of the Sig-1R have been demonstrated to form at least a portion of the binding site (Figure 1) [23, 26]. When the spatial relationship between SBDLI and SBDLII was assessed, it was discovered that there is a close juxtaposition (within 8 Å) of the SBDLI and SBDLII regions [25] (Figure 1). These data are further supported by studies that have demonstrated that an mRNA splice variant that lacks exon 3 (amino acids 119-149) of the four exon Sig-1R gene, is not able to bind (+)-[3H]-pentazocine [27] and that the region from the second transmembrane domain (SBD1) to the C-terminus may be part of the binding site [28]. Selected residues in the second transmembrane domain have also been shown to be important for ligand binding based on mutational studies [29]. In addition, a great deal of research in this field has been dedicated to the structure activity relationships of an array of ligands that bind to the Sig-1R (Tables 1) [1, 30–37]. These compounds include: benzomorphans (SKF-10047, pentazocine, dextromethorphan ); antipsychotics (haloperidol); antidepressants (fluvoxamine); steroids (progesterone); antihistamines (chlorpheniramine); nuclear hormone receptor ligands (tamoxifen); calcium channel antagonists (verapamil, emopamil); antifungals (fenpropimorph, tridemorph); and drugs of abuse (methamphetamine, cocaine, N,N′-dimethyltryptamine). Despite this wide array of ligands that bind to the Sig-1R, a basic structural pharmacophore has been identified [11, 50], which continues to provide a basis for further drug development.
Table 1.
Sigma-1 Receptor Ligands
Which ligand is an agonist and which ligand is an antagonist at Sig-1Rs has been an actively researched question. The precise molecular definition of Sig-1R agonists and antagonists is actively evolving. BD1047 and BD1063 (Table 1) have been established as Sig-1R antagonists, in part because the dystonia produced by the high affinity Sig-1R ligands, di-o-tolylguanidine (DTG) and haloperidol [47] was reduced when the compounds were administered to rats. Similar animal behavioral experiments have established haloperidol and NE-100 as antagonists, and (+)-SKF-10047, (+)-pentazocine, imipramine, fluoxetine, and DTG as agonists [47, 51–54]. The Sig-1R forms a Ca2+- regulating trimeric complex on the ER with ankyrin B and IP3receptors in NG-108 neuroblastoma cells [55]. Agonists, such as (+)-pentazocine,(+)-SKF10047, PRE084, cocaine, progesterone, and pregnenolone sulfate, dissociated ankyrin B (ANK 220) from the IP3 receptor, whereas NE-100 blocked the dissociation of ANK200 from IP3R-3 induced by (+)-pentazocine and was, therefore, defined as an antagonist [55]. Furthermore, the ability of ligands to dissociate the Sig-1R and BiP, an ER chaperone protein also known as GRP78 [4], identified (+)-pentazocine, (+)-SKF10047, PRE084, fluoxetine, cocaine, pregnenolone sulfate, and dehydroepiandosterone sulfate as agonists [4]. NE-100, progesterone, and haloperidol, however, inhibited the activation or dissociation between Sig-1R and BiP [4] and were thus classified as antagonists. Identification of N-alkylated tryptamines, such as N,N Dimethyl tryptamine ( DMT ), as endogenous agonists for the Sig-1R based on the loss of DMT-induced hypermobility responses in the Sig-1R homozygous null knock-out ( KO ) mouse [11] has additionally added to the knowledge of agonist pharmacology at the Sig-1R.
Lipid rafts and Sig-1Rs
In addition to the pharmacological investigations mentioned above, recent cell biological studies of Sig-1Rs have shed light on Sig-1Rs as an example of an unexpected link between pharmacology and cell biology. One striking finding is that Sig-1Rs have been reported to be associated with lipid-containing microdomains, where the receptor regulates the dynamics of lipids. For example, Sig-1Rs have been found in cholesterol-enriched, detergent-insoluble lipid rafts of the ER in NG108 neuroblastoma cells, in which they were shown to be important in the compartmentalization of ER-synthesized lipids [8, 9]. In the ER lipid droplets, the Sig-1R co-localized with caveolin-2, a cholesterol binding protein. In rat primary hippocampal cultures, Sig-1Rs were shown to form galactoceramide-enriched lipid rafts and promote differentiation of oligodendrocytes [56]. Additionally, the Sig-1R itself has been proposed to contain cholesterol binding domains in its C-terminal region [57]. Recent reports have also shown that the D-erythro-shingosine, sphinganine, (but not sphingosine-1 phosphate) and ceramides, endogenous lipids that in various forms are associated with lipid rafts, bind to the Sig-1R with relatively high affinity [7, 58]. Furthermore, immunohistocytochemical studies using different cell types have revealed that Sig-1Rs often cluster at submembranes of the ER [8, 9]. Recent studies demonstrate that one of the Sig-1R-enriched ER subdomains is identical to the MAM [4], which was originally discovered as an ER subcomponent physically interacting with the mitochondrial outer membrane [59]. In addition, Sig-1Rs were recently identified to reside specifically at the ceremide- and cholesterol-rich lipid microdomains at the MAM (see below). More interestingly, the cell biological localization of Sig-1Rs at the ER (or the MAM) is affected by the pharmacological manipulation of the receptor (see below).
Translocation of Sig-1Rs from the MAM
Several studies have demonstrated that Sig-1Rs are highly mobile at the ER membrane under particular conditions such as following pharmacological treatments or during cellular stress [4, 60]. Sig-1Rs can therefore redistribute to plasmalemmal ER cisternae and nuclear envelopes [7–9, 60]. Treatment of NG-108 cells with cocaine, for example, caused the translocation of Sig-1Rs from the ER to the neurite process as well as to the nucleus [8, 9]. Interestingly, Sig-1Rs were detected in the extracellular space in cocaine-treated NG-108 cells, suggesting perhaps a chaperone action of Sig-1Rs even in the extracellular space [8, 9]. In addition to ligand stimulation, stressors such as glucose deprivation [4] or depletion of ER Ca2+ by thapsigargin [4] also caused Sig-1Rs to translocate from the MAM. Overexpression of Sig-1Rs also increased the translocation of Sig-1Rs from the MAM to the plasmalemmal area [61]. Chronic ethanol consumption caused an upregulation (and perhaps translocation) of Sig-1Rs in the mouse brain [62]. More strikingly, (+)pentazocine, a Sig-1R agonist, caused a reduction of Sig-1Rs in the lipid raft fractions while apparently increasing them in the non-raft fractions [7, 8]. It is important to note that (+)pentazocine, applied at a concentration about 10 times its Ki value, caused ~ 50% of Sig-1Rs to dissociate from another ER chaperone BiP at the MAM [4; also see Fig. 2] and also caused Sig-1Rs to translocate from the MAM [8; also see Fig. 2]. Although how exactly the translocation is initiated and regulated is not clear at this moment. Evidence from recent studies suggests that lipid raft microdomains may play a part in the regulation of subcellular distribution of Sig-1Rs. In contrast to the other bulk ER membranes, the MAM can form detergent-resistant microdomains enriched with cholesterol and ceramides [7]. Solubilized Sig-1R complexes are broken into lipid raft-like fractions and have an affinity to bind these lipids [7]. Significantly, altering the composition of detergent-resistant microdomains at the ER by lowering cholesterol and/or inhibiting synthesis of ceramides caused translocation of the Sig-1R from the MAM to the ER cisternae [7].
Fig. 2. The Sig-1R chaperone as an interorganelle signaling modulator.
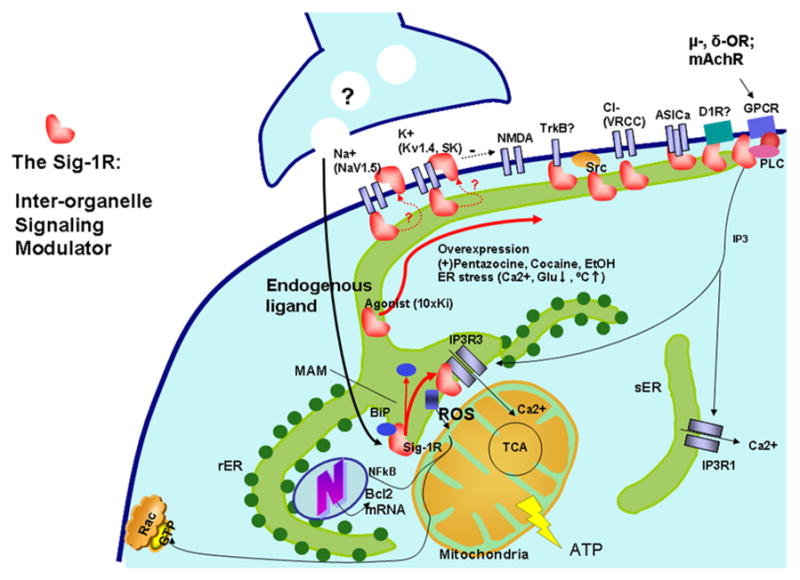
At the ER-mitochondrion interface (the MAM), Sig-1Rs, with the help of Sig-1R agonists, can dissociate themselves from another ER chaperone, BiP, and begin to chaperone conformationally unstable IP3Rs to enhance Ca2+ signaling from the ER into mitochondria [4] to increase the production of ATP in the cell through the tricarboxylic acid cycle (TCA) in the mitochondria [98]. Via an as yet unknown mechanism, Sig-1Rs at the MAM can also attenuate reactive oxidative species (ROS) at the ER to cause an increase of Bcl2 gene transcription in the nucleus to protect against cell death. Sig-1Rs attenuating the ROS level at the MAM can also, via interorganelle cross-talk, help maintain the integrity of mitochondria to prevent the release of apoptotic molecule cytochrome c that might otherwise destroy Rac•GTP on the PM, which is essential for formation of dendritic spines in neurons. If stimulated by high concentrations of agonists or impacted by extreme ER stress, Sig-1Rs translocate from the MAM to the plasmalemmal area or PM to bind various ion channels (e.g. Na+, K+, NMDA, ASIC and voltage-regulated chloride channels (VGCC)), receptors (e.g. TkB, D1R and other GPCRs) or kinases (e.g. Src). Please note that as long as those ion channels, receptors, or kinases are conformationally stable (i.e., no mutation or no redox stress imposed on the proteins due to the pathophysiology of diseases), the binding of Sig-1Rs does not affect the normal function of those conformationally correct ion channels, receptors, or kinases because chaperones exert their regulatory work only when the target proteins are “sick” and thus become clients. Many diseases are known to involve protein conformation changes. Thus, Sig-1Rs and associated ligands might represent potential therapeutic avenue for many human protein conformation diseases.
A question follows then: why do Sig-1Rs translocate?
Interactions of Sig-1Rs with functional proteins on the plasma membrane
Inasmuch as Sig-1Rs are chaperones correcting the misfolded proteins, it is possible that ligand- or stress-induced translocation of Sig-1Rs might switch their client proteins from those at the MAM to others at the plasmalemma, PM, or nuclear envelopes. Indeed, through patch clamp recordings in frog pituitary melanotrope cells, it was discovered that the Sig-1R agonist (+)-pentazocine inhibited potassium outward currents, and this effect was reversible by the Sig-1R antagonist NE-100 [63–65]. Concurrently, Jackson et al. established that the Sig-1R-mediated voltage-gated potassium ion channels could be modulated by Sig-1Rs without the utilization of G-proteins or phosphorylation [20, 66]. In addition to direct physical interaction and regulation of voltage gated potassium channels in mouse posterior pituitary nerve terminals [20], Sig-1Rs have been shown to regulate potassium channels in rat hippocampal slices, intracardiac neurons and tumor cells [67–69]. Sig-1R ligands have been shown to modulate several types of presynaptic Ca2+ channels in rat sympathetic and parasympathetic neurons [70, 71]. Sig-1Rs modulate N-methyl-D-aspartic acid (NMDA) receptor ion channels [72–75] and influence, in part, synaptic plasticity through small conductance calcium-activated potassium channels (SK channels) [76]. Recently, Sig-1Rs have been demonstrated to modulate cardiac voltage-gated sodium (Na+) channels (hNav1.5) in HEK293 cells and COS-7 cells, as well as neonatal mouse cardiac myocytes [77, 78]. Furthermore, Sig-1Rs have been demonstrated to physically associate with the acid-sensing ion channel (ASIC) 1a [79] as part of the regulation of these channels. It is probable that the chaperone functions of the Sig-1Rs are closely connected to functional ion channel regulation by providing a trafficking scaffold for ion channels [80]. Trafficking could be also regulated through endogenous molecules such as DMT and/or steroids such as progesterone [80].
Although the Sig-1R does not seem to interact with G-proteins directly [20, 66], it has been recently reported [81] that a functional and physical association of the Sig-1R occurs with cloned mu opioid receptors, which are G-Protein Coupled Receptors (GPCRs). This interaction, which occurs only through the antagonist-bound form of the Sig-1R and the mu opioid receptor, resulted in a very significant functional shift of the mu agonist (DAMGO) for G-protein activation to the left, an effect consistent with the in vivo observation that the Sig-1R antagonist potentiates morphine-induced analgesia. A similar result was observed for muscarinic acetylcholine receptor G protein activation by Sig-1Rs in mouse brain membranes, indicating a possible universal role for the Sig-1R in modulating GPCR agonist function and/or trafficking of GPCRs [82]. Indirect evidence also suggests that upregulated Sig-1Rs might interact with dopamine 1 receptors (D1Rs) in the brain of methamphetamine self-administering rats [83]. In addition to ion channels and receptors, kinases on the PM were also regulated by Sig-1Rs. For example, overexpression of Sig-1Rs increased the coupling between tyrosine kinase receptor B (TrkB) and phospholipase C (PLC) in cortical neurons [84]. Recent findings by Yao et al. [85] have demonstrated that cocaine causes Sig-1Rs to translocate from the ER to the PM lipid raft domains to activate Src kinase. It was shown that the activation of Src led to the generation of ROS and activation of nuclear transcription factor, thus leading to the induction of the chemokine MCP-1 in microglia [85].
The overall action of Sig-1Rs is depicted in Figure 2. Collectively, the data suggest a new dimension in the regulation of functional proteins on the PM; i.e., in addition to external stimuli, the functions of those proteins can be regulated via a seemingly coordinated fashion through a mobile interorganelle signaling modulator at the ER - the Sig-1R. However, it should be noted that whether Sig-1Rs interact with the PM proteins from proximity right under the PM (i.e., the plasmalemmal area) or instead from within the PM has not been totally clarified.
Sig-1Rs as therapeutic targets
In the central nervous system (CNS), Sig-1Rs play a part in complex biological processes, which include cocaine or methamphetamine addiction [83, 86], learning and memory, and depression [e.g., 10, 12]. Some reports using the molecular biological silencing approach have implicated these receptors in neurodegenerative disorders such as Alzheimer’s disease [87, 88], stroke, and neural degeneration due to HIV infection [see 10]. These findings have increased interest in Sig-1Rs as potential therapeutic targets in multiple CNS diseases, examples of which are described below.
Alzheimer’s disease (AD) is characterized by progressive neurodegenerative processes related to the presence of (a) extracellular senile plaques composed of insoluble extracellular aggregates of Aβ peptides derived by proteolytic cleavage of the amyloid precursor protein, and/or (b) neurofibrillary tangles composed of intracellular deposits of paired helical filaments of hyperphosphorylated Tau protein. Patients with AD exhibit selective synaptic and neuronal loss in brain regions involved in learning and memory [89]. The potential neuroprotective impact of Sig-1R was initially suggested by the observation that genetic polymorphisms may constitute a protective factor against AD [90]. Recent observations in preclinical AD models indeed confirmed the role of the Sig-1R as an endogenous neuroprotective system in AD. In vitro, the Sig-1R agonists PRE-084 or MR-22 attenuated the Aβ25-35-induced expression of the proapoptotic protein Bax and neuronal death in rat cortical cultures [91]. In vivo, PRE-084 prevented the appearance of oxidative stress and learning impairments induced in mice several days after the intracerebral injection of an oligomeric preparation of amyloid-β25-35 peptide, a nontransgenic rodent model of AD [92]. Interestingly, this Sig-1R mediated effect was shared by donepezil, the cholinesterase inhibitor in clinical use in AD patients, which also possesses nanomolar affinity at the Sig-1R [93]. More recently, the mixed muscarinic ligand/Sig-1R agonists, ANAVEX1-41 and ANAVEX2-73, appeared to be neuroprotective at very low (sub mg/kg) doses against the morphological damages induced by Aβ25-35, including cell death in the hippocampus, astrocytic activation, biochemical alterations (caspase-3, -9, -12 activation, oxidative stress) and learning and memory deficits [87, 88]. The exact mechanism whereby how Sig-1R agonists may attenuate mnemonic deficit is unknown at present. However, recent data suggest that this action of Sig-1Rs may involve neuronal proliferation and differentiation. Tsai et al. [5] showed that sig-1Rs promote dendritic spine formation in hippocampus, perhaps by tonically inhibiting the free radical formation at the ER [5]. Li et al. [93] first demonstrated that the intracerebral injection of Aβ25-35 in mice stimulates proliferation of progenitor cells in the hippocampal dentate gyrus (DG). However, a large population of the newborn cells died within two weeks, after birth, which happens to be a critical period for neurite growth. The group then demonstrated that the neurosteroid dehydroepiandrosterone (DHEA) dose-dependently attenuated the Aβ25-35-induced neuronal loss by activating Sig-1Rs. They also showed that the DHEA effect could be mimicked by the Sig-1R agonist PRE-084 and that the DEHA effect was blocked by the Sig-1R antagonist NE100 [93]. Interestingly, the Aβ25-35-induced decrease of dendritic spine density, and the length of doublecortin-positive cells in the DG were also attenuated by the DHEA-treatment [93].
The above observations confirm the pharmacological potential of Sig-1R agonists as cognition-enhancing agents and demonstrate that Sig-1R agonists do so perhaps by their ability to increase dendrites and dendritic spine formation that are critical for neuronal communications in the brain.
Sig-1R compounds may also have therapeutic potential in other neurodegenerative disorders. Numerous Sig-1R ligands have been tested in stroke models and appeared to be neuroprotective. For example, the Sig-1R ligand 4-phenyl-1-(4-phenylbutyl)piperidine (PPBP) was examined for its neuroprotective property in rats by using the middle cerebral artery occlusion (MCAO) technique [94]. PPBP significantly reduced the infarct volume in the cortex [94], and its neuroprotective property was related to its attenuation of nitric oxide production in the brain [95]. The most striking example of an Sig-1R ligand acting as a neuroprotective agent came from a study by Ajmo et al. [96] who reported that, a subcutaneous injection of Sig-1R agonist DTG (15 mg/kg) given 24 hrs after MCAO in rats reduced infarct areas in both cortical/striatal and cortical/hippocampal regions by more than 80% when compared to controls [96]. DTG was found to attenuate the rise in the intracellular Ca2+ concentration caused by ischemia via Sig-1Rs because the Sig-1R antagonist BD1047 blocked this protective effect of DTG [97]. The efficacy of DTG may also be due to its ability to reduce the inflammatory response in the brain [96]. Other Sig-1R ligands have also been shown to be neuroprotective. PRE-084 and the antitussive Sig-1R agent dimemorphan were neuroprotective in MCAO rats [98]. Both drugs ameliorated the size of infarcts by 50% and 70% respectively, and their protective effects were blocked by BD1047 [98]. Further support for a role of Sig-1Rs in neuroprotection came from brain slice studies. When examining the effect of drugs on spreading depression, which is a profound but transient neuronal/glial depolarization that transmits across cortical and subcortical gray during ischemia, Anderson & Andrew [99] found that pretreatment of brain slices with Sig-1R agonist dextromethorphan or carbetapentane significantly blocked the spreading depression and that the Sig-1R antagonist BD1063 nullified the blocking effect of those ligands [99].
The Sig-1R ligands could also have therapeutic implication in multiple sclerosis (MS) although more experimental evidence is certainly required. Haiman et al. [100] examined the brain activity and associated cortical structures involved in the pseudobulbar effect that is often seen in patients with MS and is characterized by uncontrollable episodes of laughing and/or crying. They found that a combined medication of quinidine and the Sig-1R ligand dextromethorphan resulted in the attenuation of the pseudobulbar effect, as well as a normalization of electrophysiological measures in those patients [100]. This study provides the first evidence for potential involvement of Sig-1Rs in MS.
Sig-1Rs and associated ligands may have an important role in neuroinflammation, especially in cocaine-HIV-related CNS inflammation. Recent efforts have aimed at unraveling the role of Sig-1R in HIV-1-associated neurological disorder (HAND) among cocaine abusers and have thus investigated the effect of cocaine on the virus-producing cells of the CNS, the microglia [85, 101, 102]. Neuropathological correlates for HAND include glial activation, neuroinflammation, chemokine induction, increased monocyte extravasation, and neural death/loss. Research in this area has implications for HIV-1-infected cocaine abusers who are known to have an increased risk of stroke and CNS-associated neuroinflammation. Additionally, the role of Sig-1Rs in cocaine-induced immune alteration and HIV-1 expression has also been examined in several model systems, including the humanized mice. In this model of severe combined immunodeficiency (SCID) mice, cocaine exposure was shown to increase the expression of HIV co-receptors C-C chemokine receptor type 5 (CCR5) and chemokine (CXC motif) receptor 4 (CXCR4) in human peripheral blood mononuclear cells, thereby facilitating increased viremia. Interestingly, pretreatment of these mice with the Sig-1R antagonist-BD1047 significantly dampened the viral infection induced by cocaine [85, 101, 102]. Consistent with these findings, cocaine-mediated increase of HIV replication by microglial cells was also significantly abrogated by BD-1047 [85]. The results suggest that Sig-1R antagonists may reduce cocaine-HIV-related neuroinflammation and dementia.
The Sig-1R also has become a target for modulating cell growth and immune responses because SR31747, a potent immunosuppressant, was found to bind the Sig-1R [103–105]. Indeed, an important discovery has been that Sig-1Rs (and sigma-2 receptors) are overexpressed in many human and non-human tumors [57, 106, 107]. As a result, the pharmacological study of small molecule Sig-1R ligands for potential clinical treatment and imaging applications has developed into an active area of cancer research [e.g., 108].
Overall, these findings point to new and emerging roles of Sig-1Rs in disease pathogenesis. Intriguingly, most of these functions of Sig-1Rs are apparently mediated by an intricate mechanism involving their translocation to the plasma membrane [85, 101, 102] or by translocation to unique ER cisternae that are proximal to the plasma membrane, e.g., as has been discovered by the Ruoho laboratory in the ventral horn motor neuron cell bodies of mice implicated in ALS [82]. However, the involvement of Sig-1Rs in ALS however has not been demonstrated so far.
Pharmacological and therapeutic selectivity concerning Sig-1R ligands
Given the wide-ranging effects of Sig-1R activation discussed above, an important issue arises concerning the potential for Sig-1R targeting in the treatment of disease states. At the MAM, Sig-1Rs might have only a limited number of client proteins to interact with (e.g., IP3R3). Therefore, because Sig-1Rs can chaperone IP3R3 to increase the Ca2+ flow from the ER to mitochondria to increase ATP production, one can increase bioenergetics in cells by simply adding Sig-1R agonists to the living system. However, when Sig-1Rs translocate, either by agonist stimulation or extreme ER stress, Sig-1Rs may interact with many different target proteins. If those target proteins are related to diseases, how might it be possible to control the specificity of Sig-1R-related pharmacotherapy? We argue that the specificity can be achieved because of the intrinsic nature of the Sig-1R as a molecular chaperone.
One characteristic feature of molecular chaperones is that even though they may bind conformationally correct proteins, they may increase their affinity and exert their chaperone activity on the proteins only when the target proteins are conformationally misfolded or are prone to be misfolded under certain experimental conditions. According to this speculation, the target ion channels, receptors, or kinases at the PM might have been affected by Sig-1Rs or Sig-1R ligands only because they were conformationally unstable under the experimental conditions, thus allowing Sig-1Rs to exert their regulation on those proteins. In other words, under normal physiological conditions, most ion channels or receptors are not affected by Sig-1Rs or ligands. Only when caused by diseases (for example due to gene mutations or intra-protein oxidative stress) could specific ion channels or receptors demand the assistance of Sig-1R chaperones. Therefore, it is possible that it is the nature of the disease that provides the selectivity for Sig-1R pharmacotherapy. Sig-1R-based pharmacotherapy may particularly benefit patients suffering from protein conformation diseases.
Conclusion
Over the past 2–3 decades, there has been considerable research focus on Sig-1R ligands and their binding activity. With the cloning of the receptor in 1996 [13], the development of the Sig-1R KO mouse in 2003 [66], as well as the pharmacological findings and conceptualizations put forth here, the field is well poised to elucidate the important inter-organelle signaling property of the Sig-1R in cellular homeostasis and physiological functions. Because Sig-1Rs are present in the CNS as well as in the lung, liver, pancreas, spleen, and adrenal gland [e.g., 4, 55, 67], pharmacological interventions of this interorganelle signaling modulator, the Sig-1R, at the MAM, the plasmalemmal area, or the PM may represent new avenues for therapeutic developments in combating many human CNS and peripheral diseases including particularly protein conformation diseases in this regard.
Acknowledgments
The authors thank the financial supports from the following sources: NIDA Intramural Research Program (TPS, TH); INSERM (TM); MH-068212, DA-020392, DA-023397, DA-024442 (SB); MH-065503, T32 GM08688, F31DA-022932, Gates Millenium Foundation Award (AER). We also thank the following collaborators: S.-Y. Tsai, M. Fujimoto, J. Meunier (TPS, TH); J. Meunier (TM); H.H. Yao (SB); U. Chu, D. Fontanilla, L. Guo (AER).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Su TP, et al. Steroid binding at sigma receptors suggest a link between endocrine, nervous, and immune systems. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- 2.Snyder SH, Largent BL. Receptor mechanisms in antipsychotic drug action: focus on sigma receptors. J Neuropsychiatry Clin Neurosci. 1989;1:7–15. doi: 10.1176/jnp.1.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Quirion R, et al. A proposal for the classification of sigma binding sites. Trend Pharmacol Sci. 1992;13:85–87. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 5.Tsai SY, et al. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive Rac.GTP pathway. Proc Natl Acad Sci USA. 2009;106:22468–22473. doi: 10.1073/pnas.0909089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meunier J, Hayashi T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J Pharmacol Exp Ther. 2010;332:388–397. doi: 10.1124/jpet.109.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayashi T, Fujimoto M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol. 2010;77:517–28. doi: 10.1124/mol.109.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi T, Su TP. Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108–15 cells. J Pharmacol Exp Ther. 2003;306:726–733. doi: 10.1124/jpet.103.051292. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Su TP. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J Pharmacol Exp Ther. 2003;306:718–725. doi: 10.1124/jpet.103.051284. [DOI] [PubMed] [Google Scholar]
- 10.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacology & Therapeutics. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fontanilla D, et al. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fishback JA, et al. Sigma receptors: Potential targets for a new class of antidepressant drugs. Pharmacol Ther. 2010;127:271–282. doi: 10.1016/j.pharmthera.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanner M, et al. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kekuda R, et al. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1) Biochem Biophys Res Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 15.Prasad PD, et al. Exon-intron structure, analysis of promoter region, and chromosomal localization of the human type 1 sigma receptor gene. J Neurochem. 1998;70:443–451. doi: 10.1046/j.1471-4159.1998.70020443.x. [DOI] [PubMed] [Google Scholar]
- 16.Seth P, et al. Cloning and functional characterization of a sigma receptor from rat brain. J Neurochem. 1998;70:922–931. doi: 10.1046/j.1471-4159.1998.70030922.x. [DOI] [PubMed] [Google Scholar]
- 17.Mei J, Pasternak GW. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem Pharmacol. 2001;62:349–355. doi: 10.1016/s0006-2952(01)00666-9. [DOI] [PubMed] [Google Scholar]
- 18.Moebius FF, et al. High affinity of sigma 1-binding sites for sterol isomerization inhibitors: evidence for a pharmacological relationship with the yeast sterol C8-C7 isomerase. Br J Pharmacol. 1997;121:1–6. doi: 10.1038/sj.bjp.0701079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silve S, et al. Emopamil-binding protein, a mammalian protein that binds a series of structurally diverse neuroprotective agents, exhibits delta8-delta7 sterol isomerase activity in yeast. J Biol Chem. 1996;271:22434–22440. doi: 10.1074/jbc.271.37.22434. [DOI] [PubMed] [Google Scholar]
- 20.Aydar E, et al. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron. 2002;34:399–410. doi: 10.1016/s0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran S, et al. Purification and characterization of the guinea pig sigma-1 receptor functionally expressed in Escherichia coli. Protein Expr Purif. 2007;51:283–292. doi: 10.1016/j.pep.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, et al. Characterization of the cocaine binding site on the sigma-1 receptor. Biochemistry. 2007;46:3532–3542. doi: 10.1021/bi061727o. [DOI] [PubMed] [Google Scholar]
- 23.Pal A, et al. Identification of regions of the sigma-1 receptor ligand binding site using a novel photoprobe. Mol Pharmacol. 2007;72:921–933. doi: 10.1124/mol.107.038307. [DOI] [PubMed] [Google Scholar]
- 24.Guo LW, et al. Sulfhydryl-reactive, cleavable, and radioiodinatable benzophenone photoprobes for study of protein-protein interaction. Bioconjug Chem. 2005;16:685–693. doi: 10.1021/bc050016k. [DOI] [PubMed] [Google Scholar]
- 25.Pal A, et al. Juxtaposition of the steroid binding domain-like I and II regions constitutes a ligand binding site in the sigma-1 receptor. J Biol Chem. 2008;283:19646–19656. doi: 10.1074/jbc.M802192200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontanilla D, et al. Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry. 2008;47:7205–7217. doi: 10.1021/bi800564j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganapathy ME, et al. Molecular and ligand-binding characterization of the sigma-receptor in the Jurkat human T lymphocyte cell line. J Pharmacol Exp Ther. 1999;289:251–260. [PubMed] [Google Scholar]
- 28.Seth P, et al. Expression pattern of the type 1 sigma receptor in the brain and identity of critical anionic amino acid residues in the ligand-binding domain of the receptor. Biochim Biophys Acta. 2001;1540:59–67. doi: 10.1016/s0167-4889(01)00117-3. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto H, et al. Amino acid residues in the transmembrane domain of the type 1 sigma receptor critical for ligand binding. FEBS Lett. 1999;445:19–22. doi: 10.1016/s0014-5793(99)00084-8. [DOI] [PubMed] [Google Scholar]
- 30.Tam SW. (+)-[3H]SKF 10,047, (+)-[3H]ethylketocyclazocine, mu, kappa, delta and phencyclidine binding sites in guinea pig brain membranes. Eur J Pharmacol. 1985;109:33–41. doi: 10.1016/0014-2999(85)90536-9. [DOI] [PubMed] [Google Scholar]
- 31.Tam SW, Cook L. Sigma opiates and certain antipsychotic drugs mutually inhibit (+)-[3H] SKF 10,047 and [3H]haloperidol binding in guinea pig brain membranes. Proc Natl Acad Sci U S A. 1984;81:5618–5621. doi: 10.1073/pnas.81.17.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber E, et al. 1,3-Di(2-[5–3H]tolyl)guanidine: a selective ligand that labels sigma-type receptors for psychotomimetic opiates and antipsychotic drugs. Proc Natl Acad Sci U S A. 1986;83:8784–8788. doi: 10.1073/pnas.83.22.8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tam SW. Naloxone-inaccessible sigma receptor in rat central nervous system. Proc Natl Acad Sci U S A. 1983;80:6703–6707. doi: 10.1073/pnas.80.21.6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Costa BR, et al. Alterations in the stereochemistry of the kappa-selective opioid agonist U50,488 result in high-affinity sigma ligands. J Med Chem. 1989;32:1996–2002. doi: 10.1021/jm00128a050. [DOI] [PubMed] [Google Scholar]
- 35.de Costa BR, et al. Synthesis and evaluation of optically pure [3H]-(+)-pentazocine, a highly potent and selective radioligand for sigma receptors. FEBS Lett. 1989;251:53–58. doi: 10.1016/0014-5793(89)81427-9. [DOI] [PubMed] [Google Scholar]
- 36.Ablordeppey SY, et al. Is a nitrogen atom an important pharmacophoric element in sigma ligand binding? Bioorg Med Chem. 2000;8:2105–2111. doi: 10.1016/s0968-0896(00)00148-6. [DOI] [PubMed] [Google Scholar]
- 37.Glennon RA, et al. Structural features important for sigma 1 receptor binding. J Med Chem. 1994;37:1214–1219. doi: 10.1021/jm00034a020. [DOI] [PubMed] [Google Scholar]
- 38.McCann DJ, et al. Sigma-1 and sigma-2 sites in rat brain: comparison of regional, ontogenetic, and subcellular patterns. Synapse. 1994;17:182–189. doi: 10.1002/syn.890170307. [DOI] [PubMed] [Google Scholar]
- 39.Moebius FF, et al. Yeast sterol C8-C7 isomerase: identification and characterization of a high-affinity binding site for enzyme inhibitors. Biochemistry. 1996;35:16871–16878. doi: 10.1021/bi961996m. [DOI] [PubMed] [Google Scholar]
- 40.Gray NM, et al. H1 antihistamines interact with central sigma receptors. Life Sci. 1990;47:175–180. doi: 10.1016/0024-3205(90)90231-f. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen EC, et al. Involvement of sigma receptors in the acute actions of methamphetamine: receptor binding and behavioral studies. Neuropharmacol. 2005;49:638–645. doi: 10.1016/j.neuropharm.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 42.Sharkey J, et al. Cocaine binding at sigma receptors. Eur J Pharmacol. 1988;149:171–174. doi: 10.1016/0014-2999(88)90058-1. [DOI] [PubMed] [Google Scholar]
- 43.Narita N, et al. Interactions of selective serotonin reuptake inhibitors with subtypes of sigma receptors in rat brain. Eur J Pharmacol. 1996;307:117–119. doi: 10.1016/0014-2999(96)00254-3. [DOI] [PubMed] [Google Scholar]
- 44.Cagnotto A, et al. [3H](+)pentazocine binding to rat brain sigma-1 receptors. Eur J Pharmacol. 1994;266:131–138. doi: 10.1016/0922-4106(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 45.Ramachandran S, et al. The sigma-1 receptor interacts with N-alkylamines and endogenous sphingolipids. Eur J Pharmacol. 2009;609:19–26. doi: 10.1016/j.ejphar.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su TP, et al. Sigma compounds derived from phencyclidine: identification of PRE-084, a new, selective sigma ligand. J Pharmacol Exp Ther. 1991;259:543–550. [PubMed] [Google Scholar]
- 47.Matsumoto RR, et al. Characterization of two novel sigma receptor ligands: antidystonic effects in rats suggest sigma receptor antagonism. Eur J Pharmacol. 1995;280:301–310. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 48.Ferris RM, et al. Evidence that the potential antipsychotic agnet rimcazole (BW234U) is a specific, competitive antagonist of sigma sites in brain. Life Sci. 1986;38:2329–2337. doi: 10.1016/0024-3205(86)90640-5. [DOI] [PubMed] [Google Scholar]
- 49.Okuyama S, et al. NE-100, a novel sigma receptor ligand: in vivo tests. Life Sci. 1993;53:PL285–290. doi: 10.1016/0024-3205(93)90588-t. [DOI] [PubMed] [Google Scholar]
- 50.Glennon RA. Pharmacophore identification for sigma-1 (sigma1) receptor binding: application of the “deconstruction-reconstruction-elaboration” approach. Mini Rev Med Chem. 2005;5:927–940. doi: 10.2174/138955705774329519. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, et al. Interactions of metaphit with phencyclidine and sigma agonist actions in rat cerebellum: determination of specificity and selectivity. J Pharmacol Exp Ther. 1987;241:321–327. [PubMed] [Google Scholar]
- 52.Maurice T, et al. Behavioral evidence for a modulating role of sigma ligands in memory processes. I. Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res 6. 1994;47:44–56. doi: 10.1016/0006-8993(94)91397-8. [DOI] [PubMed] [Google Scholar]
- 53.Maurice T, et al. Sigma1 (σ1) receptor agonists and neurosteroids attenuate β25–35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience. 1998;83:413–28. doi: 10.1016/s0306-4522(97)00405-3. [DOI] [PubMed] [Google Scholar]
- 54.Okuyama S, et al. Effect of NE-100, a novel sigma receptor ligand, on phencyclidine- induced delayed cognitive dysfunction in rats. Neurosci Lett. 1995;189:60–62. doi: 10.1016/0304-3940(95)11440-8. [DOI] [PubMed] [Google Scholar]
- 55.Hayashi T, Su TP. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc Natl Acad Sci U S A. 2001;98:491–496. doi: 10.1073/pnas.98.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi T, Su TP. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc Natl Acad Sci U S A. 2004;101:14949–14954. doi: 10.1073/pnas.0402890101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palmer CP, et al. Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 2007;67:11166–11175. doi: 10.1158/0008-5472.CAN-07-1771. [DOI] [PubMed] [Google Scholar]
- 58.Ramachandran S, et al. The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur J Pharmacol. 2009 doi: 10.1016/j.ejphar.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rusinol AE, et al. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494–502. [PubMed] [Google Scholar]
- 60.Mavlyutov TA, Ruoho AE. Ligand-dependent localization and intracellular stability of sigma-1 receptors in CHO-K1 cells. J Mol Signal. 2007;2:8. doi: 10.1186/1750-2187-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayashi T, Su T-P. Subcellular localization and intracellular dynamics of σ1 receptors. In: Matsumoto RR, Bowen WD, Su T-P, editors. Sigma receptors: Chemistry, Cell Biolocy and Clinical Implications. Springer; New York: 2007. pp. 151–164. [Google Scholar]
- 62.Meunier, et al. Compensatory effect by sigma-1 receptor stimulation during alcohol withdrawal in mice performing an object recognition task. Beh Brain Res. 2006;166:166–176. doi: 10.1016/j.bbr.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 63.Soriani O, et al. A-Current down-modulated by sigma receptor in frog pituitary melanotrope cells through a G protein-dependent pathway. J Pharmacol Exp Ther. 1999;289:321–328. [PubMed] [Google Scholar]
- 64.Soriani O, et al. The sigma-ligand (+)-pentazocine depresses M current and enhances calcium conductances in frog melanotrophs. Am J Physiol. 1999;277:E73–80. doi: 10.1152/ajpendo.1999.277.1.E73. [DOI] [PubMed] [Google Scholar]
- 65.Hayashi T, Su T. The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr Neuropharmacol. 2005;3:267–280. doi: 10.2174/157015905774322516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilke RA, et al. Sigma receptor photolabeling and sigma receptor-mediated modulation of potassium channels in tumor cells. J Biol Chem. 1999;274:18387–18392. doi: 10.1074/jbc.274.26.18387. [DOI] [PubMed] [Google Scholar]
- 67.Kennedy C, Henderson G. Inhibition of potassium currents by the sigma receptor ligand (+)-3-(3-hydroxyphenyl)-N-(1-propyl)piperidine in sympathetic neurons of the mouse isolated hypogastric ganglion. Neuroscience. 1990;35:725–733. doi: 10.1016/0306-4522(90)90343-3. [DOI] [PubMed] [Google Scholar]
- 68.Monassier L, et al. Sigma(2)-receptor ligand-mediated inhibition of inwardly rectifying K(+) channels in the heart. J Pharmacol Exp Ther. 2007;322:341–350. doi: 10.1124/jpet.107.122044. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Cuevas J. Sigma Receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J Pharmacol Exp Ther. 2005;313:1387–1396. doi: 10.1124/jpet.105.084152. [DOI] [PubMed] [Google Scholar]
- 70.Zhang H, Cuevas J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol. 2002;87:2867–2879. doi: 10.1152/jn.2002.87.6.2867. [DOI] [PubMed] [Google Scholar]
- 71.Hayashi T, et al. Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J Pharmacol Exp Ther. 2000;293:788. [PubMed] [Google Scholar]
- 72.Monnet FP, et al. N-methyl-D-aspartate-induced neuronal activation is selectively modulated by sigma receptors. Eur J Pharmacol. 1990;179:441–445. doi: 10.1016/0014-2999(90)90186-a. [DOI] [PubMed] [Google Scholar]
- 73.Monnet FP, et al. Protein kinase C-dependent potentiation of intracellular calcium influx by sigma1 receptor agonists in rat hippocampal neurons. J Pharmacol Exp Ther. 2003;307:705–712. doi: 10.1124/jpet.103.053447. [DOI] [PubMed] [Google Scholar]
- 74.Kume T, et al. Antagonism of NMDA receptors by sigma receptor ligands attenuates chemical ischemia-induced neuronal death in vitro. Eur J Pharmacol. 2002;455:91–100. doi: 10.1016/s0014-2999(02)02582-7. [DOI] [PubMed] [Google Scholar]
- 75.Hayashi T, et al. Modulation by sigma ligands of intracellular free Ca++ mobilization by N-methyl-D-aspartate in primary culture of rat frontal cortical neurons. J Pharmacol Exp Ther. 1995;275:207–214. [PubMed] [Google Scholar]
- 76.Martina M, et al. The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J Physiol. 2007;578:143–157. doi: 10.1113/jphysiol.2006.116178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langa F, et al. Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice. Eur J Neurosci. 2003;18:2188–2196. doi: 10.1046/j.1460-9568.2003.02950.x. [DOI] [PubMed] [Google Scholar]
- 78.Johannessen MA, et al. Voltage-gated sodium channel modulation by sigma receptors in cardiac myocytes and heterologous systems. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00431.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carnally SM, et al. Demonstration of a direct interaction between sigma-1 receptors and acid-sensing ion channels. Biophys J. 98:1182–1191. doi: 10.1016/j.bpj.2009.12.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Su TP, et al. When the endogenous hallucinogenic trace amine N,N-dimethyltryptamine meets the sigma-1 receptor. Sci Signal. 2009;2:pe12. doi: 10.1126/scisignal.261pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim FJ, et al. Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol. 2009;77:695–703. doi: 10.1124/mol.109.057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mavlyutov TA, et al. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience. 2010;167:247–55. doi: 10.1016/j.neuroscience.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hayashi T, et al. Regulation of sigma-1 receptors and endoplasmic reticulum chaperones in the brain of methamphetamine self-administering rats. J Pharmacol Exp Ther. 2010;332:1054–1063. doi: 10.1124/jpet.109.159244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yagasaki Y, et al. Chronic antidepressants potentiate via sigma-1 receptors the brain-derived neurotrophic factor-induced signaling for glutamate release. J Biol Chem. 2006;281:12941–12949. doi: 10.1074/jbc.M508157200. [DOI] [PubMed] [Google Scholar]
- 85.Yao H, et al. Molecular mechanisms involving sigma receptor-mediated induction of MCP-1: implication for increased monocyte transmigration. Blood. 2010;115:4951–4962. doi: 10.1182/blood-2010-01-266221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hiratani T, et al. Reinforcing effects of sigma-receptor agonists in rats trained to self-administer cocaine. J Pharmacol Exp Ther. 2010;332:515–524. doi: 10.1124/jpet.109.159236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Villard V, et al. Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative anavex1–41 against amyloid β25–35-induced toxicity in mice. Neuropsychopharmacology. 2009;34:1552–1566. doi: 10.1038/npp.2008.212. [DOI] [PubMed] [Google Scholar]
- 88.Villard V, et al. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 (σ1) ligand ANAVEX2–73, a novel aminotetrahydrofuran derivative. J Psychopharmacology. 2010 doi: 10.1177/0269881110379286. in press. [DOI] [PubMed] [Google Scholar]
- 89.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 90.Uchida N, et al. A variant of the sigma receptor type-1 gene is a protective factor for Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:1062–1066. doi: 10.1176/appi.ajgp.13.12.1062. [DOI] [PubMed] [Google Scholar]
- 91.Marrazzo A, et al. Neuroprotective effects of sigma-1 receptor agonists against β-amyloid-induced toxicity. Neuroreport. 2005;16:1223–1226. doi: 10.1097/00001756-200508010-00018. [DOI] [PubMed] [Google Scholar]
- 92.Meunier J, et al. The anti-amnesic and neuroprotective effects of donepezil against amyloid β25–35 peptide-induced toxicity in mice involve an interaction with the σ1 receptor. Br J Pharmacol. 2006;149:998–1012. doi: 10.1038/sj.bjp.0706927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li L, et al. DHEA prevents Aβ25–35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology. 2010 Feb 16; doi: 10.1016/j.neuropharm.2010.02.009. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 94.Harukuni I, et al. sigma1-receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine affords neuroprotection from focal ischemia with prolonged reperfusion. Stroke. 2000;31:976–982. doi: 10.1161/01.str.31.4.976. [DOI] [PubMed] [Google Scholar]
- 95.Goyagi T, et al. Neuroprotective effect of sigma1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is linked to reduced neuronal nitric oxide production. Stroke. 2001;32:1613–1620. doi: 10.1161/01.str.32.7.1613. [DOI] [PubMed] [Google Scholar]
- 96.Ajmo CT, Jr , et al. Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats. Curr Neurovasc Res. 2006;3:89–98. doi: 10.2174/156720206776875849. [DOI] [PubMed] [Google Scholar]
- 97.Katnik C, et al. Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J Pharmacol Exp Ther. 2006;319:1355–1365. doi: 10.1124/jpet.106.107557. [DOI] [PubMed] [Google Scholar]
- 98.Shen YC, Wang YH, Chou YC, Liou KT, Yen JC, Wang WY, Liao JF. Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-mediated mechanisms by decreasing glutamate accumulation. J Neurochem. 2008;104:558–572. doi: 10.1111/j.1471-4159.2007.05058.x. [DOI] [PubMed] [Google Scholar]
- 99.Anderson TR, Andrew RD. Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol. 2002;88:2713–2725. doi: 10.1152/jn.00321.2002. [DOI] [PubMed] [Google Scholar]
- 100.Haiman G, et al. Effects of dextromethorphan/quinidine on auditory event-related potentials in multiple sclerosis patients with pseudobulbar affect. J Clin Psychopharmacol. 2009;29:444–452. doi: 10.1097/JCP.0b013e3181b5ae5c. [DOI] [PubMed] [Google Scholar]
- 101.Gekker GS, et al. Cocaine-induced HIV-1 expression in microglia involves sigma-1 receptors and transforming growth factor-beta1. Int Immunopharmacol. 2006;6:1029–1033. doi: 10.1016/j.intimp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 102.Roth MD, et al. Cocaine and sigma-1 receptors modulate HIV infection, chemokine receptors, and the HPA axis in the huPBL-SCID model. J Leukoc Biol. 2005;78:1198–1203. doi: 10.1189/jlb.0405219. [DOI] [PubMed] [Google Scholar]
- 103.Berthois Y, et al. SR31747A is a sigma receptor ligand exhibiting antitumoural activity both in vitro and in vivo. Br J Cancer. 2003;88:438–446. doi: 10.1038/sj.bjc.6600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jbilo O, et al. Purification and characterization of the human SR 31747A-binding protein. A nuclear membrane protein related to yeast sterol isomerase. J Biol Chem. 1997;272:27107–27115. doi: 10.1074/jbc.272.43.27107. [DOI] [PubMed] [Google Scholar]
- 105.Casellas P, et al. Immunopharmacological profile of SR 31747: in vitro and in vivo studies on humoral and cellular responses. J Neuroimmunol. 1994;52:193–203. doi: 10.1016/0165-5728(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 106.Vilner BJ, et al. Cytotoxic effects of sigma ligands: sigma receptor-mediated alterations in cellular morphology and viability. J Neurosci. 1995;15:117–134. doi: 10.1523/JNEUROSCI.15-01-00117.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang B, et al. Expression of sigma 1 receptor in human breast cancer. Breast Cancer Res Treat. 2004;87:205–214. doi: 10.1007/s10549-004-6590-0. [DOI] [PubMed] [Google Scholar]
- 108.Spruce BA, et al. Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res. 2004;64:4875–4886. doi: 10.1158/0008-5472.CAN-03-3180. [DOI] [PubMed] [Google Scholar]
- 109.Cardenas C, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]